Abstract
Dipivaloylketene (2) is obtained by flash vacuum pyrolysis of furan-2,3-dione 6 and dimerizes to 1,3-dioxin-4-one 3, which is a stable but reactive ketene. The transannular addition and rearrangement of enols formed by the addition of nucleophiles to the ketene function in 3 generates axially chiral 2,6,9-trioxabicyclo[3.3.1]nonadienes (bisdioxines) 4. When arylamines are used as the nucleophiles under neutral conditions, decarboxylation occurs during the formation of bisdioxines 8. However, when water or alcohols are added to 3 under acidic conditions, bisdioxine-carboxylic acids and esters 10 and 11 are obtained. Acid hydrolysis of the bisdioxines proceeds through the addition of water to a C=C double bond and results in a second transannular oxa-Michael-type reaction and generation of tetraoxaadamantanes 5. This reaction is decarboxylative when free carboxylic acid functions are present in the bisdioxines, thus forming 21 and 22, but carboxylic acid derivatives are preserved to yield compounds 20, 23, 25, 28, and 29. A hydrogenolysis of the dibenzyl ester 23 yields the free dicarboxylic acid 24. The tetraoxaadamantanes are formed in high yields (65–95%) in most cases, but the addition of water to the concave inside of the bisdioxines becomes severely hindered in cyclic derivatives, so that the 38-membered ring compound 32 requires microwave heating at 170 °C to form tetraoxaadamantane 33, and the catenated compound 36 and calix[6]arene derivative 37 did not form tetraoxaadamantanes. The reaction mechanisms of bisdioxine and tetraoxaadamantane formation are discussed.

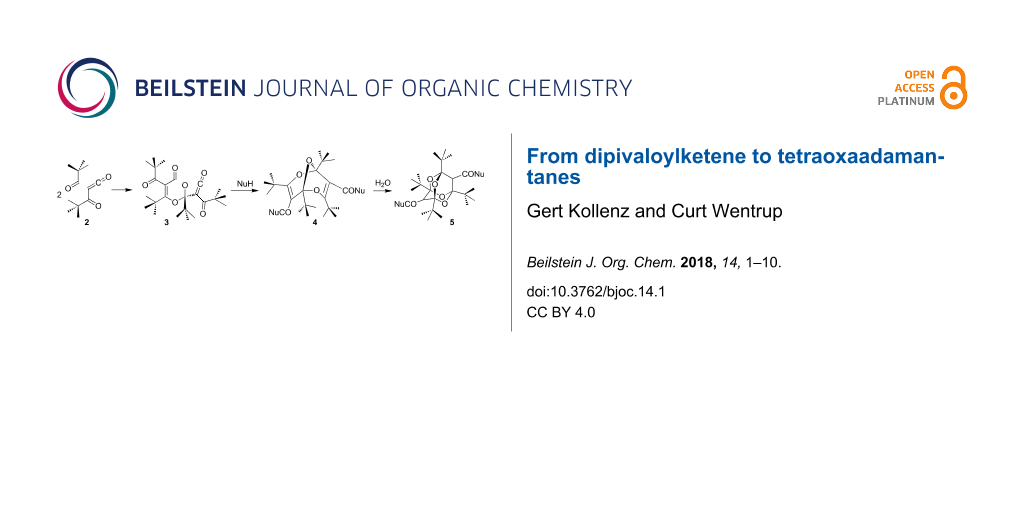
Graphical Abstract
Introduction
The tetraoxaadamantane ring system is relatively unknown and no functional group derivatives had been reported prior to our work. The first methyl and phenyl-substituted tetraoxaadamantanes 1a and 1b were obtained by Arnold [1] and Almqvist [2] through the dimerization of β-ketoaldehydes (reactions 1 and 2 in Scheme 1). This was confirmed by Opitz et al., but the attempted synthesis of further analogs by these procedures failed [3].
![[1860-5397-14-1-i1]](/bjoc/content/inline/1860-5397-14-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to 2,4,6,8-tetraoxaadamantanes.
Scheme 1: Synthetic routes to 2,4,6,8-tetraoxaadamantanes.
Takeda et al. performed the dimerization of β-ketoaldehydes with SO2Cl2 (reaction 3 in Scheme 1) [4], and the X-ray crystal structure of 1b was reported [5]. Chekalov et al. described the preparation of the tetramethyl derivative 1d by dimerization of acetylacetone induced by SiF4 (reaction 4 in Scheme 1) [6] and also reported the compound’s X-ray crystal structure. This transformation can also be achieved with MoOCl4 [7]. Dersch and Reichardt obtained the tetrafluoro derivative 1e in the attempted preparation of difluoromalonic dialdehyde (Scheme 1, reaction 5) [8], and Guseinov et al. reported the synthesis of the corresponding tetrachlorotetraoxaadamantanes together with an X-ray crystal structure of the diphenyltetrachloro derivative 1g (reaction 6 in Scheme 1) [9]. A tetraarsatetraoxaadamantane [10] also has been reported, and 13C NMR spectral characteristics of tetraoxa-, tetrathia-, and tetraselenaadamantanes have been discussed [11], but due to the lack of functional groups, tetraoxaadamantanes have remained largely laboratory curiosities.
It is worth noting that the 2,4,10-trioxaadamantanes, which are orthoesters, are well known [12,13], and the natural product muamvatin [14] is a 2,6,9-trioxaadamantane derivative. The dioxaadamantanes are best known in the form of the highly toxic tetrodotoxins occurring in the puffer fish, certain newt species and several other aquatic animals [15].
In our laboratories we have developed an efficient and high-yielding synthesis of tetraoxaadamantanes 5 by employing two unusual reaction steps: (i) the conversion of the dimer 3 of dipivaloylketene (2) to bisdioxines (2,6,9-trioxabicyclo[3.3.1]nona-3,7-dienes) 4, by the addition of nucleophiles, and (ii) the facile acid-catalyzed hydrolysis of 4 with concomitant transannular cyclization. Following this route a wide variety of compounds 5 containing functional groups is accessible (Scheme 2).
![[1860-5397-14-1-i2]](/bjoc/content/inline/1860-5397-14-1-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Conversion of dipivaloylketene (2) to bisdioxines (2,6,9-trioxabicyclo[3.3.1]nona-3,7-dienes) 4 and tetraoxaadamantanes 5.
Scheme 2: Conversion of dipivaloylketene (2) to bisdioxines (2,6,9-trioxabicyclo[3.3.1]nona-3,7-dienes) 4 and...
In this review we will start with an overview of the syntheses and chemistry of the bisdioxines leading on to the syntheses of tetraoxaadamantanes.
Review
Bisdioxines
The synthesis of bisdioxines 8–13 starts with furandione 6, which, upon flash vacuum pyrolysis (FVP) at 350–500 °C at 10−3–10−4 hPa, eliminates a molecule of CO to generate dipivaloylketene (2) in over 90% yield (Scheme 3).
![[1860-5397-14-1-i3]](/bjoc/content/inline/1860-5397-14-1-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: 2,6,9-Trioxabicyclo[3.3.1]nonadienes (bisdioxines, 9–13) derived from dipivaloylketene (2).
Scheme 3: 2,6,9-Trioxabicyclo[3.3.1]nonadienes (bisdioxines, 9–13) derived from dipivaloylketene (2).
Usually, α-oxoketenes are not isolable, but due to the steric hindrance exerted by the pivaloyl groups ketene 2 is kinetically stable at up to −20 °C. However, it dimerizes at room temperature to afford an 88% yield of the thermally very stable dimer 3, which still carries a ketene function [16]. Compound 3 is formed through a [2 + 4] cycloaddition between one molecule of the α-oxoketene 2 and the carbonyl C=O bond of a second molecule. It is noteworthy that in the presence of DMSO a different dimer 7 is formed, again in high yield, originating from a [2 + 4] cycloaddition between a molecule of the α-oxoketene and the ketene C=O bond of the second molecule (Scheme 3) [17].
The treatment of the dimeric ketene 3 with nucleophiles allowed the preparation of numerous derivatives of the unique 2,6,9-trioxabicyclo[3.3.1]nonadiene (bisdioxine) system 8–13, namely the monoamides 8, the diacid 11, the diacid dichloride 12, and the esters 9, 10 and 13 [18,19] (Scheme 3). The mechanism of formation of these derivatives is summarized in Scheme 4.
![[1860-5397-14-1-i4]](/bjoc/content/inline/1860-5397-14-1-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanisms of formation of bisdioxine acid derivatives from dimer 3.
Scheme 4: Mechanisms of formation of bisdioxine acid derivatives from dimer 3.
The amides 8, formed by the addition of arylamines are invariably decarboxylated. We interpret this in terms of the sequence 3 → 14 → 15 → 16, which in the absence of an acid decarboxylates. In contrast, the addition of water and alcohols to 3 requires the addition of acid (p-TsOH) and results in isolable enol intermediates of type 9 [18]. The carboxylate cannot form in the presence of an acid and therefore decarboxylation does not take place (17 → 18 → 10). The mono- and dicarboxylic acids 10 and 11 are stable and do not decarboxylate easily.
However, aromatic amines carrying strongly electron-withdrawing groups (nitroanilines) do not form stable bisdioxines such as 8. Instead, a cleavage of the dioxinone ring in 3 with formation of dipivaloylacetamides takes place. Surprisingly, the more basic aliphatic amines also cause cleavage to dipivaloylacetamides. On the other hand, neutral thiols do not add to 3, but in the presence of triethylamine ring opening again takes place [18]. Further chemistry of the ketenes obtained by FVP of 5-tert-butyl-4-pivaloylfurandione (6) and 5-tert-butyl-4-methoxyfurandione and their dimers has been reported [20].
It is worth noting that the bisdioxines exhibit axial chirality [21], as has been demonstrated by 1H NMR spectroscopy using the optically active shift reagent Eu(hfc)3 [19]. The enantiomers of the dicarboxylic acid 11 have been separated by flash chromatography of their diastereomeric salts with 1-phenethylamine, and the structures of the acids and ethyl esters were determined by X-ray crystallography [19]. The X-ray structure of the Pt(II) chelate of tetramethyl 2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene obtained from tris-acetylacetonato platinum(II) was determined previously [22,23], and the separation of the enantiomers of the free ligand was achieved by fractional crystallization [24]. Chirality of the bisdioxine dicarbaldehyde, 2,6,9-trioxabicyclo[3.3.l]nona-3,7-diene-4,8-dicarbaldehyde, obtained by extrusion of water from triformylmethane, has also been demonstrated [25], and X-ray crystallography confirmed the structure of this molecule, too [26].
There are few other reports of bisdioxines in the literature. The synthesis of dimethyl bisdioxinedicarboxylate has been described [27], and recently a new synthesis of chromenobisdioxines based on a mild base-mediated reaction of 4-chloro-3-formylcoumarin and o-hydroxyacetophenones has been reported (Scheme 5) [28].
![[1860-5397-14-1-i5]](/bjoc/content/inline/1860-5397-14-1-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Recently reported synthesis of chromenobisdioxines.
Scheme 5: Recently reported synthesis of chromenobisdioxines.
Formation of tetraoxaadamantanes
Although the bisdioxine skeleton is a thermodynamically stable moiety, allowing numerous derivatives to be synthesized, it was soon discovered that in the presence of strong acids, very efficient addition of water and cyclization to tetraoxaadamantanes 20–25 took place (Scheme 6) [29].
![[1860-5397-14-1-i6]](/bjoc/content/inline/1860-5397-14-1-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of tetraoxaadamantanes.
Scheme 6: Formation of tetraoxaadamantanes.
The reaction is usually carried out at room temperature in dichloromethane in the presence of concentrated HCl and glacial acetic acid, and the yields are mostly in the range 65–95%. Depending on the starting material, diesters, monoesters or the fully decarboxylated tetraoxaadamantane can be obtained. It is noteworthy that, when a free carboxylic acid moiety is present in the bisdioxine, it is invariably lost during the tetraoxaadamantane formation. The decarboxylation is likely to take place in the acrylic acid moieties in the trioxanonadienes during the reaction (Scheme 7), and not in the final products, which are not prone to decarboxylation: the stable bis-carboxylic acid 24 can be obtained by hydrogenolysis of the dibenzyl ester 23 (Scheme 6) [30]. The reaction may be seen as a decarboxylative [31,32] oxa-Michael addition (Scheme 7) and may be related to the recently described acid-catalyzed decarboxylation of vinylic and aromatic carboxylic acids [33].
![[1860-5397-14-1-i7]](/bjoc/content/inline/1860-5397-14-1-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Decarboxylative hydrolysis and oxa-Michael-type ring closure.
Scheme 7: Decarboxylative hydrolysis and oxa-Michael-type ring closure.
The amide derivatives 8 react in the same way as monoesters, forming arylaminotetraoxaadamantanes 25 (Scheme 6) [29,34]. The X-ray crystal structure of 25 (Ar = p-methoxyphenyl) has been published [29]. It should be noted that both the bisdioxines [19] and the tetraoxaadamantanes [29] exhibit axial chirality as confirmed by 1H NMR spectroscopy with the Eu(hfc)3 chiral shift reagent.
Bisdioxine oxime and hydrazine derivatives 26 and 27 (Scheme 8) are formed from 3 at room temperature without the need for acid catalysis. As in the case of the addition of arylamines, monodecarboxylation takes place, and in the presence of a strong acid, they are converted to the tetraoxaadamantanes 28 and 29 (65–93%) [35].
![[1860-5397-14-1-i8]](/bjoc/content/inline/1860-5397-14-1-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Oxime and hydrazine derivatives of bisdioxines and tetraoxaadamantanes.
Scheme 8: Oxime and hydrazine derivatives of bisdioxines and tetraoxaadamantanes.
Several bisdioxine derivatives of aromatic di- and triamines as well as crown-ether derivatives were prepared from 3 in order to examine their host–guest properties by ESI mass spectrometry and NMR spectroscopy. Some tetraoxaadamantanes were also examined in this way. For example, compound 30 (Figure 1) was found to have a particular affinity for complexation with choline [26,36], and the crown-5 derivative 31 showed an enhanced ability to extract Na+ and K+ ions from water into CHCl3 (22 and 21%, respectively, within 10 minutes using equimolar amounts of salt and ligand) [37].
![[1860-5397-14-1-1]](/bjoc/content/figures/1860-5397-14-1-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bistetraoxaadamantane derivatives.
Figure 1: Bistetraoxaadamantane derivatives.
In the concave structures of the bisdioxines, the functional groups such as esters, amides, carbamates, urethanes and isocyanates point inward (Scheme 9).
![[1860-5397-14-1-i9]](/bjoc/content/inline/1860-5397-14-1-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Inward-pointing isocyanate, urethane and carbamate groups in bisdioxines. The diisocyanate is obtained by Curtius and Hofmann rearrangements of the diazides and diamides [38,39].
Scheme 9: Inward-pointing isocyanate, urethane and carbamate groups in bisdioxines. The diisocyanate is obtai...
This observation was confirmed by X-ray crystallography as well as calculations at the B3LYP/6-31G** level [38,39]. The tert-butyl groups provide steric protection to the exo sides of the molecules, making the diisocyanate stable at ordinary temperatures. However, it readily reacts with amines and alcohols to form ureas and urethanes, respectively. Taking advantage of this type of concave structure, several cyclic derivatives were synthesized, and some of them were converted to tetraoxaadamantanes [37]. However, when two bisdioxine units are present in a cyclic structure as in Scheme 10, the formation of tetraoxaadamantanes requires the addition of water from the concave inside. Thus, the bisdioxine 32 did not form a tetraoxaadamantane 33 under the usual reaction conditions, but this was finally achieved in 35% yield by microwave irradiation at 170 °C for 40 min [40].
![[1860-5397-14-1-i10]](/bjoc/content/inline/1860-5397-14-1-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Microwave-assisted tetraoxaadamantane formation.
Scheme 10: Microwave-assisted tetraoxaadamantane formation.
This subject was investigated further by synthesizing the cyclic bisdioxine ester derivatives 34 and 36 (Scheme 11 and Figure 2) [41]. The 1,4-catenated dinitro compound 34 is readily converted to the mono-tetraoxaadamantane derivative 35 (Scheme 11). However, all attempts to convert the second bisdioxine unit were fruitless, presumably due to steric hindrance: the cavity in 34 is large enough to form one tetraoxaadamantane derivative, but this reduces the available space, so that the attack by another water molecule on the second bisdioxine unit from the concave inside in 35 was not observed. Force-field calculations indicated that the internal cavity is significantly smaller in the 1,3-catenated nitro compound 36 than in 34, and in fact it was not possible to prepare any tetraoxaadamantane derivative from 36 (Figure 2).
![[1860-5397-14-1-i11]](/bjoc/content/inline/1860-5397-14-1-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Cyclic bisdioxine ester derivative 34 forming a single mono-tetraoxaadamantane.
Scheme 11: Cyclic bisdioxine ester derivative 34 forming a single mono-tetraoxaadamantane.
![[1860-5397-14-1-2]](/bjoc/content/figures/1860-5397-14-1-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyclic bisdioxine derivative not forming a tetraoxaadamantane due to reduced cavity size.
Figure 2: Cyclic bisdioxine derivative not forming a tetraoxaadamantane due to reduced cavity size.
An even higher hindrance is induced by the p-tert-butylcalix[6]arene moiety in 37 (Figure 3), which is obtained from the bisdioxine diacid dichloride 12 and calixarene [42]. The wider upper rim is clearly seen in the X-ray structure and the compound demonstrates a pronounced ability to extract Cs+ ions from water into chloroform by forming endohedral complexes, which is typical for capped calixarenes [43]. However, the lower rim is very congested, thereby hindering the endohedral addition of water to the bisdioxine unit, and in fact a tetraoxaadamantane derivative was not formed.
![[1860-5397-14-1-3]](/bjoc/content/figures/1860-5397-14-1-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The bisdioxine-calix[6]arene derivative 37 complexes Cs+ but does not form a tetraoxaadamantane derivative.
Figure 3: The bisdioxine-calix[6]arene derivative 37 complexes Cs+ but does not form a tetraoxaadamantane der...
Conclusion
The stable 1,3-dioxin-4-one ketene derivative 3 is obtained by dimerization of dipivaloylketene (2), itself obtained in high yield by FVP of furan-2,3-dione 6 (Scheme 3). Ketene 3 reacts with a variety of nucleophiles in an addition reaction to the ketene function, thereby transforming the ketene to enol derivatives 14 or 9 of 1,3-dioxin-4-ones. Subsequently, these enols can undergo transannular cyclizations to yield initial intermediates 15 and 17. However, the latter compounds rearrange, whereby an O–C=O moiety in the 1,3-dioxinone becomes a carboxylic acid function in the resulting, axially chiral 2,6,9-trioxabicyclo[3.3.1]nonadienes (bisdioxines) 8, 10, and 11 (Scheme 4). When this reaction is carried out under neutral conditions (with arylamines), decarboxylation of the carboxylic acid function occurs, yielding 8, but under acidic conditions (with alcohols and water) the acid function is preserved, yielding 10 and 11.
Addition of water to one of the acrylic-type double bonds in the bisdioxines under acidic conditions generates a tertiary alcohol, which again undergoes a transannular oxa-Michael-type ring closure forming a tetraoxaadamantane. Free carboxylic acid functions are decarboxylated in this process (Scheme 7), but amide and ester functions are preserved in products 20, 23, 25, 28, and 29. The dibenzyl ester 23 can be hydrogenated to yield the free dicarboxylic acid 24 (Scheme 6). The tetraoxaadamantane-forming reaction is very efficient and high-yielding, taking place in a variety of open-chain and catenated bisdioxine derivatives. However, the 38-membered ring 32 requires forcing conditions to form a tetraoxaadamantane, and compounds 36 and 37 did not form tetraoxaadamantanes at all.
Acknowledgements
This work was supported by the Karl-Franzens Universität Graz and The University of Queensland. We are indebted to the students, co-workers and colleagues mentioned in the references for their engaged collaboration. G.K. is also indebted to Maria-Theresia Gräfin von Meran, for technical assistance.
References
-
Dolejš, L.; Arnold, Z. Collect. Czech. Chem. Commun. 1966, 31, 4187–4189. doi:10.1135/cccc19664187
Return to citation in text: [1] -
Almqvist, S.-O. Acta Chem. Scand. 1968, 22, 1367–1368. doi:10.3891/acta.chem.scand.22-1367
Return to citation in text: [1] -
Kuhlmey, S.-R.; Adolph, H.; Rieth, K.; Opitz, G. Liebigs Ann. Chem. 1979, 617–627. doi:10.1002/jlac.197919790506
Return to citation in text: [1] -
Tsuboi, S.; Ono, T.; Takeda, A. Heterocycles 1986, 24, 2007–2014. doi:10.3987/R-1986-07-2007
Return to citation in text: [1] -
Kashino, S.; Haisa, M.; Tsuboi, S.; Ono, T.; Takeda, A. Acta Crystallogr., Sect. C 1983, 39, 103–105. doi:10.1107/S0108270183003753
Return to citation in text: [1] -
Chekalov, A. K.; Yufit, D. S.; Prokof'ev, A. I.; Vol'eva, V. B.; Struchkov, Y. T.; Kabachnik, M. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1989, 2375–2377.
Return to citation in text: [1] -
Drew, M. G. B.; Fowles, G. W. A.; Rice, D. A.; Shanton, K. J. J. Chem. Soc., Chem. Commun. 1974, 614–615. doi:10.1039/c39740000614
Return to citation in text: [1] -
Dersch, R.; Reichardt, C. Liebigs Ann. Chem. 1982, 1348–1358. doi:10.1002/jlac.198219820711
Return to citation in text: [1] -
Guseinov, F. I.; Yudina, N. A.; Burangulova, R. N.; Klimentova, G. Y.; Ismailov, V. M.; Gubaidullin, A. T.; Litvinov, I. A.; Zefirov, N. S. Dokl. Akad. Nauk 1998, 359, 778–781.
Return to citation in text: [1] -
Marx, M. B. L.; Pritzkow, H.; Keppler, B. K. Z. Anorg. Allg. Chem. 1996, 622, 1097–1100. doi:10.1002/zaac.19966220627
Return to citation in text: [1] -
Shabab, Y. A. Org. Magn. Reson. 1977, 9, 580–583. doi:10.1002/mrc.1270091006
Return to citation in text: [1] -
Diaper, C. M. Product Class 16: Other Tetraheterosubstituted Methanes. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Knight, J. K., Ed.; Thieme: Stuttgart, 2005; Vol. 18.16, pp 1203–1282.
Return to citation in text: [1] -
Chandrasekhar, S.; Mukherjee, S. J. Mol. Struct. 2015, 1083, 426–429. doi:10.1016/j.molstruc.2014.10.077
Return to citation in text: [1] -
Ward, D. E.; Zahedi, M. M. Org. Lett. 2012, 14, 6246–6249. doi:10.1021/ol303004k
Return to citation in text: [1] -
Yasumoto, T.; Yotsu, M.; Murata, M.; Naoki, H. J. Am. Chem. Soc. 1988, 110, 2344–2345. doi:10.1021/ja00215a078
Return to citation in text: [1] -
Kappe, C. O.; Evans, R. A.; Kennard, C. H. L.; Wentrup, C. J. Am. Chem. Soc. 1991, 113, 4234–4237. doi:10.1021/ja00011a027
Return to citation in text: [1] -
Kappe, C. O.; Färber, G.; Wentrup, C.; Kollenz, G. J. Org. Chem. 1992, 57, 7078–7083. doi:10.1021/jo00052a018
Return to citation in text: [1] -
Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024
Return to citation in text: [1] [2] [3] -
Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109
Return to citation in text: [1] [2] [3] [4] -
Wallfisch, B. C.; Belaj, F.; Wentrup, C.; Kappe, C. O.; Kollenz, G. J. Chem. Soc., Perkin Trans. 1 2002, 599–605. doi:10.1039/b111143d
Return to citation in text: [1] -
Schreier, P.; Bernreuther, A.; Huffer, M., Eds. Analysis of Chiral Organic Molecules: Methodology and Applications; De Gruyter: Berlin, 1995. doi:10.1515/9783110867855
Return to citation in text: [1] -
Gibson, D.; Oldham, C.; Lewis, J.; Lawton, D.; Mason, R.; Robertson, G. B. Nature 1965, 208, 580–581. doi:10.1038/208580a0
Return to citation in text: [1] -
Gibson, D.; Lewis, J.; Oldham, C. J. Chem. Soc. A 1967, 72–77. doi:10.1039/J19670000072
Return to citation in text: [1] -
de Renzi, A.; Panuzi, A.; Paolillo, L.; Vitagliano, A. J. Organomet. Chem. 1977, 124, 221–228. doi:10.1016/S0022-328X(00)90969-0
Return to citation in text: [1] -
Arnold, Z.; Budesinsky, M. J. Org. Chem. 1988, 53, 5352–5353. doi:10.1021/jo00257a029
Return to citation in text: [1] -
Podlaha, J.; Podlahová, J.; Arnold, Z.; Malý, K.; Petrícek, V. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1988, C44, 1966–1968. doi:10.1107/S010827018800753X
Return to citation in text: [1] [2] -
Marx, P. Investigations in the synthesis of secoiridoid derivatives through photocycloaddition of diformylacetic acid methyl ester with open-chain C6-olefins. Ph.D. Thesis, University of Göttingen, Germany, 1981.
Return to citation in text: [1] -
Jaggavarapu, S. R.; Kamalakaran, A. S.; Gayatri, G.; Shukla, M.; Dorai, K.; Gaddamanugu, G. Tetrahedron 2013, 69, 2142–2149. doi:10.1016/j.tet.2013.01.002
Return to citation in text: [1] -
Heilmayer, W.; Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Gruber, K.; Sterk, H.; Kollenz, G. J. Chem. Soc., Chem. Commun. 1995, 797–798. doi:10.1039/c39950000797
Return to citation in text: [1] [2] [3] [4] -
Smounig, R. Darstellung, Eigenschaften und Komplexierungsverhalten neuartiger Wirt – Systeme. Ph.D. Thesis, Karl-Franzens Universität Graz, Austria, 1998.
Return to citation in text: [1] -
Xu, L.; Shao, Z.; Wang, L.; Xiao, J. Org. Lett. 2014, 16, 796–799. doi:10.1021/ol403541g
Return to citation in text: [1] -
Shao, Z.; Xu, L.; Wang, L.; Wei, H.; Xiao, J. Org. Biomol. Chem. 2014, 12, 2185–2188. doi:10.1039/C3OB42582G
Return to citation in text: [1] -
Howe, G. W.; Vandersteen, A. A.; Kluger, R. J. Am. Chem. Soc. 2016, 138, 7568–7573. doi:10.1021/jacs.6b01770
Return to citation in text: [1] -
Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y
Return to citation in text: [1] -
Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Heterocycles 1998, 48, 1841–1850. doi:10.3987/COM-98-8218
Return to citation in text: [1] -
Heilmayer, W.; Smounig, R.; Gruber, K.; Fabian, W. M. F.; Reidlinger, C.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Tetrahedron 2004, 60, 2857–2867. doi:10.1016/j.tet.2004.01.058
Return to citation in text: [1] -
Wallfisch, B. C.; Egger, T.; Heilmayer, W.; Kappe, C. O.; Wentrup, C.; Gloe, K.; Belaj, F.; Klintschar, G.; Kollenz, G. Supramol. Chem. 2002, 14, 383–397. doi:10.1080/1061027021000003430
Return to citation in text: [1] [2] -
Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h
Return to citation in text: [1] [2] -
Kollenz, G.; Smounig, R.; Belaj, F.; Kvaskoff, D.; Wentrup, C. Beilstein J. Org. Chem. 2015, 11, 1–8. doi:10.3762/bjoc.11.1
Return to citation in text: [1] [2] -
Chebanov, V. A.; Reidlinger, C.; Kanaani, H.; Wentrup, C.; Kappe, C. O.; Kollenz, G. Supramol. Chem. 2004, 16, 121–127. doi:10.1080/10610270310001614197
Return to citation in text: [1] -
Leber, S.; Kollenz, G.; Wentrup, C. Beilstein J. Org. Chem. 2012, 8, 738–743. doi:10.3762/bjoc.8.83
Return to citation in text: [1] -
Haberz, P.; Belaj, F.; Gloe, K.; Wentrup, C.; Kollenz, G. Supramol. Chem. 2012, 24, 279–284. doi:10.1080/10610278.2012.658393
Return to citation in text: [1] -
Jose, P.; Menon, S. Bioinorg. Chem. Appl. 2007, No. 65815. doi:10.1155/2007/65815
Return to citation in text: [1]
| 27. | Marx, P. Investigations in the synthesis of secoiridoid derivatives through photocycloaddition of diformylacetic acid methyl ester with open-chain C6-olefins. Ph.D. Thesis, University of Göttingen, Germany, 1981. |
| 28. | Jaggavarapu, S. R.; Kamalakaran, A. S.; Gayatri, G.; Shukla, M.; Dorai, K.; Gaddamanugu, G. Tetrahedron 2013, 69, 2142–2149. doi:10.1016/j.tet.2013.01.002 |
| 29. | Heilmayer, W.; Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Gruber, K.; Sterk, H.; Kollenz, G. J. Chem. Soc., Chem. Commun. 1995, 797–798. doi:10.1039/c39950000797 |
| 1. | Dolejš, L.; Arnold, Z. Collect. Czech. Chem. Commun. 1966, 31, 4187–4189. doi:10.1135/cccc19664187 |
| 5. | Kashino, S.; Haisa, M.; Tsuboi, S.; Ono, T.; Takeda, A. Acta Crystallogr., Sect. C 1983, 39, 103–105. doi:10.1107/S0108270183003753 |
| 16. | Kappe, C. O.; Evans, R. A.; Kennard, C. H. L.; Wentrup, C. J. Am. Chem. Soc. 1991, 113, 4234–4237. doi:10.1021/ja00011a027 |
| 29. | Heilmayer, W.; Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Gruber, K.; Sterk, H.; Kollenz, G. J. Chem. Soc., Chem. Commun. 1995, 797–798. doi:10.1039/c39950000797 |
| 4. | Tsuboi, S.; Ono, T.; Takeda, A. Heterocycles 1986, 24, 2007–2014. doi:10.3987/R-1986-07-2007 |
| 17. | Kappe, C. O.; Färber, G.; Wentrup, C.; Kollenz, G. J. Org. Chem. 1992, 57, 7078–7083. doi:10.1021/jo00052a018 |
| 35. | Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Heterocycles 1998, 48, 1841–1850. doi:10.3987/COM-98-8218 |
| 3. | Kuhlmey, S.-R.; Adolph, H.; Rieth, K.; Opitz, G. Liebigs Ann. Chem. 1979, 617–627. doi:10.1002/jlac.197919790506 |
| 14. | Ward, D. E.; Zahedi, M. M. Org. Lett. 2012, 14, 6246–6249. doi:10.1021/ol303004k |
| 29. | Heilmayer, W.; Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Gruber, K.; Sterk, H.; Kollenz, G. J. Chem. Soc., Chem. Commun. 1995, 797–798. doi:10.1039/c39950000797 |
| 2. | Almqvist, S.-O. Acta Chem. Scand. 1968, 22, 1367–1368. doi:10.3891/acta.chem.scand.22-1367 |
| 15. | Yasumoto, T.; Yotsu, M.; Murata, M.; Naoki, H. J. Am. Chem. Soc. 1988, 110, 2344–2345. doi:10.1021/ja00215a078 |
| 19. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 9. | Guseinov, F. I.; Yudina, N. A.; Burangulova, R. N.; Klimentova, G. Y.; Ismailov, V. M.; Gubaidullin, A. T.; Litvinov, I. A.; Zefirov, N. S. Dokl. Akad. Nauk 1998, 359, 778–781. |
| 33. | Howe, G. W.; Vandersteen, A. A.; Kluger, R. J. Am. Chem. Soc. 2016, 138, 7568–7573. doi:10.1021/jacs.6b01770 |
| 8. | Dersch, R.; Reichardt, C. Liebigs Ann. Chem. 1982, 1348–1358. doi:10.1002/jlac.198219820711 |
| 12. | Diaper, C. M. Product Class 16: Other Tetraheterosubstituted Methanes. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Knight, J. K., Ed.; Thieme: Stuttgart, 2005; Vol. 18.16, pp 1203–1282. |
| 13. | Chandrasekhar, S.; Mukherjee, S. J. Mol. Struct. 2015, 1083, 426–429. doi:10.1016/j.molstruc.2014.10.077 |
| 29. | Heilmayer, W.; Dalvi, T. S.; Kappe, C. O.; Wentrup, C.; Gruber, K.; Sterk, H.; Kollenz, G. J. Chem. Soc., Chem. Commun. 1995, 797–798. doi:10.1039/c39950000797 |
| 34. | Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y |
| 7. | Drew, M. G. B.; Fowles, G. W. A.; Rice, D. A.; Shanton, K. J. J. Chem. Soc., Chem. Commun. 1974, 614–615. doi:10.1039/c39740000614 |
| 30. | Smounig, R. Darstellung, Eigenschaften und Komplexierungsverhalten neuartiger Wirt – Systeme. Ph.D. Thesis, Karl-Franzens Universität Graz, Austria, 1998. |
| 6. | Chekalov, A. K.; Yufit, D. S.; Prokof'ev, A. I.; Vol'eva, V. B.; Struchkov, Y. T.; Kabachnik, M. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1989, 2375–2377. |
| 10. | Marx, M. B. L.; Pritzkow, H.; Keppler, B. K. Z. Anorg. Allg. Chem. 1996, 622, 1097–1100. doi:10.1002/zaac.19966220627 |
| 31. | Xu, L.; Shao, Z.; Wang, L.; Xiao, J. Org. Lett. 2014, 16, 796–799. doi:10.1021/ol403541g |
| 32. | Shao, Z.; Xu, L.; Wang, L.; Wei, H.; Xiao, J. Org. Biomol. Chem. 2014, 12, 2185–2188. doi:10.1039/C3OB42582G |
| 18. | Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024 |
| 18. | Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024 |
| 19. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 26. | Podlaha, J.; Podlahová, J.; Arnold, Z.; Malý, K.; Petrícek, V. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1988, C44, 1966–1968. doi:10.1107/S010827018800753X |
| 36. | Heilmayer, W.; Smounig, R.; Gruber, K.; Fabian, W. M. F.; Reidlinger, C.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Tetrahedron 2004, 60, 2857–2867. doi:10.1016/j.tet.2004.01.058 |
| 18. | Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024 |
| 37. | Wallfisch, B. C.; Egger, T.; Heilmayer, W.; Kappe, C. O.; Wentrup, C.; Gloe, K.; Belaj, F.; Klintschar, G.; Kollenz, G. Supramol. Chem. 2002, 14, 383–397. doi:10.1080/1061027021000003430 |
| 38. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 39. | Kollenz, G.; Smounig, R.; Belaj, F.; Kvaskoff, D.; Wentrup, C. Beilstein J. Org. Chem. 2015, 11, 1–8. doi:10.3762/bjoc.11.1 |
| 25. | Arnold, Z.; Budesinsky, M. J. Org. Chem. 1988, 53, 5352–5353. doi:10.1021/jo00257a029 |
| 26. | Podlaha, J.; Podlahová, J.; Arnold, Z.; Malý, K.; Petrícek, V. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1988, C44, 1966–1968. doi:10.1107/S010827018800753X |
| 22. | Gibson, D.; Oldham, C.; Lewis, J.; Lawton, D.; Mason, R.; Robertson, G. B. Nature 1965, 208, 580–581. doi:10.1038/208580a0 |
| 23. | Gibson, D.; Lewis, J.; Oldham, C. J. Chem. Soc. A 1967, 72–77. doi:10.1039/J19670000072 |
| 42. | Haberz, P.; Belaj, F.; Gloe, K.; Wentrup, C.; Kollenz, G. Supramol. Chem. 2012, 24, 279–284. doi:10.1080/10610278.2012.658393 |
| 24. | de Renzi, A.; Panuzi, A.; Paolillo, L.; Vitagliano, A. J. Organomet. Chem. 1977, 124, 221–228. doi:10.1016/S0022-328X(00)90969-0 |
| 43. | Jose, P.; Menon, S. Bioinorg. Chem. Appl. 2007, No. 65815. doi:10.1155/2007/65815 |
| 19. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 40. | Chebanov, V. A.; Reidlinger, C.; Kanaani, H.; Wentrup, C.; Kappe, C. O.; Kollenz, G. Supramol. Chem. 2004, 16, 121–127. doi:10.1080/10610270310001614197 |
| 19. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 41. | Leber, S.; Kollenz, G.; Wentrup, C. Beilstein J. Org. Chem. 2012, 8, 738–743. doi:10.3762/bjoc.8.83 |
| 20. | Wallfisch, B. C.; Belaj, F.; Wentrup, C.; Kappe, C. O.; Kollenz, G. J. Chem. Soc., Perkin Trans. 1 2002, 599–605. doi:10.1039/b111143d |
| 38. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 39. | Kollenz, G.; Smounig, R.; Belaj, F.; Kvaskoff, D.; Wentrup, C. Beilstein J. Org. Chem. 2015, 11, 1–8. doi:10.3762/bjoc.11.1 |
| 21. | Schreier, P.; Bernreuther, A.; Huffer, M., Eds. Analysis of Chiral Organic Molecules: Methodology and Applications; De Gruyter: Berlin, 1995. doi:10.1515/9783110867855 |
| 37. | Wallfisch, B. C.; Egger, T.; Heilmayer, W.; Kappe, C. O.; Wentrup, C.; Gloe, K.; Belaj, F.; Klintschar, G.; Kollenz, G. Supramol. Chem. 2002, 14, 383–397. doi:10.1080/1061027021000003430 |
© 2018 Kollenz and Wentrup; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)