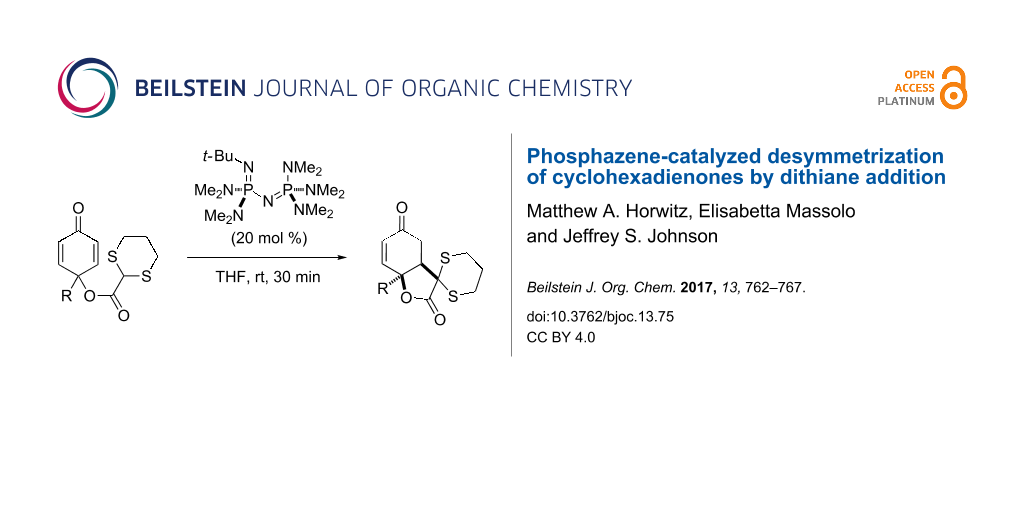
Abstract
We report a desymmetrization of cyclohexadienones by intramolecular conjugate addition of a tethered dithiane nucleophile. Mild reaction conditions allow the formation of diversely functionalized fused bicyclic lactones. The products participate in facially selective additions from the convex surface, leading to allylic alcohol derivatives.

Graphical Abstract
Findings
Desymmetrization has become a well-developed strategy for the construction of complex molecular frameworks [1-6]. Cyclohexadienones are multipurpose synthetic building blocks that have found a central role in desymmetrization methodologies. The functional groups present in these symmetrical molecules allow for a wide array of downstream transformations and they are all formed through a single reaction from cheap and readily available aromatic feedstocks [7-11]. These substrates have been successfully employed in a number of stereoselective desymmetrization reaction manifolds. Intramolecular Michael additions via enamine intermediates have been studied by the Gaunt [12] and Johnson groups [13]. The You group has disclosed methods for the intramolecular addition of amine [14] and bisphenylsulfonyl [15] nucleophiles using bifunctional cinchona alkaloid catalysts. The Sasai and Enders groups used a phosphinothiourea to enable a Rauhut–Currier reaction to form bicyclic enones [16]. The Tian and Lin group used alkyne-tethered cyclohexadienones in an arylrhodation/conjugate addition sequence that enantioselectively delivered oxabicyclo[4.3.0]nonanes [17]; the Lautens and Lan groups have also contributed to the further development of this reaction [18,19]. The Rovis group employed cyclohexadienone hydroperoxides in a chiral phosphoric acid-catalyzed [1,2]/[1,4]-addition cascade [20]. The same group also developed an acyl anion addition promoted by N-heterocyclic carbenes (NHC) that furnished bicyclic furanones via Stetter addition [21]; later, the You group developed an extension of this theme using the same catalytic manifold [22]. More recently, the Corey group has enabled the enantioselective conjugate reduction of prochiral cyclohexadienones using copper hydride generated in situ [23]. Inspired by these advances, we sought to develop an alternative and complementary method invoking the dithiane moiety as an established and easily accessible glyoxylate anion surrogate [24-29]. This would in principle provide access to highly functionalized products with orthogonally protected carbonyl groups in a novel glycolic acid scaffold.
We envisioned utilizing para-quinol derivatives featuring a tethered nucleophile as desymmetrization substrates, with the intention of implementing a Brønsted base organocatalyzed addition (Scheme 1). This reaction would lead to bicyclic systems with the salient attribute of having a convex-concave facial differentiation, allowing subsequent diastereoselective transformations. With the aim of using a dithiane nucleophile, we selected 1,3-dithiane-2-carboxylic acid because of its relatively low pKa (compared with non-carboxylate substituted analogs) and the possibility of using an ester linkage as a tether. We found that the heretofore unknown dicyclohexylcarbodiimide (DCC) mediated coupling between para-quinols and 1,3-dithiane-2-carboxylic acid proceeds in a straightforward manner in cases where R is unbranched (though it does work for R = Ph). Using this method, we were able to easily generate diversely functionalized dithiane-linked para-quinols to study the intramolecular cyclization.
![[1860-5397-13-75-i1]](/bjoc/content/inline/1860-5397-13-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Desymmetrization of cyclohexadienone by tethered nucleophile.
Scheme 1: Desymmetrization of cyclohexadienone by tethered nucleophile.
Based on a prior report [30] demonstrating the efficacy of phosphazene bases in deprotonating carboxylate dithianes, we selected the commercially available achiral superbase P2-t-Bu phosphazene to initiate the ring closure (Scheme 2) [31,32]. We found that in the simplest case, with the methyl-substituted para-quinol ester (1a), the reaction was complete in 30 min at ambient temperature with 20 mol % catalyst [33]. Extending the length of the alkyl chain, the reaction proceeded similarly, even in the presence of a methyl ester or a TBS-protected primary alcohol (1b–d); a comparable result was observed with a phenyl substituent (1e). We considered that if a nucleophilic group were appended to the para-quinol, it would be possible to construct a 5–6–5 fused ring system. Indeed, when R = CH2CH2NHBoc (1f), the desired tricyclic product 2f was obtained. In all cases only a single diastereomer was observed. In a substrate where a β-methyl group is present on the cyclohexadienone (1g), the reaction proved to be completely regioselective, only allowing conjugate addition to the less substituted position.
![[1860-5397-13-75-i2]](/bjoc/content/inline/1860-5397-13-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
We attempted to render the reaction enantioselective using chiral iminophosphoranes (Figure 1) structurally related to P2-t-Bu phosphazene, which are known to be substantially more basic than trialkylamines [34]. With C1 [34-38] and C2 [39-55], we observed no product formation, presumably due to insufficient basicity. Though C3 [30,56,57] led to partial conversion of starting material, no appreciable enantioselectivity was observed.
![[1860-5397-13-75-1]](/bjoc/content/figures/1860-5397-13-75-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chiral iminophosphorane catalysts surveyed.
Figure 1: Chiral iminophosphorane catalysts surveyed.
To investigate the feasibility of a convex-facial addition, we subjected 2a to Luche reduction conditions (Scheme 3). We found this transformation to be completely diastereoselective, and an X-ray diffraction study [58] of the product confirmed our hypothesis regarding the facial selectivity, as the hydride was delivered to the convex face. An analogous reaction occurs when 2a is treated with AlMe3, affording the 1,2-addition product (Scheme 3) [59-63].
![[1860-5397-13-75-i3]](/bjoc/content/inline/1860-5397-13-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
We next sought to establish the glyoxylate anion equivalency of the dithiane substructure in our system. In order to reveal the masked carbonyl functionality, we rigorously applied reported dithiane deprotection conditions to 2a (Table 1). Despite extensive investigations, none of our efforts were fruitful, resulting in either no conversion, side reactions [64], or decomposition. We rationalized these disappointing results considering: 1) the crowded steric environment surrounding the dithiane moiety on the concave face of the bicycle, 2) the sensitive nature of this class of compounds, which stems from the highly reactive functional groups present, and 3) the strained character of the five-membered α-ketolactone (5) that would result from deprotection.
Table 1: Carbonyl deprotection conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-75-i6.svg?max-width=637&scale=1.0)
|
||
| Entry | Conditions | Result |
|---|---|---|
| 1 [65] | NBS, MeCN/H2O, 0 °C, 10 mina | side reaction |
| 2 [66] | NBS, AgNO3, MeCN/H2O, 0 °C, 5 minb | side reaction |
| 3 [67] | PhI(OAc)2, MeCN/CH2Cl2/H2O, 50 °C, 18 h | no reactionc |
| 4 [68] | Hg(ClO4)2, MeOH/CH2Cl2, rt, 2 h | side reaction |
| 5 [69] | HgCl2,HgO, MeOH/H2O, 55 °C, 18 hd | no reaction |
| 6 [70] | MeI, MeCN/H2O, reflux, 18 h | no reaction |
| 7 [71] | m-CPBAe, MeCN, rt, 18 h, then 1 M HCl, reflux, 4 h | decomposition |
| 8 [72] | SbCl5, CH2Cl2, 0 °C, 1 h | decomposition |
| 9 [73] | I2, NaHCO3, acetone/H2O, 50 °C, 18 h | no reaction |
| 10 [74] | CANf, acetone/H2O, 50 °C, 18 h | no reaction |
| 11 [75] | Chloramine T, MeOH/H2O, 70 °C, 18 h | no reaction |
aDifferent solvent systems, such as acetone/H2O and DMSO were used, stoichiometry was varied and the reaction was run also at rt and for longer times (4 and 18 h) but in none of the cases was the desired product obtained. bThe reaction was also run at rt for 18 h, but the desired product was not obtained. cDecomposition products were also observed. dThe MeCN/H2O solvent system was also used and the reaction was also run at rt and reflux, but in none of the cases was the desired product obtained. em-CPBA= meta-chloroperbenzoic acid. fCAN = ceric ammonium nitrate.
We attempted to synthesize 5 via an alternative route using Cu(II)-catalyzed aerobic oxidative deacylation [76] of the β-keto ester 6 (Scheme 4) [77]. The fact that this reaction also leads to decomposition of the starting material is cause for general concern about the feasibility of easily reaching the target substructure. In order to minimize the observed side reactions, we sought to apply the deprotection conditions to allylic alcohol 3. However, both the use of NBS and HgCl2/HgO were unsuccessful.
![[1860-5397-13-75-i4]](/bjoc/content/inline/1860-5397-13-75-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Attempted oxidative deacylation.
Scheme 4: Attempted oxidative deacylation.
We further investigated the removal of the dithiane moiety via Raney nickel-promoted desulfurization (Scheme 5). To observe any substrate conversion, it was necessary to use a hydrogen atmosphere. Under those conditions, though the dithiane function was removed, degradation occurred.
![[1860-5397-13-75-i5]](/bjoc/content/inline/1860-5397-13-75-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempted desulfurization with Raney nickel.
Scheme 5: Attempted desulfurization with Raney nickel.
Conclusion
In conclusion, we have developed a desymmetrizing intramolecular conjugate addition of a tethered dithiane moiety to para-cresol-derived cyclohexadienones. The substrates are easily accessible from cheap starting materials and the reaction provides functionalized bicyclic lactones as a single diastereomer. The products of the reaction were able to undergo diastereoselective convex-facial additions. The carbonyl deprotection was unsuccessful and we hope that our efforts can serve as a cautionary tale for future synthetic planning involving related structures.
References
-
Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002
Return to citation in text: [1] -
Magnuson, S. R. Tetrahedron 1995, 51, 2167–2213. doi:10.1016/0040-4020(94)01070-G
Return to citation in text: [1] -
García-Urdiales, E.; Alfonso, I.; Gotor, V. Chem. Rev. 2005, 105, 313–354. doi:10.1021/cr040640a
Return to citation in text: [1] -
Wang, M.; Feng, M.; Tang, B.; Jiang, X. Tetrahedron Lett. 2014, 55, 7147–7155. doi:10.1016/j.tetlet.2014.10.152
Return to citation in text: [1] -
Borissov, A.; Davies, T. Q.; Ellis, S. R.; Fleming, T. A.; Richardson, M. S. W.; Dixon, D. J. Chem. Soc. Rev. 2016, 45, 5474–5540. doi:10.1039/C5CS00015G
Return to citation in text: [1] -
Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330–7396. doi:10.1021/acs.chemrev.6b00094
Return to citation in text: [1] -
Felpin, F.-X. Tetrahedron Lett. 2007, 48, 409–412. doi:10.1016/j.tetlet.2006.11.073
Return to citation in text: [1] -
McKillop, A.; McLaren, L.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 1994, 2047–2048. doi:10.1039/P19940002047
Return to citation in text: [1] -
Jepsen, T. H.; Jensen, A. A.; Lund, M. H.; Glibstrup, E.; Kristensen, J. L. ACS Med. Chem. Lett. 2014, 5, 766–770. doi:10.1021/ml500094c
Return to citation in text: [1] -
Canesi, S.; Bouchu, D.; Ciufolini, M. A. Org. Lett. 2005, 7, 175–177. doi:10.1021/ol048094v
Return to citation in text: [1] -
Brown, P. D.; Willis, A. C.; Sherburn, M. S.; Lawrence, A. L. Org. Lett. 2012, 14, 4537–4539. doi:10.1021/ol302042u
Return to citation in text: [1] -
Vo, N. T.; Pace, R. D. M.; O’Hara, F.; Gaunt, M. J. J. Am. Chem. Soc. 2008, 130, 404–405. doi:10.1021/ja077457u
Return to citation in text: [1] -
Corbett, M. T.; Johnson, J. S. Chem. Sci. 2013, 4, 2828–2832. doi:10.1039/C3SC51022K
Return to citation in text: [1] -
Gu, Q.; You, S.-L. Chem. Sci. 2011, 2, 1519–1522. doi:10.1039/C1SC00083G
Return to citation in text: [1] -
Gu, Q.; You, S.-L. Org. Lett. 2011, 13, 5192–5195. doi:10.1021/ol202073p
Return to citation in text: [1] -
Takizawa, S.; Nguyen, T. M.-N.; Grossman, A.; Enders, D.; Sasai, H. Angew. Chem., Int. Ed. 2012, 51, 5423–5426. doi:10.1002/anie.201201542
Return to citation in text: [1] -
He, Z.-T.; Tian, B.; Fukui, Y.; Tong, X.; Tian, P.; Lin, G.-Q. Angew. Chem., Int. Ed. 2013, 52, 5314–5318. doi:10.1002/anie.201300137
Return to citation in text: [1] -
Keilitz, J.; Newman, S. G.; Lautens, M. Org. Lett. 2013, 15, 1148–1151. doi:10.1021/ol400363f
Return to citation in text: [1] -
Yu, Z.; Qi, X.; Li, Y.; Liu, S.; Lan, Y. Org. Chem. Front. 2016, 3, 209–216. doi:10.1039/C5QO00334B
Return to citation in text: [1] -
Rubush, D. M.; Morges, M. A.; Rose, B. J.; Thamm, D. H.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 13554–13557. doi:10.1021/ja3052427
Return to citation in text: [1] -
Liu, Q.; Rovis, T. Org. Process Res. Dev. 2007, 11, 598–604. doi:10.1021/op600278f
Return to citation in text: [1] -
Jia, M.-Q.; Liu, C.; You, S.-L. J. Org. Chem. 2012, 77, 10996–11001. doi:10.1021/jo3022555
Return to citation in text: [1] -
Han, Y.; Breitler, S.; Zheng, S.-L.; Corey, E. J. Org. Lett. 2016, 18, 6172–6175. doi:10.1021/acs.orglett.6b03186
Return to citation in text: [1] -
Eliel, E. L.; Hartmann, A. A. J. Org. Chem. 1972, 37, 505–506. doi:10.1021/jo00968a043
Return to citation in text: [1] -
Takahashi, T.; Okano, T.; Harada, T.; Imamura, K.; Yamada, H. Synlett 1994, 121–122. doi:10.1055/s-1994-22762
Return to citation in text: [1] -
Takahashi, T.; Tsukamoto, H.; Kurosaki, M.; Yamada, H. Synlett 1997, 1065–1066. doi:10.1055/s-1997-1549
Return to citation in text: [1] -
Enders, D.; Bonten, M. H.; Raabe, G. Synlett 2007, 885–888. doi:10.1055/s-2007-970787
Return to citation in text: [1] -
Enders, D.; Bonten, M. H.; Raabe, G. Angew. Chem., Int. Ed. 2007, 46, 2314–2316. doi:10.1002/anie.200604802
Return to citation in text: [1] -
Liu, Q.; Perreault, S.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14066–14067. doi:10.1021/ja805680z
Return to citation in text: [1] -
Kondoh, A.; Oishi, M.; Takeda, T.; Terada, M. Angew. Chem., Int. Ed. 2015, 54, 15836–15839. doi:10.1002/anie.201508178
Return to citation in text: [1] [2] -
Massolo, E.; Benaglia, M.; Genoni, A.; Annunziata, R.; Celentano, G.; Gaggero, N. Org. Biomol. Chem. 2015, 13, 5591–5596. doi:10.1039/C5OB00492F
A significant increase in the dithiane carboxylate acidity is observed when the substrate is derivatized to an activated thioester.
Return to citation in text: [1] -
Massolo, E.; Brenna, D.; Cozzi, F.; Raimondi, L.; Gaggero, N.; Benaglia, M. Synlett 2016, 27, 2716–2720. doi:10.1055/s-0036-1588306
Return to citation in text: [1] -
Under the same reaction conditions, we observed that triethylamine was unable to promote the transformation and tetramethylguanidine provided only trace amounts of desired product after 13 h.
Return to citation in text: [1] -
Núñez, M. G.; Farley, A. J. M.; Dixon, D. J. J. Am. Chem. Soc. 2013, 135, 16348–16351. doi:10.1021/ja409121s
Return to citation in text: [1] [2] -
Goldys, A. M.; Dixon, D. J. Macromolecules 2014, 47, 1277–1284. doi:10.1021/ma402258y
Return to citation in text: [1] -
Farley, A. J. M.; Sandford, C.; Dixon, D. J. J. Am. Chem. Soc. 2015, 137, 15992–15995. doi:10.1021/jacs.5b10226
Return to citation in text: [1] -
Robertson, G. P.; Farley, A. J. M.; Dixon, D. J. Synlett 2016, 27, 21–24. doi:10.1055/s-0035-1560530
Return to citation in text: [1] -
Horwitz, M. A.; Zavesky, B. P.; Martinez-Alvarado, J. I.; Johnson, J. S. Org. Lett. 2016, 18, 36–39. doi:10.1021/acs.orglett.5b03127
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 14088–14089. doi:10.1021/ja806311e
Return to citation in text: [1] -
Uraguchi, D.; Ito, T.; Ooi, T. J. Am. Chem. Soc. 2009, 131, 3836–3837. doi:10.1021/ja810043d
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. Science 2009, 326, 120–123. doi:10.1126/science.1176758
Return to citation in text: [1] -
Uraguchi, D.; Ito, T.; Nakamura, S.; Sakaki, S.; Ooi, T. Chem. Lett. 2009, 38, 1052–1053. doi:10.1246/cl.2009.1052
Return to citation in text: [1] -
Uraguchi, D.; Ito, T.; Nakamura, S.; Ooi, T. Chem. Sci. 2010, 1, 488–490. doi:10.1039/C0SC00268B
Return to citation in text: [1] -
Uraguchi, D.; Nakamura, S.; Ooi, T. Angew. Chem., Int. Ed. 2010, 49, 7562–7565. doi:10.1002/anie.201004072
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. Angew. Chem., Int. Ed. 2011, 50, 3681–3683. doi:10.1002/anie.201007752
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. Chem. Sci. 2012, 3, 842–845. doi:10.1039/C1SC00678A
Return to citation in text: [1] -
Corbett, M. T.; Uraguchi, D.; Ooi, T.; Johnson, J. S. Angew. Chem., Int. Ed. 2012, 51, 4685–4689. doi:10.1002/anie.201200559
Return to citation in text: [1] -
Uraguchi, D.; Yoshioka, K.; Ueki, Y.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 19370–19373. doi:10.1021/ja310209g
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Sugiyama, A.; Ooi, T. Chem. Sci. 2013, 4, 1308–1311. doi:10.1039/C2SC22027J
Return to citation in text: [1] -
Uraguchi, D.; Tsutsumi, R.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 8161–8164. doi:10.1021/ja403491j
Return to citation in text: [1] -
Uraguchi, D.; Tsutsumi, R.; Ooi, T. Tetrahedron 2014, 70, 1691–1701. doi:10.1016/j.tet.2013.12.086
Return to citation in text: [1] -
Tsutsumi, R.; Kim, S.; Uraguchi, D.; Ooi, T. Synthesis 2014, 46, 871–878. doi:10.1055/s-0033-1340818
Return to citation in text: [1] -
Uraguchi, D.; Nakamura, S.; Sasaki, H.; Konakade, Y.; Ooi, T. Chem. Commun. 2014, 50, 3491–3493. doi:10.1039/C3CC49477B
Return to citation in text: [1] -
Uraguchi, D.; Yamada, K.; Ooi, T. Angew. Chem., Int. Ed. 2015, 54, 9954–9957. doi:10.1002/anie.201503928
Return to citation in text: [1] -
Horwitz, M. A.; Tanaka, N.; Yokosaka, T.; Uraguchi, D.; Johnson, J. S.; Ooi, T. Chem. Sci. 2015, 6, 6086–6090. doi:10.1039/C5SC02170G
Return to citation in text: [1] -
Takeda, T.; Terada, M. J. Am. Chem. Soc. 2013, 135, 15306–15309. doi:10.1021/ja408296h
Return to citation in text: [1] -
Takeda, T.; Kondoh, A.; Terada, M. Angew. Chem., Int. Ed. 2016, 55, 4734–4737. doi:10.1002/anie.201601352
Return to citation in text: [1] -
CCDC1511800 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Carreño, M. C.; González, M. P.; Ribagorda, M. J. Org. Chem. 1996, 61, 6758–6759. doi:10.1021/jo961126a
Return to citation in text: [1] -
Carreño, M. C.; González, M. P.; Ribagorda, M. J. Org. Chem. 1998, 63, 3687–3693. doi:10.1021/jo980084i
Return to citation in text: [1] -
Carreño, M. C.; Ribagorda, M.; Somoza, Á.; Urbano, A. Angew. Chem., Int. Ed. 2002, 41, 2755–2757. doi:10.1002/1521-3773(20020802)41:15<2755::AID-ANIE2755>3.0.CO;2-A
Return to citation in text: [1] -
Carreño, M. C.; Merino, E.; Ribagorda, M.; Somoza, Á.; Urbano, A. Org. Lett. 2005, 7, 1419–1422. doi:10.1021/ol050287a
Return to citation in text: [1] -
Carreño, M. C.; Merino, E.; Ribagorda, M.; Somoza, Á.; Urbano, A. Chem. – Eur. J. 2007, 13, 1064–1077. doi:10.1002/chem.200601330
Return to citation in text: [1] -
When NBS was used, the unpurified 1H NMR spectrum was suggestive of bromination at the ketone α-position, although the product was not isolable by column chromatography. With Hg(ClO4)2, an unidentified rearomatized product was observed.
Return to citation in text: [1] -
Corey, E. J.; Erickson, B. W. J. Org. Chem. 1971, 36, 3553–3560. doi:10.1021/jo00822a019
Return to citation in text: [1] -
Burks, E. A.; Johnson, W. H., Jr.; Whitman, C. P. J. Am. Chem. Soc. 1998, 120, 7665–7675. doi:10.1021/ja9808402
Return to citation in text: [1] -
Gao, A. X.; Hamada, T.; Snyder, S. A. Angew. Chem., Int. Ed. 2016, 55, 10301–10306. doi:10.1002/anie.201604744
Return to citation in text: [1] -
Tsuboi, K.; Ichikawa, Y.; Jiang, Y.; Naganawa, A.; Isobe, M. Tetrahedron 1997, 53, 5123–5142. doi:10.1016/S0040-4020(97)00230-5
Return to citation in text: [1] -
Nicewicz, D. A.; Yates, C. M.; Johnson, J. S. Angew. Chem., Int. Ed. 2004, 43, 2652–2655. doi:10.1002/anie.200353354
Return to citation in text: [1] -
Fetizon, M.; Jurion, M. J. Chem. Soc., Chem. Commun. 1972, 382–383. doi:10.1039/C39720000382
Return to citation in text: [1] -
Smith, A. B.; Dorsey, B. D.; Visnick, M.; Maeda, T.; Malamas, M. S. J. Am. Chem. Soc. 1986, 108, 3110–3112. doi:10.1021/ja00271a054
Return to citation in text: [1] -
Kamata, M.; Otogawa, H.; Hasegawa, E. Tetrahedron Lett. 1991, 32, 7421–7424. doi:10.1016/0040-4039(91)80123-N
Return to citation in text: [1] -
Nicolaou, K. C.; Bunnage, M. E.; McGarry, D. G.; Shi, S.; Somers, P. K.; Wallace, P. A.; Chu, X.-J.; Agrios, K. A.; Gunzner, J. L.; Yang, Z. Chem. – Eur. J. 1999, 5, 599–617. doi:10.1002/(SICI)1521-3765(19990201)5:2<599::AID-CHEM599>3.0.CO;2-N
Return to citation in text: [1] -
La Claire, J. J.; Lansbury, P. T.; Zhi, B.-x.; Hoogsteen, K. J. Org. Chem. 1995, 60, 4822–4833. doi:10.1021/jo00120a027
Return to citation in text: [1] -
Emerson, D. W.; Wynberg, H. Tetrahedron Lett. 1971, 12, 3445–3448. doi:10.1016/S0040-4039(01)97201-6
Return to citation in text: [1] -
Steward, K. M.; Johnson, J. S. Org. Lett. 2011, 13, 2426–2429. doi:10.1021/ol200649u
Return to citation in text: [1] -
Tello-Aburto, R.; Kalstabakken, K. A.; Volp, K. A.; Harned, A. M. Org. Biomol. Chem. 2011, 9, 7849–7859. doi:10.1039/C1OB06125A
Return to citation in text: [1]
| 68. | Tsuboi, K.; Ichikawa, Y.; Jiang, Y.; Naganawa, A.; Isobe, M. Tetrahedron 1997, 53, 5123–5142. doi:10.1016/S0040-4020(97)00230-5 |
| 69. | Nicewicz, D. A.; Yates, C. M.; Johnson, J. S. Angew. Chem., Int. Ed. 2004, 43, 2652–2655. doi:10.1002/anie.200353354 |
| 70. | Fetizon, M.; Jurion, M. J. Chem. Soc., Chem. Commun. 1972, 382–383. doi:10.1039/C39720000382 |
| 1. | Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002 |
| 2. | Magnuson, S. R. Tetrahedron 1995, 51, 2167–2213. doi:10.1016/0040-4020(94)01070-G |
| 3. | García-Urdiales, E.; Alfonso, I.; Gotor, V. Chem. Rev. 2005, 105, 313–354. doi:10.1021/cr040640a |
| 4. | Wang, M.; Feng, M.; Tang, B.; Jiang, X. Tetrahedron Lett. 2014, 55, 7147–7155. doi:10.1016/j.tetlet.2014.10.152 |
| 5. | Borissov, A.; Davies, T. Q.; Ellis, S. R.; Fleming, T. A.; Richardson, M. S. W.; Dixon, D. J. Chem. Soc. Rev. 2016, 45, 5474–5540. doi:10.1039/C5CS00015G |
| 6. | Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330–7396. doi:10.1021/acs.chemrev.6b00094 |
| 30. | Kondoh, A.; Oishi, M.; Takeda, T.; Terada, M. Angew. Chem., Int. Ed. 2015, 54, 15836–15839. doi:10.1002/anie.201508178 |
| 77. | Tello-Aburto, R.; Kalstabakken, K. A.; Volp, K. A.; Harned, A. M. Org. Biomol. Chem. 2011, 9, 7849–7859. doi:10.1039/C1OB06125A |
| 13. | Corbett, M. T.; Johnson, J. S. Chem. Sci. 2013, 4, 2828–2832. doi:10.1039/C3SC51022K |
| 31. |
Massolo, E.; Benaglia, M.; Genoni, A.; Annunziata, R.; Celentano, G.; Gaggero, N. Org. Biomol. Chem. 2015, 13, 5591–5596. doi:10.1039/C5OB00492F
A significant increase in the dithiane carboxylate acidity is observed when the substrate is derivatized to an activated thioester. |
| 32. | Massolo, E.; Brenna, D.; Cozzi, F.; Raimondi, L.; Gaggero, N.; Benaglia, M. Synlett 2016, 27, 2716–2720. doi:10.1055/s-0036-1588306 |
| 12. | Vo, N. T.; Pace, R. D. M.; O’Hara, F.; Gaunt, M. J. J. Am. Chem. Soc. 2008, 130, 404–405. doi:10.1021/ja077457u |
| 23. | Han, Y.; Breitler, S.; Zheng, S.-L.; Corey, E. J. Org. Lett. 2016, 18, 6172–6175. doi:10.1021/acs.orglett.6b03186 |
| 75. | Emerson, D. W.; Wynberg, H. Tetrahedron Lett. 1971, 12, 3445–3448. doi:10.1016/S0040-4039(01)97201-6 |
| 7. | Felpin, F.-X. Tetrahedron Lett. 2007, 48, 409–412. doi:10.1016/j.tetlet.2006.11.073 |
| 8. | McKillop, A.; McLaren, L.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 1994, 2047–2048. doi:10.1039/P19940002047 |
| 9. | Jepsen, T. H.; Jensen, A. A.; Lund, M. H.; Glibstrup, E.; Kristensen, J. L. ACS Med. Chem. Lett. 2014, 5, 766–770. doi:10.1021/ml500094c |
| 10. | Canesi, S.; Bouchu, D.; Ciufolini, M. A. Org. Lett. 2005, 7, 175–177. doi:10.1021/ol048094v |
| 11. | Brown, P. D.; Willis, A. C.; Sherburn, M. S.; Lawrence, A. L. Org. Lett. 2012, 14, 4537–4539. doi:10.1021/ol302042u |
| 24. | Eliel, E. L.; Hartmann, A. A. J. Org. Chem. 1972, 37, 505–506. doi:10.1021/jo00968a043 |
| 25. | Takahashi, T.; Okano, T.; Harada, T.; Imamura, K.; Yamada, H. Synlett 1994, 121–122. doi:10.1055/s-1994-22762 |
| 26. | Takahashi, T.; Tsukamoto, H.; Kurosaki, M.; Yamada, H. Synlett 1997, 1065–1066. doi:10.1055/s-1997-1549 |
| 27. | Enders, D.; Bonten, M. H.; Raabe, G. Synlett 2007, 885–888. doi:10.1055/s-2007-970787 |
| 28. | Enders, D.; Bonten, M. H.; Raabe, G. Angew. Chem., Int. Ed. 2007, 46, 2314–2316. doi:10.1002/anie.200604802 |
| 29. | Liu, Q.; Perreault, S.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14066–14067. doi:10.1021/ja805680z |
| 76. | Steward, K. M.; Johnson, J. S. Org. Lett. 2011, 13, 2426–2429. doi:10.1021/ol200649u |
| 18. | Keilitz, J.; Newman, S. G.; Lautens, M. Org. Lett. 2013, 15, 1148–1151. doi:10.1021/ol400363f |
| 19. | Yu, Z.; Qi, X.; Li, Y.; Liu, S.; Lan, Y. Org. Chem. Front. 2016, 3, 209–216. doi:10.1039/C5QO00334B |
| 21. | Liu, Q.; Rovis, T. Org. Process Res. Dev. 2007, 11, 598–604. doi:10.1021/op600278f |
| 73. | Nicolaou, K. C.; Bunnage, M. E.; McGarry, D. G.; Shi, S.; Somers, P. K.; Wallace, P. A.; Chu, X.-J.; Agrios, K. A.; Gunzner, J. L.; Yang, Z. Chem. – Eur. J. 1999, 5, 599–617. doi:10.1002/(SICI)1521-3765(19990201)5:2<599::AID-CHEM599>3.0.CO;2-N |
| 17. | He, Z.-T.; Tian, B.; Fukui, Y.; Tong, X.; Tian, P.; Lin, G.-Q. Angew. Chem., Int. Ed. 2013, 52, 5314–5318. doi:10.1002/anie.201300137 |
| 22. | Jia, M.-Q.; Liu, C.; You, S.-L. J. Org. Chem. 2012, 77, 10996–11001. doi:10.1021/jo3022555 |
| 74. | La Claire, J. J.; Lansbury, P. T.; Zhi, B.-x.; Hoogsteen, K. J. Org. Chem. 1995, 60, 4822–4833. doi:10.1021/jo00120a027 |
| 16. | Takizawa, S.; Nguyen, T. M.-N.; Grossman, A.; Enders, D.; Sasai, H. Angew. Chem., Int. Ed. 2012, 51, 5423–5426. doi:10.1002/anie.201201542 |
| 71. | Smith, A. B.; Dorsey, B. D.; Visnick, M.; Maeda, T.; Malamas, M. S. J. Am. Chem. Soc. 1986, 108, 3110–3112. doi:10.1021/ja00271a054 |
| 20. | Rubush, D. M.; Morges, M. A.; Rose, B. J.; Thamm, D. H.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 13554–13557. doi:10.1021/ja3052427 |
| 72. | Kamata, M.; Otogawa, H.; Hasegawa, E. Tetrahedron Lett. 1991, 32, 7421–7424. doi:10.1016/0040-4039(91)80123-N |
| 34. | Núñez, M. G.; Farley, A. J. M.; Dixon, D. J. J. Am. Chem. Soc. 2013, 135, 16348–16351. doi:10.1021/ja409121s |
| 35. | Goldys, A. M.; Dixon, D. J. Macromolecules 2014, 47, 1277–1284. doi:10.1021/ma402258y |
| 36. | Farley, A. J. M.; Sandford, C.; Dixon, D. J. J. Am. Chem. Soc. 2015, 137, 15992–15995. doi:10.1021/jacs.5b10226 |
| 37. | Robertson, G. P.; Farley, A. J. M.; Dixon, D. J. Synlett 2016, 27, 21–24. doi:10.1055/s-0035-1560530 |
| 38. | Horwitz, M. A.; Zavesky, B. P.; Martinez-Alvarado, J. I.; Johnson, J. S. Org. Lett. 2016, 18, 36–39. doi:10.1021/acs.orglett.5b03127 |
| 33. | Under the same reaction conditions, we observed that triethylamine was unable to promote the transformation and tetramethylguanidine provided only trace amounts of desired product after 13 h. |
| 34. | Núñez, M. G.; Farley, A. J. M.; Dixon, D. J. J. Am. Chem. Soc. 2013, 135, 16348–16351. doi:10.1021/ja409121s |
| 66. | Burks, E. A.; Johnson, W. H., Jr.; Whitman, C. P. J. Am. Chem. Soc. 1998, 120, 7665–7675. doi:10.1021/ja9808402 |
| 67. | Gao, A. X.; Hamada, T.; Snyder, S. A. Angew. Chem., Int. Ed. 2016, 55, 10301–10306. doi:10.1002/anie.201604744 |
| 64. | When NBS was used, the unpurified 1H NMR spectrum was suggestive of bromination at the ketone α-position, although the product was not isolable by column chromatography. With Hg(ClO4)2, an unidentified rearomatized product was observed. |
| 65. | Corey, E. J.; Erickson, B. W. J. Org. Chem. 1971, 36, 3553–3560. doi:10.1021/jo00822a019 |
| 58. | CCDC1511800 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 59. | Carreño, M. C.; González, M. P.; Ribagorda, M. J. Org. Chem. 1996, 61, 6758–6759. doi:10.1021/jo961126a |
| 60. | Carreño, M. C.; González, M. P.; Ribagorda, M. J. Org. Chem. 1998, 63, 3687–3693. doi:10.1021/jo980084i |
| 61. | Carreño, M. C.; Ribagorda, M.; Somoza, Á.; Urbano, A. Angew. Chem., Int. Ed. 2002, 41, 2755–2757. doi:10.1002/1521-3773(20020802)41:15<2755::AID-ANIE2755>3.0.CO;2-A |
| 62. | Carreño, M. C.; Merino, E.; Ribagorda, M.; Somoza, Á.; Urbano, A. Org. Lett. 2005, 7, 1419–1422. doi:10.1021/ol050287a |
| 63. | Carreño, M. C.; Merino, E.; Ribagorda, M.; Somoza, Á.; Urbano, A. Chem. – Eur. J. 2007, 13, 1064–1077. doi:10.1002/chem.200601330 |
| 39. | Uraguchi, D.; Ueki, Y.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 14088–14089. doi:10.1021/ja806311e |
| 40. | Uraguchi, D.; Ito, T.; Ooi, T. J. Am. Chem. Soc. 2009, 131, 3836–3837. doi:10.1021/ja810043d |
| 41. | Uraguchi, D.; Ueki, Y.; Ooi, T. Science 2009, 326, 120–123. doi:10.1126/science.1176758 |
| 42. | Uraguchi, D.; Ito, T.; Nakamura, S.; Sakaki, S.; Ooi, T. Chem. Lett. 2009, 38, 1052–1053. doi:10.1246/cl.2009.1052 |
| 43. | Uraguchi, D.; Ito, T.; Nakamura, S.; Ooi, T. Chem. Sci. 2010, 1, 488–490. doi:10.1039/C0SC00268B |
| 44. | Uraguchi, D.; Nakamura, S.; Ooi, T. Angew. Chem., Int. Ed. 2010, 49, 7562–7565. doi:10.1002/anie.201004072 |
| 45. | Uraguchi, D.; Ueki, Y.; Ooi, T. Angew. Chem., Int. Ed. 2011, 50, 3681–3683. doi:10.1002/anie.201007752 |
| 46. | Uraguchi, D.; Ueki, Y.; Ooi, T. Chem. Sci. 2012, 3, 842–845. doi:10.1039/C1SC00678A |
| 47. | Corbett, M. T.; Uraguchi, D.; Ooi, T.; Johnson, J. S. Angew. Chem., Int. Ed. 2012, 51, 4685–4689. doi:10.1002/anie.201200559 |
| 48. | Uraguchi, D.; Yoshioka, K.; Ueki, Y.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 19370–19373. doi:10.1021/ja310209g |
| 49. | Uraguchi, D.; Ueki, Y.; Sugiyama, A.; Ooi, T. Chem. Sci. 2013, 4, 1308–1311. doi:10.1039/C2SC22027J |
| 50. | Uraguchi, D.; Tsutsumi, R.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 8161–8164. doi:10.1021/ja403491j |
| 51. | Uraguchi, D.; Tsutsumi, R.; Ooi, T. Tetrahedron 2014, 70, 1691–1701. doi:10.1016/j.tet.2013.12.086 |
| 52. | Tsutsumi, R.; Kim, S.; Uraguchi, D.; Ooi, T. Synthesis 2014, 46, 871–878. doi:10.1055/s-0033-1340818 |
| 53. | Uraguchi, D.; Nakamura, S.; Sasaki, H.; Konakade, Y.; Ooi, T. Chem. Commun. 2014, 50, 3491–3493. doi:10.1039/C3CC49477B |
| 54. | Uraguchi, D.; Yamada, K.; Ooi, T. Angew. Chem., Int. Ed. 2015, 54, 9954–9957. doi:10.1002/anie.201503928 |
| 55. | Horwitz, M. A.; Tanaka, N.; Yokosaka, T.; Uraguchi, D.; Johnson, J. S.; Ooi, T. Chem. Sci. 2015, 6, 6086–6090. doi:10.1039/C5SC02170G |
| 30. | Kondoh, A.; Oishi, M.; Takeda, T.; Terada, M. Angew. Chem., Int. Ed. 2015, 54, 15836–15839. doi:10.1002/anie.201508178 |
| 56. | Takeda, T.; Terada, M. J. Am. Chem. Soc. 2013, 135, 15306–15309. doi:10.1021/ja408296h |
| 57. | Takeda, T.; Kondoh, A.; Terada, M. Angew. Chem., Int. Ed. 2016, 55, 4734–4737. doi:10.1002/anie.201601352 |
© 2017 Horwitz et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)