Abstract
Depending on the reaction conditions, N,N’,N’’-tris(benzylamino)guanidinium salts can react with carboxylic acid chlorides to form either symmetrical N,N’,N’’-tris(N-acyl-N-benzylamido)guanidines 6 or mesoionic 4-amino-1,2,4-triazolium-3-hydrazinides 7. The latter were converted into 1,2,4-triazolium salts by protonation or methylation at the hydrazinide nitrogen atom. Neutral 1,2,4-triazoles 10 were obtained by catalytic hydrogenation of an N-benzyl derivative. Crystal structure analyses of a 4-benzylamino-1,2,4-triazolium-3-hydrazinide and of two derived 1,2,4-triazolium salts are presented.

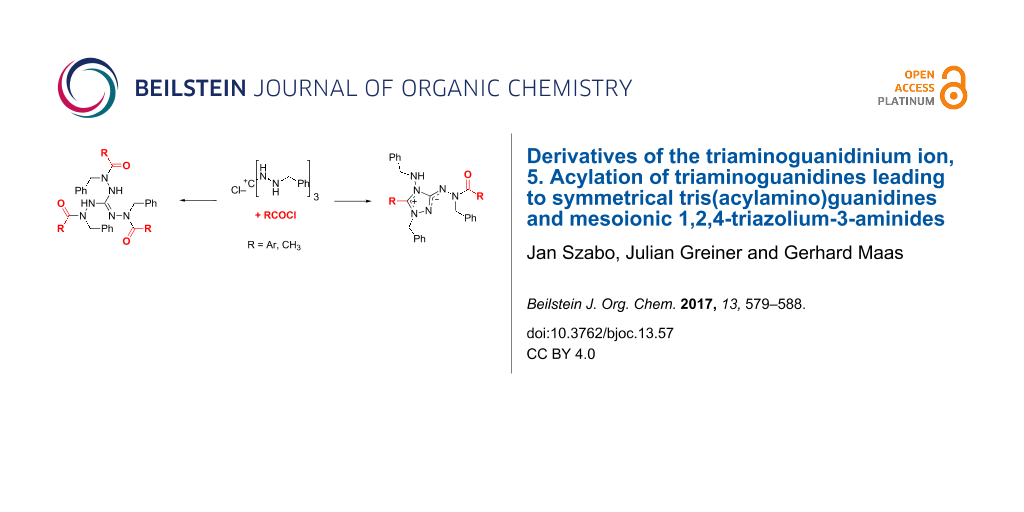
Graphical Abstract
Introduction
Easily accessible by the reaction of guanidinium chloride with hydrazine [2], triaminoguanidinium chloride (TAG-Cl) offers the opportunity to serve as a C3-symmetrical platform for the synthesis of triaminoguanidines with multiple functionalization (see [3] and references cited there). In most studies reported so far, triaminoguanidinium salts or the neutral triaminoguanidine have been reacted with carbonyl compounds. Reactions with aldehydes [3-7] or ketones [8-10] yielded the corresponding tris(iminyl)guanidines; cyclocondensation of pentane-2,4-dione at one hydrazinyl branch of TAG-Cl afforded a pyrazole which went on to a 3,6-di(pyrazol-1-yl)-1,2,4,5-tetrazine [11], and with 1,1,1-trifluoro-2,4-pentanedione two branches of TAG-Cl were converted into pyrazoline moieties and the third one into an enaminone [12].
Not much is known about the acylation of triaminoguanidines. Reactions with carboxylic acids have been addressed only rarely, reactions with acid chlorides appear to be unknown. We have recently reported on the threefold carbamoylation of N,N’,N’’-tris(benzylamino)guanidinium salts with aryl isocyanates [3]. Concerning the reaction with carboxylic acids, it is known that TAG-Cl and formic acid on heating yield 3-hydrazinyl-4-amino-4H-1,2,4-triazole hydrochloride (I, R = H) (Scheme 1) [13]. With the higher homologs of formic acid, the authors of that study observed the formation of resinous materials only. In a recent paper, however, evidence for the formation of the corresponding derivatives of I (R = Me, CF3, Ph, ClCH2) in good yields was presented, although they were transformed further without isolation [14]. An old observation with a long-lasting impact was made by M. Busch who studied the reaction of formic acid with triphenylaminoguanidine: the originally assumed bicyclic constitution of the reaction product, called “nitron” [15,16], was much later recognized [17] and structurally confirmed [18] as the mesoionic 1,2,4-triazolium-3-aminide II (Scheme 1). “Nitron” became known as an analytical reagent for the quantitative determination of nitrate [16,19], perchlorate, and some metals [20].
![[1860-5397-13-57-i1]](/bjoc/content/inline/1860-5397-13-57-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of aminoguanidines with carboxylic acids and acid chlorides. The structural formulae shown for II and III, suggesting a positively charged heteroaromatic ring system, are discontinued nowadays, because they do not represent the bond structure appropriately (vide infra).
Scheme 1: Reactions of aminoguanidines with carboxylic acids and acid chlorides. The structural formulae show...
In subsequent studies, Busch and co-workers found that a variety of 5-substituted 1,2,4-triazolium-3-aminides III (Scheme 1) can be prepared either in one step from triphenylaminoguanidine and acid chlorides at elevated temperatures or in two steps using an aldehyde and subsequent oxidation [21,22]. Several decades later, a range of diversely substituted 1,2,4-triazolium-3-aminides were prepared by condensation of N-amino-N-R-N’-phenylbenzamidines with phenylisocyanide dichloride [23] or 4-aryl-1,1-dibromo-2,3-diazabutadienes [24].
The 1,2,4-triazolium-3-aminides II and III belong to the family of mesoionic five-membered heterocycles, of which the sydnones (1,2,3-oxadiazolium-5-olates), sydnone imines (1,2,3-oxadiazolium-5-aminides), and 1,3,4-thiadiazolium derived mesoionics are perhaps the best known members [25,26]. While not much is known about congeners of II, diverse biological activities have been reported for some of the other types of mesoionic heterocycles. For example, antitumor [27-29], antileishmanial [30] and trypanocidal [31] activities, as well as reduction of the phosphorylation efficiency of rat liver mitochondria [32], have been described for 1,3,4-thiadiazolium-2-phenylaminides.
We present now the results of our studies on the acylation of triaminoguanidines with carboxylic acid chlorides. Further, we show that N,N’,N’’-tris(benzylamino)guanidine reacts with acid chlorides to afford either the threefold N-acylation product or a mesoionic 1,2,4-triazolium-3-aminide, depending on the reaction conditions. Because of the long-standing interest in the bond structure of mesoionic compounds, we have also conducted some structural and spectroscopic studies of the new 1,2,4-triazolium-3-aminides and the 3-amino-1,2,4-triazolium salts derived thereof [33].
Results and Discussion
Acylation of triaminoguanidine
Triaminoguanidine is highly soluble in water [34], and triaminoguanidinium chloride (TAG-Cl, 1) is soluble in hot water or water/ethanol mixtures, but both are insoluble or sparingly soluble in common organic solvents. Since aqueous media cannot be avoided, these properties pose an obstacle to acylation reactions, which therefore are expected to succeed only with acylating reagents less sensitive to hydrolysis. We found indeed, that triaminoguanidine, generated in situ by deprotonation of 1 in strongly alkaline aqueous solution, could be acylated effectively with 3,4,5-trimethoxybenzoyl chloride (2b) to form the N,N’,N’’-tris(acylamino)guanidinium chloride 3 (Scheme 2). Hydrolysis of the acyl chloride is not a competitive reaction under these conditions, and formation of a 1,2,4-triazole, as observed for the reaction of 1 with formic acid at reflux conditions (see Scheme 1) also does not occur.
![[1860-5397-13-57-i2]](/bjoc/content/inline/1860-5397-13-57-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Threefold N-acylation of triaminoguanidinium chloride (1) with acyl chloride 2b.
Scheme 2: Threefold N-acylation of triaminoguanidinium chloride (1) with acyl chloride 2b.
On the other hand, the analogous acylation reaction could not be performed with benzoyl chloride and acetyl chloride under various conditions, including the application of weaker bases (NEt3, Na2CO3) in dioxane and other aprotic-polar solvents.
Reaction of 1,2,3-tris(benzylamino)guanidinium chloride with acyl chlorides
As an example of N,N’,N’’-tris(alkylamino)guanidines, we have recently described the synthesis of N,N’,N’’-tris(benzylamino)guanidinium chloride (4) [3]. Since the direct alkylation of TAG-Cl (1) was not possible due to the solubility problem mentioned above, a two-step protocol – conversion of 1 into N,N’,N’’-tris(benzylideneamino)guanidinium chloride followed by catalytic hydrogenation of the imine groups – was developed. The fluorophenyl-substituted salt 5 was prepared analogously. Depending on the reaction conditions, the guanidinium salts 4 and 5 were found to react with acid chlorides 2 in two different ways (Scheme 3). When the acid chloride was added slowly to a solution of the guanidinium salt in chloroform in the presence of solid sodium carbonate, the expected N,N’,N’’-tris(acylamino)guanidines 6 were formed as the major products. Both the use of a weak base and a mild reaction temperature favor the formation of 6 at the expense of competitively formed mesoionic compounds 7. This can be seen for the acylation of 4 with benzoyl chloride, where an elevated temperature was required for a sufficiently fast conversion and a mixture of 6a and 7a was obtained (Scheme 3 and Table 1).
![[1860-5397-13-57-i3]](/bjoc/content/inline/1860-5397-13-57-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 1,2,3-tris(benzylamino)guanidinium salts 4 and 5 with acyl chlorides to give 1,2,3-tris(acylamino)guanidines 6 and mesoionic 1,2,4-triazolium-3-aminides 7. See Table 1 for Ar, R and yields of 7.
Scheme 3: Reaction of 1,2,3-tris(benzylamino)guanidinium salts 4 and 5 with acyl chlorides to give 1,2,3-tris...
Table 1: 1,2,4-Triazolium-3-aminides 7 synthesized (see Scheme 3).
| Product | Ar | R | Method | Yield (%) |
|---|---|---|---|---|
| 7a | Ph | Ph | A | 47a |
| 7b | Ph | 3,4,5-(OMe)3-C6H2 |
A
B |
84
58 |
| 7c | Ph | 4-NO2-C6H4 |
Ab
B |
41b
50 |
| 7d | Ph | CH3 |
A
B |
48
70 |
| 7e | 4-F-C6H4 | CH3 | A | 75 |
aWhen the reaction 4 + 2a was performed in CHCl3 at 70 °C with Na2CO3 as the base, 7a could be isolated in ≈7% yield. bK2CO3 instead of NaOH; 6c (21%) was also obtained.
On the other hand, when the acylation reactions were conducted at elevated temperature in the presence of a stronger base (aqueous NaOH), mesoionic (1H-1,2,4-triazol-4-ium-3-yl)hydrazin-1-ides 7a–e were formed in moderate to good yields (Scheme 3, method A; Table 1). With easily hydrolyzing 4-nitrobenzoyl chloride (2c), the alkaline medium had to be replaced by solid potassium carbonate, which resulted, however, in a separable mixture of 6c and 7c. It should be emphasized that, although the two procedures for the acylation of guanidinium salts 4 and 5 have been optimized to give either 6 or 7 selectively, the undesired product could be detected in some cases by characteristic signals in the 1H NMR spectra of the crude product mixtures.
It is reasonable to assume that the formation of betaines 7 according to method A (Scheme 3) proceeds via the neutral guanidine derivatives 6 as intermediates. In fact, the latter compounds are converted into betaines 7 under the same reaction conditions (method B). In terms of yields, however, the direct, one-step route (method A) turned out to be the better choice, as was found for betaines 7b–d (Table 1).
All reactions shown in Scheme 3 were performed in the absence of air, since salts 4 and 5 suffer a partial oxidative degradation in the presence of oxygen under alkaline conditions. In fact, 3,4,5-trimethoxybenzamide was isolated in 20% yield when the reaction of 4 and 2b (aq NaOH 5 M, CH3CN, 70 °C) was performed in the presence of air.
Similar to structurally related guanidines (N-ureido instead of N-acyl substitution [3]) and to N,N’,N’’-tris(isopropylideneamino)guanidine [10], the 1H NMR signals of tris(acylamino)guanidines 6 are in coalescence over a wide temperature range, without reaching the fast exchange regime up to 350 K. As an example, a 1H NMR spectrum of 6b, recorded at 298 K, is shown in Figure S1 (Supporting Information File 1). This is a result of several dynamic processes, namely conformational and prototropic equilibria. The 13C NMR spectra of 6a–d, on the other hand, indicate a static, unsymmetrical molecule around 300 K, showing for example separate signals for the three CH2 and Ccarbonyl nuclei.
A single-crystal X-ray structure determination was performed for 6b, which gave suitable crystals of 6b·2C2H5OH when crystallized from ethanol. The guanidine structure could so be confirmed (Figure 1). It is interesting to note the unsymmetrical shape of the molecule: two benzyl groups are found above and one below the plane defined by the CN3 core of the guanidine. This is in contrast to the structure of the related 1,2,3-tris(1-benzyl-3-phenylureido)guanidine [3], where all three benzyl groups are placed on one side and the three polar ureido branches, interconnected by N–H···O hydrogen bonds, on the other side of the CN3 plane. In 6b, on the other hand, the two guanidine N–H bonds and one of the carbonyl oxygen atoms are involved in hydrogen bonds to ethanol molecules.
![[1860-5397-13-57-1]](/bjoc/content/figures/1860-5397-13-57-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 6b·2C2H5OH in the solid state, with numbering of atoms (ORTEP plot). Selected bond lengths (Å): C1–N1 1.297(4), C1–C2 1.390(4), C1–N3 1.365(4). Selected bond angles (°): N1–C1–N2 117.3(3), N2–C1–N3 115.6(2), N1–C1–N3 126.9(3). Hydrogen bonds to the two ethanol molecules: N2–H···O13: N2···O13 3.060(1) Å, NH···O13 1.96(2) Å; N3–H···O14(1.5−x, y−0.5, 0.5−z): N3···O14 2.917(4) Å, NH···O14 2.11(2) Å; O13–H···O5(−x+1, -y, −z+1): O13···O5 2.733(3) Å, (O13–)H···O5 1.97 Å; O14–H···O1: O14···O1 2.846(4) Å, (O14–)H···O1 2.07(6) Å.
Figure 1: Molecular structure of 6b·2C2H5OH in the solid state, with numbering of atoms (ORTEP plot). Selecte...
The mesoionic 1,2,4-triazolium-3-aminides 7a–e are high-melting solids, the colors of which range from colorless (7e) to orange-red (7c). Their bond structure and characteristic spectroscopic data are discussed in the next section. A charge distribution as shown in the molecular formula (Scheme 3) is generally considered as being typical for these and related mesoionic compounds (see the next chapter). It was therefore expected that betaines 7 would be attacked by electrophiles at the exocyclic nitrogen atom bearing excess π-electron density. In fact the protonation of 7b,c with hydrochloric acid gave rise to the 3-hydrazinyl-1,2,4-triazolium chlorides 8b,c (Scheme 4) as well-crystallizing colorless or pale yellow solids. Analogously, the reaction of 7b with methyl triflate afforded the 3-(1-methylhydrazinyl)-1,2,4-triazolium triflate 9b by methylation of the exocyclic aminide nitrogen atom of the betaine; in contrast to other 1,2,4-triazolium-3-aminides [21,23], methylation with methyl iodide was not successful. Salt 9b forms colorless hygroscopic crystals which assume an orange surface color when exposed to air for some minutes. The structures of 1,2,4-triazolium salts 8b and 9b were confirmed by single-crystal X-ray diffraction (vide infra).
![[1860-5397-13-57-i4]](/bjoc/content/inline/1860-5397-13-57-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Protonation and methylation of 1,2,4-triazolium-3-aminides 7b,c.
Scheme 4: Protonation and methylation of 1,2,4-triazolium-3-aminides 7b,c.
Catalytic hydrogenation of 1,2,4-triazolium-3-aminides 7 with H2 and Pd/C in methanol selectively cleaves the N1–Cbenzyl bond and yields the neutral N-benzyl-N’-(4-benzylamino-4H-1,2,4-triazol-3-yl)benzohydrazides 10 in high yields (Scheme 5). In the case of 7c, the nitro groups are concomitantly reduced to NH2 groups and the bis(aminophenyl) derivative 10c is obtained. Thus, highly substituted and functionalized 1,2,4-triazoles can easily be prepared from TAG-Cl (1) in four steps.
![[1860-5397-13-57-i5]](/bjoc/content/inline/1860-5397-13-57-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Catalytic hydrogenation/debenzylation of betaines 7.
Scheme 5: Catalytic hydrogenation/debenzylation of betaines 7.
Solid-state structures of salts 8b and 9b
The crystal structure of salt 8b (Figure 2) shows a nearly planar core of the molecule (the triazole ring, N4, N5, N6). One face of this core is occupied by the three benzylic substituents, leaving the other face open for the chloride anion which is held in place by two N–H···Cl hydrogen bonds, one of them being very short (d(H···Cl) = 2.11(2) Å, d(N···Cl) = 3.060(1) Å) and linear and the other one much longer and non-linear. Short N–H···Cl hydrogen bonds have also been found in the crystal structures of related compounds, such as II·HCl·CH3OH (d(H···Cl) = 2.36 Å, d(N···Cl) = 3.15 Å [35]) and a 2-phenylamino-1,3,4-thiadiazolium chloride (2.05/3.00 Å [36]). In the packing arrangement of the crystal, the chloride ions are found in anion channels oriented along the 21 screw axis parallel to the crystallographic a-axis of the orthorhombic unit cell (space group P212121).
![[1860-5397-13-57-2]](/bjoc/content/figures/1860-5397-13-57-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Left: molecular structure of 8b in the solid state (OLEX2 plot). Right: crystal structure viewed along the crystallographic a-axis. Selected bond lengths (Å): C1–N1 1.317(2), C1–N3 1.372(2), C1–N5 1.349(2), N1–N2 1.388(2), C2–N2 1.319(2), C2–N3 1.352(2), N3–N4 1.412(2). Hydrogen bonds: N5–H···Cl: N5···Cl 3.060(1) Å, NH···Cl 2.11(2) Å, <(N5–H···Cl) 176(2)°; N4–H···Cl, N4···Cl 3.320(2) Å, NH···Cl 2.53(2) Å, <(N4–H···Cl) 148(2)°.
Figure 2: Left: molecular structure of 8b in the solid state (OLEX2 plot). Right: crystal structure viewed al...
The molecular shape of the cation of 1,2,4-triazolium triflate 9b in the solid state (Figure 3) is similar to that of 8b. However, the uptake of one water molecule per formula unit has resulted in different hydrogen bond patterns. A centrosymmetric hydrogen-bonded dimer (CF3SO3·H2O)2 is present which is connected to two cations through hydrogen bonds with N4–H as the donor and the oxygen atom of water as the acceptor (Figure 3, right).
![[1860-5397-13-57-3]](/bjoc/content/figures/1860-5397-13-57-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Left: solid-state structure of 9b·H2O (ORTEP plot). Right: centrosymmetric hydrogen-bonded dimer of (CF3SO3)−·H2O (Mercury plot); the substituents at the triazole ring are not complete. Selected bond lengths (Å): C1–N1 1.309(3), C1–N3 1.376(3), C1–N5 1.376(3), N1–N2 1.378(3), C2–N2 1.314(3), C2–N3 1.366(3), N3–N4 1.400(3). Hydrogen bonds: N4–H···O11, N4–H 0.89(4) Å, N···O11 2.946(3) Å, H···O11 2.22(4) Å, <(N4–H···O11) 139(3)°; O11–Hb···O8, O11–Hb 1.01 Å, O11···O8 3.023(4) Å, Hb···O8 2.02 Å, <(O11–Hb···O8) 170°; O11–Ha···O8 (1−x, 1−y, 1−z), O11–Ha 0.95 Å, O11···O8 2.979(3) Å, Ha···O8 2.05 Å, <(O11–Ha···O8) 169°.
Figure 3: Left: solid-state structure of 9b·H2O (ORTEP plot). Right: centrosymmetric hydrogen-bonded dimer of...
Structural and spectroscopic characterization of 1,2,4-triazolium-3-aminides 7
The bond structure of mesoionic compounds such as 7 can be described as resonance hybrids of quite a number of canonical structures. Depending on the definition of mesoionic compounds, two resonance hybrid formulations are frequently found in the literature: the one displayed in Scheme 1, which assumes the aromatic electron delocalization over the positively charged heterocyclic ring [37], and the one used in the rest of this paper, which is based on a more recent definition [38] and that divides the mesoionic system into two oppositely charged resonance-stabilized moieties (here: an amidinium/amidinate combination). These two moieties are separated by bonds having high single-bond character. The latter concept is supported inter alia by recent theoretical studies using DFT calculations, Natural Bond Orbital analysis and Natural Resonance Theory calculations for mesoionic systems of the 1,3-oxazole, 1,3-diazole and 1,3-thiazole type [39].
As 1,2,4-triazolium-3-aminides have not yet been studied in depth with respect to their structural and electronic properties, we herein present some relevant data of the 1,2,4-triazolium-hydrazinides 7. Thus, the molecular structure of 7a in the solid state was determined by X-ray diffraction analysis and is shown in Figure 4. Two symmetrically independent molecules are present in the triclinic unit cell, being associated by two N–H···N hydrogen bonds in which benzylamino NH bonds act as donors and aminide nitrogen atoms as acceptors.
![[1860-5397-13-57-4]](/bjoc/content/figures/1860-5397-13-57-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Solid-state structure of mesoionic compound 7a (ORTEP plot); thermal displacement ellipsoids are drawn at the 30% probability level. Selected bond lengths (Å): C1–N1 1.343(1), C1–N3 1.398(1), C1–N5 1.322(1), N1–N2 1.389(1), C2–N2 1.317(1), C2–N3 1.357(1); selected torsion angles (°): N2–N1–C1–N5 177.7(1), N3–C2–C10–C11 -42.8(2). Right: Hydrogen bonds between two symmetrically not related molecules of 7a are shown; N10···N5 2.939(2) Å, N10H···N5 2.057(1) Å, <(N10–H···N5) 167.0(2)°; N4···N11 2.969(2) Å, N4H···N11 2.061(1) Å; <(N4–H···N11) 168.7(2)°.
Figure 4: Solid-state structure of mesoionic compound 7a (ORTEP plot); thermal displacement ellipsoids are dr...
As expected, the triazolium-aminide moiety in 7a is planar. The comparison of the bond lengths in this moiety with those in nitron II reveals quite similar values for corresponding bonds (Table 2). The data are in agreement with the description of mesoionic compounds 7 as a combination of an amidinium part with delocalized positive charge and an amidinate part incorporating the negative charge [38]. The separation between these two moieties is indicated by the long C–N bond “c” (as compared to the other C–N bonds in the triazole ring) and the endocyclic N–N bond, both of which have a high single-bond character. The influence of the substituents on the triazole ring of II and 7a on the bond lengths is moderate. The bond geometry of the parent 1,2,4-triazolium-3-aminide has been calculated by quantum chemical ab initio methods [40]. The bond length differences are in qualitative agreement with the experimentally determined values for 7a. On protonation or methylation of the exocyclic aminide nitrogen atom, the mesoionic moiety is converted into a 3-hydrazinyl-substituted 1,2,4-triazolium ion with a delocalized 6π-electron aromatic system. This affects significantly (≥0.22 Å) the three bonds around ring atom C-3, where the two endocyclic bonds are shortened and the exocyclic one is expanded.
Table 2: Selected bond lengths (Å) in 1,2,4-triazolium-3-aminides II and 7a and 1,2,4-triazolium salts 8b and 9b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-57-i6.svg?max-width=637&scale=1.0)
|
||||
| Bond | II a | 7a | 8b | 9b |
| a | 1.40–1.41 | 1.389(1) | 1.388(2) | 1.378(2) |
| b | 1.35–1.36 | 1.343(1) | 1.317(2) | 1.309(3) |
| c | 1.41–1.43 | 1.398(1) | 1.372(2) | 1.376(3) |
| d | 1.32–1.35 | 1.357(1) | 1.352(2) | 1.366(3) |
| e | 1.33 | 1.317(1) | 1.319(2) | 1.314(3) |
| f | 1.31–1.33 | 1.322(1) | 1.349(2) | 1.376(3) |
a Lit. [18]; two symmetrically independent molecules.
In the 13C NMR spectra of 1,2,4-triazolium-3-aminides 7a–e, the chemical shifts of ring atoms C-3 and C-5 are of interest (Table 3; for ab initio GIAO-CHF calculations of chemical shifts of the parent 1,2,4-triazolium-3-aminide, see lit. [40]). The C-3 chemical shifts of 7a–e are equal within 1.14 ppm and are in the typical range for guanidines and guanidinium salts. Notably, the chemical shifts of the amidinate carbon atom C-3 in 7a–d appear at δ-values that are higher by 2.98–7.07 ppm than those of the corresponding neutral guanidine derivatives 6a–d, in spite of the formal negative charge in the amidinate unit. In contrast, the δ(C-5) values vary strongly with the substituent attached to this carbon atom. Whereas the values for 7a–c reflect the different interaction of the phenyl, 3,4,5-trimethoxyphenyl and 4-nitrophenyl substituents with the positive charge density at C-5 through mesomeric and inductive effects, the δ(C-5) value in 7d corresponds to the deshielding α-effect of a methyl group, if one takes the δ-value of the unsubstituted C-5 carbon nucleus in II as a reference.
Table 3: 13C chemical shifts of C-3 and C-5 (triazole ring) of mesoionic compounds 7a–e and II, triazolium salt 8b and triazole 10ba.
| Comp. | R | δ(13C) [ppm] | |
|---|---|---|---|
| C-3 ((CN3)−) | C-5 ((NCN)+) | ||
| 7a | Ph | 159.57 | 120.69 |
| 7b | 3,4,5-(CH3O)3-C6H2 | 159.44 | 115.62 |
| 7c | 4-NO2-C6H4 | 159.83 | 126.39 |
| 7d | CH3 | 158.69 | 145.23 |
| 7e | CH3 | 158.84 | 144.96 |
| IIb | 153.9 | 133.5 | |
| 8b | 152.54 | 112.92 | |
| 10b | 153.60 | 122.66 | |
aSpectra were measured in [D6]DMSO solution. bLit. [41].
The UV–vis spectra of 7a–d are presented in Figure 5. One notes an increasingly bathochromic shift of the long-wavelength absorption maximum in the series R = CH3 < phenyl ≈ 3,4,5-trimethoxyphenyl < 4-nitrophenyl. For 7a–c, this absorption band could result from a charge transfer between the amidinate moiety, representing the HOMO of the mesoionic system, and an unoccupied π-orbital of the C(=O)Ar group.
![[1860-5397-13-57-5]](/bjoc/content/figures/1860-5397-13-57-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: UV–vis spectra of 7a–d in chloroform (c = 0.04 mmol L−1); λmax [nm] (ε [L mol−1 cm−1]): 7a: 350 (4710); 7b: 350 (5080); 7c: 430 (3510); 7d: 300 (2670).
Figure 5: UV–vis spectra of 7a–d in chloroform (c = 0.04 mmol L−1); λmax [nm] (ε [L mol−1 cm−1]): 7a: 350 (47...
Upon N-protonation or N-methylation, the mesoionic system loses its betainic character and a 1,2,4-triazolium ion is formed. This is accompanied by the disappearance of the long-wavelength absorption in the electronic spectra, leaving colorless salts 8.
Conclusion
We have found that triaminoguanidine, generated from its hydrochloride (TAG-Cl) in aqueous alkaline solution, can be triply acylated to give 1,2,3-tris(acylamino)guanidinium salts only with acyl chlorides that are not easily hydrolyzed. On the other hand, 1,2,3-tris(benzylamino)guanidinium chloride underwent a threefold N-acylation under carefully controlled conditions (typically with a weak base and at room temperature) with aroyl chlorides and with acetyl chloride. Under different conditions (higher temperature, alkaline medium), mesoionic 1,2,4-triazolium-3-hydrazinides were obtained as the major products. The latter compounds were converted into 3-hydrazinyl-1,2,4-triazolium salts by protonation or methylation at the anionic hydrazinide nitrogen atom and into highly substituted and functionalized 1,2,4-triazoles by N-debenzylation through catalytic hydrogenation. Thus, the reaction of triaminoguanidine and its 1,2,3-tribenzyl derivative with acid chlorides gives access to diverse chemical structures which could be considered for further studies, e.g., the biological activity of the triazole-based compounds and the use of threefold symmetrically substituted triaminoguanidines as novel hosts in supramolecular chemistry or as ligands in coordination chemistry.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data for synthesized compounds and the data for the X-ray crystal structure determinations. | ||
| Format: PDF | Size: 704.0 KB | Download |
References
-
Szabo, J.; Maas, G. Z. Naturforsch. 2016, 71b, 697–703. doi:10.1515/znb-2016-0035
-
Weiss, S.; Krommer, H. Verfahren zur herstellung von Triaminoguanidin-Salzen. DE3341645A1, May 30, 1985.
Chem. Abstr. 1986, 104, 206730.
Return to citation in text: [1] -
Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Stollé, R. Ber. Dtsch. Chem. Ges. 1904, 37, 3548–3549. doi:10.1002/cber.190403703183
Return to citation in text: [1] -
Scott, F. L.; Cashman, M.; Reilly, J. J. Am. Chem. Soc. 1952, 74, 5802. doi:10.1021/ja01142a603
Return to citation in text: [1] -
Müller, I. M.; Robson, R. Angew. Chem. 2000, 112, 4527–4530. doi:10.1002/1521-3757(20001201)112:23<4527::AID-ANGE4527>3.0.CO;2-A
Angew. Chem. Int. Ed. 2000, 39, 4357-4359. doi:10.1002/1521-3773(20001201)39:23<4357::AID-ANIE4357>3.0.CO;2-0
Return to citation in text: [1] -
Müller, I. M.; Möller, D.; Föcker, K. Chem. – Eur. J. 2005, 11, 3318–3324. doi:10.1002/chem.200401260
Return to citation in text: [1] -
Plass, W.; El-Tabl, A. S.; Pohlmann, A. J. Coord. Chem. 2009, 62, 358–372. doi:10.1080/00958970802279790
Return to citation in text: [1] -
Zelenin, K. N.; Saminskaya, A. G.; Kuznetsova, O. B. Zh. Obshch. Khim. 1996, 66, 141–146.
Return to citation in text: [1] -
Szabo, J.; Maas, G. Z. Naturforsch. 2013, 68b, 207–213. doi:10.5560/znb.2013-3023
Return to citation in text: [1] [2] -
Coburn, M. D.; Buntain, G. A.; Harris, B. W.; Hiskey, M. A.; Lee, K.-Y.; Ott, D. G. J. Heterocycl. Chem. 1991, 28, 2049–2050. doi:10.1002/jhet.5570280844
Return to citation in text: [1] -
Tolshchina, S. G.; Ignatenko, N. K.; Slepukhin, P. A.; Ishmetova, R. I.; Rusinov, G. L. Chem. Heterocycl. Compd. 2010, 46, 691–698. doi:10.1007/s10593-010-0571-5
Return to citation in text: [1] -
Kröger, C.-F.; Etzold, G.; Beyer, H. Justus Liebigs Ann. Chem. 1963, 664, 146–155. doi:10.1002/jlac.19636640112
Return to citation in text: [1] -
Cardillo, P.; Dellavedova, M.; Gigante, L.; Lunghi, A.; Pasturenzi, C.; Salatelli, E.; Zanirato, P. Eur. J. Org. Chem. 2012, 2012, 1195–1201. doi:10.1002/ejoc.201101450
Return to citation in text: [1] -
Busch, M. Ber. Dtsch. Chem. Ges. 1905, 38, 856–860. doi:10.1002/cber.190503801148
Return to citation in text: [1] -
Busch, M. Ber. Dtsch. Chem. Ges. 1905, 38, 861–866. doi:10.1002/cber.190503801149
Return to citation in text: [1] [2] -
Baker, W.; Ollis, W. D. Q. Rev., Chem. Soc. 1957, 11, 15–29. doi:10.1039/qr9571100015
Return to citation in text: [1] -
Cannon, J. R.; Raston, C. L.; White, A. H. Aust. J. Chem. 1980, 18, 2237–2247. doi:10.1071/CH9802237
Return to citation in text: [1] [2] -
Gutbier, A. Angew. Chem. 1905, 18, 494–499. doi:10.1002/ange.19050181305
Return to citation in text: [1] -
The Merck Index, 10th ed.; Merck & Co, Inc.: Rahway, NJ, U.S.A., 1983.
No. 6459.
Return to citation in text: [1] -
Busch, M.; Mehrtens, G. Ber. Dtsch. Chem. Ges. 1905, 38, 4049–4068. doi:10.1002/cber.19050380477
Return to citation in text: [1] [2] -
Busch, M. J. Prakt. Chem. 1906, 74, 533–549. doi:10.1002/prac.19060740136
Return to citation in text: [1] -
Ollis, W. D.; Ramsden, C. A. J. Chem. Soc., Perkin Trans. 1 1974, 638–642. doi:10.1039/p19740000638
Return to citation in text: [1] [2] -
Cawkill, E.; Ollis, W. D.; Ramsden, C. A.; Rowson, G. P. J. Chem. Soc., Perkin Trans. 1 1979, 724–731. doi:10.1039/P19790000724
Return to citation in text: [1] -
Ollis, W. D.; Ramsden, C. A. Adv. Heterocycl. Chem. 1976, 19, 1–121. doi:10.1016/S0065-2725(08)60230-5
Return to citation in text: [1] -
Kawase, M.; Sakagami, H.; Motohashi, N. The Chemistry of Bioactive Mesoionic Heterocycles. In Bioactive Heterocycles VII; Motohashi, N., Ed.; Topics in Heterocyclic Chemistry, Vol. 16; Springer: Berlin, Germany, 2009; pp 135–152. doi:10.1007/7081_2007_096
Return to citation in text: [1] -
Grynberg, N.; Gomes, R.; Shinzato, T.; Echevarria, A.; Miller, J. Anticancer Res. 1992, 12, 1025–1028.
Return to citation in text: [1] -
Grynberg, N.; Santos, A. C.; Echevarria, A. Anti-Cancer Drugs 1997, 8, 88–91. doi:10.1097/00001813-199701000-00012
Return to citation in text: [1] -
Senff-Ribeiro, A.; Echevarria, A.; Silva, E. F.; Franco, C. R. C.; Veiga, S. S.; Oliveira, M. B. M. Br. J. Cancer 2004, 91, 297–304. doi:10.1038/sj.bjc.6601946
Return to citation in text: [1] -
Rodrigues, R. F.; da Silva, E. F.; Echevarria, A.; Fajardo-Bonin, R.; Amaral, V. F.; Leon, L. L.; Canto-Cavalheiro, M. M. Eur. J. Med. Chem. 2007, 42, 1039–1043. doi:10.1016/j.ejmech.2006.12.026
Return to citation in text: [1] -
da Silva Ferreira, W.; Freire-de-Lima, L.; Saraiva, V. B.; Alisson-Silva, F.; Mendonça-Previato, L.; Previato, J. O.; Echevarria, A.; Freire de Lima, M. E. Bioorg. Med. Chem. 2008, 16, 2984–2991. doi:10.1016/j.bmc.2007.12.049
Return to citation in text: [1] -
Cadena, S. M. S. C.; Carnieri, E. G. S.; Echevarria, A.; de Oliveira, M. B. M. FEBS Lett. 1998, 440, 46–50. doi:10.1016/S0014-5793(98)01427-6
Return to citation in text: [1] -
Part of this work has been published: Szabo, J., Studien zur Reaktivität von Tris(benzylamino)guanidinium-Salzen mit Carbonylverbindungen und Isocyanaten; Doctoral Thesis, Ulm University, Ulm, 2014.
Return to citation in text: [1] -
Benjamin, L. E. J. Org. Chem. 1964, 29, 3729–3730. doi:10.1021/jo01035a528
Return to citation in text: [1] -
Hitzel, S.; Färber, C.; Bruhn, C.; Siemeling, U. Organometallics 2014, 33, 425–428. doi:10.1021/om401058e
Return to citation in text: [1] -
Cheung, K.-K.; Echevarria, A.; Galembeck, S.; Aparecida, M. A. M.; Miller, J.; Rumjanek, V. M.; Simas, A. M. Acta Crystallogr. 1992, C48, 1471–1474. doi:10.1107/S0108270192000283
Return to citation in text: [1] -
Potts, K. T. Lect. Heterocycl. Chem. 1978, 4, 35–46.
Return to citation in text: [1] -
Simas, A. M.; Miller, J.; de Athayde Filho, P. F. Can. J. Chem. 1998, 76, 869–872. doi:10.1139/v98-065
Return to citation in text: [1] [2] -
Anjos, I. C.; Vasconcellos, M. L. A. A.; Rocha, G. B. Theor. Chem. Acc. 2012, 131, 1294–1302. doi:10.1007/s00214-012-1294-8
Return to citation in text: [1] -
Wiench, J. W.; Stefaniak, L.; Tabaszewska, A.; Webb, G. A. Electron. J. Theor. Chem. 1997, 2, 71–84. doi:10.1002/ejtc.35
Return to citation in text: [1] [2] -
Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K
Return to citation in text: [1]
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 21. | Busch, M.; Mehrtens, G. Ber. Dtsch. Chem. Ges. 1905, 38, 4049–4068. doi:10.1002/cber.19050380477 |
| 23. | Ollis, W. D.; Ramsden, C. A. J. Chem. Soc., Perkin Trans. 1 1974, 638–642. doi:10.1039/p19740000638 |
| 35. | Hitzel, S.; Färber, C.; Bruhn, C.; Siemeling, U. Organometallics 2014, 33, 425–428. doi:10.1021/om401058e |
| 2. |
Weiss, S.; Krommer, H. Verfahren zur herstellung von Triaminoguanidin-Salzen. DE3341645A1, May 30, 1985.
Chem. Abstr. 1986, 104, 206730. |
| 11. | Coburn, M. D.; Buntain, G. A.; Harris, B. W.; Hiskey, M. A.; Lee, K.-Y.; Ott, D. G. J. Heterocycl. Chem. 1991, 28, 2049–2050. doi:10.1002/jhet.5570280844 |
| 21. | Busch, M.; Mehrtens, G. Ber. Dtsch. Chem. Ges. 1905, 38, 4049–4068. doi:10.1002/cber.19050380477 |
| 22. | Busch, M. J. Prakt. Chem. 1906, 74, 533–549. doi:10.1002/prac.19060740136 |
| 18. | Cannon, J. R.; Raston, C. L.; White, A. H. Aust. J. Chem. 1980, 18, 2237–2247. doi:10.1071/CH9802237 |
| 8. | Plass, W.; El-Tabl, A. S.; Pohlmann, A. J. Coord. Chem. 2009, 62, 358–372. doi:10.1080/00958970802279790 |
| 9. | Zelenin, K. N.; Saminskaya, A. G.; Kuznetsova, O. B. Zh. Obshch. Khim. 1996, 66, 141–146. |
| 10. | Szabo, J.; Maas, G. Z. Naturforsch. 2013, 68b, 207–213. doi:10.5560/znb.2013-3023 |
| 23. | Ollis, W. D.; Ramsden, C. A. J. Chem. Soc., Perkin Trans. 1 1974, 638–642. doi:10.1039/p19740000638 |
| 40. | Wiench, J. W.; Stefaniak, L.; Tabaszewska, A.; Webb, G. A. Electron. J. Theor. Chem. 1997, 2, 71–84. doi:10.1002/ejtc.35 |
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 4. | Stollé, R. Ber. Dtsch. Chem. Ges. 1904, 37, 3548–3549. doi:10.1002/cber.190403703183 |
| 5. | Scott, F. L.; Cashman, M.; Reilly, J. J. Am. Chem. Soc. 1952, 74, 5802. doi:10.1021/ja01142a603 |
| 6. |
Müller, I. M.; Robson, R. Angew. Chem. 2000, 112, 4527–4530. doi:10.1002/1521-3757(20001201)112:23<4527::AID-ANGE4527>3.0.CO;2-A
Angew. Chem. Int. Ed. 2000, 39, 4357-4359. doi:10.1002/1521-3773(20001201)39:23<4357::AID-ANIE4357>3.0.CO;2-0 |
| 7. | Müller, I. M.; Möller, D.; Föcker, K. Chem. – Eur. J. 2005, 11, 3318–3324. doi:10.1002/chem.200401260 |
| 16. | Busch, M. Ber. Dtsch. Chem. Ges. 1905, 38, 861–866. doi:10.1002/cber.190503801149 |
| 19. | Gutbier, A. Angew. Chem. 1905, 18, 494–499. doi:10.1002/ange.19050181305 |
| 38. | Simas, A. M.; Miller, J.; de Athayde Filho, P. F. Can. J. Chem. 1998, 76, 869–872. doi:10.1139/v98-065 |
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 20. |
The Merck Index, 10th ed.; Merck & Co, Inc.: Rahway, NJ, U.S.A., 1983.
No. 6459. |
| 40. | Wiench, J. W.; Stefaniak, L.; Tabaszewska, A.; Webb, G. A. Electron. J. Theor. Chem. 1997, 2, 71–84. doi:10.1002/ejtc.35 |
| 14. | Cardillo, P.; Dellavedova, M.; Gigante, L.; Lunghi, A.; Pasturenzi, C.; Salatelli, E.; Zanirato, P. Eur. J. Org. Chem. 2012, 2012, 1195–1201. doi:10.1002/ejoc.201101450 |
| 17. | Baker, W.; Ollis, W. D. Q. Rev., Chem. Soc. 1957, 11, 15–29. doi:10.1039/qr9571100015 |
| 38. | Simas, A. M.; Miller, J.; de Athayde Filho, P. F. Can. J. Chem. 1998, 76, 869–872. doi:10.1139/v98-065 |
| 13. | Kröger, C.-F.; Etzold, G.; Beyer, H. Justus Liebigs Ann. Chem. 1963, 664, 146–155. doi:10.1002/jlac.19636640112 |
| 18. | Cannon, J. R.; Raston, C. L.; White, A. H. Aust. J. Chem. 1980, 18, 2237–2247. doi:10.1071/CH9802237 |
| 39. | Anjos, I. C.; Vasconcellos, M. L. A. A.; Rocha, G. B. Theor. Chem. Acc. 2012, 131, 1294–1302. doi:10.1007/s00214-012-1294-8 |
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 36. | Cheung, K.-K.; Echevarria, A.; Galembeck, S.; Aparecida, M. A. M.; Miller, J.; Rumjanek, V. M.; Simas, A. M. Acta Crystallogr. 1992, C48, 1471–1474. doi:10.1107/S0108270192000283 |
| 12. | Tolshchina, S. G.; Ignatenko, N. K.; Slepukhin, P. A.; Ishmetova, R. I.; Rusinov, G. L. Chem. Heterocycl. Compd. 2010, 46, 691–698. doi:10.1007/s10593-010-0571-5 |
| 15. | Busch, M. Ber. Dtsch. Chem. Ges. 1905, 38, 856–860. doi:10.1002/cber.190503801148 |
| 16. | Busch, M. Ber. Dtsch. Chem. Ges. 1905, 38, 861–866. doi:10.1002/cber.190503801149 |
| 27. | Grynberg, N.; Gomes, R.; Shinzato, T.; Echevarria, A.; Miller, J. Anticancer Res. 1992, 12, 1025–1028. |
| 28. | Grynberg, N.; Santos, A. C.; Echevarria, A. Anti-Cancer Drugs 1997, 8, 88–91. doi:10.1097/00001813-199701000-00012 |
| 29. | Senff-Ribeiro, A.; Echevarria, A.; Silva, E. F.; Franco, C. R. C.; Veiga, S. S.; Oliveira, M. B. M. Br. J. Cancer 2004, 91, 297–304. doi:10.1038/sj.bjc.6601946 |
| 24. | Cawkill, E.; Ollis, W. D.; Ramsden, C. A.; Rowson, G. P. J. Chem. Soc., Perkin Trans. 1 1979, 724–731. doi:10.1039/P19790000724 |
| 41. | Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K |
| 25. | Ollis, W. D.; Ramsden, C. A. Adv. Heterocycl. Chem. 1976, 19, 1–121. doi:10.1016/S0065-2725(08)60230-5 |
| 26. | Kawase, M.; Sakagami, H.; Motohashi, N. The Chemistry of Bioactive Mesoionic Heterocycles. In Bioactive Heterocycles VII; Motohashi, N., Ed.; Topics in Heterocyclic Chemistry, Vol. 16; Springer: Berlin, Germany, 2009; pp 135–152. doi:10.1007/7081_2007_096 |
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 10. | Szabo, J.; Maas, G. Z. Naturforsch. 2013, 68b, 207–213. doi:10.5560/znb.2013-3023 |
| 3. | Szabo, J.; Karger, K.; Bucher, N.; Maas, G. Beilstein J. Org. Chem. 2014, 10, 2255–2262. doi:10.3762/bjoc.10.234 |
| 32. | Cadena, S. M. S. C.; Carnieri, E. G. S.; Echevarria, A.; de Oliveira, M. B. M. FEBS Lett. 1998, 440, 46–50. doi:10.1016/S0014-5793(98)01427-6 |
| 33. | Part of this work has been published: Szabo, J., Studien zur Reaktivität von Tris(benzylamino)guanidinium-Salzen mit Carbonylverbindungen und Isocyanaten; Doctoral Thesis, Ulm University, Ulm, 2014. |
| 30. | Rodrigues, R. F.; da Silva, E. F.; Echevarria, A.; Fajardo-Bonin, R.; Amaral, V. F.; Leon, L. L.; Canto-Cavalheiro, M. M. Eur. J. Med. Chem. 2007, 42, 1039–1043. doi:10.1016/j.ejmech.2006.12.026 |
| 31. | da Silva Ferreira, W.; Freire-de-Lima, L.; Saraiva, V. B.; Alisson-Silva, F.; Mendonça-Previato, L.; Previato, J. O.; Echevarria, A.; Freire de Lima, M. E. Bioorg. Med. Chem. 2008, 16, 2984–2991. doi:10.1016/j.bmc.2007.12.049 |
© 2017 Szabo et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)