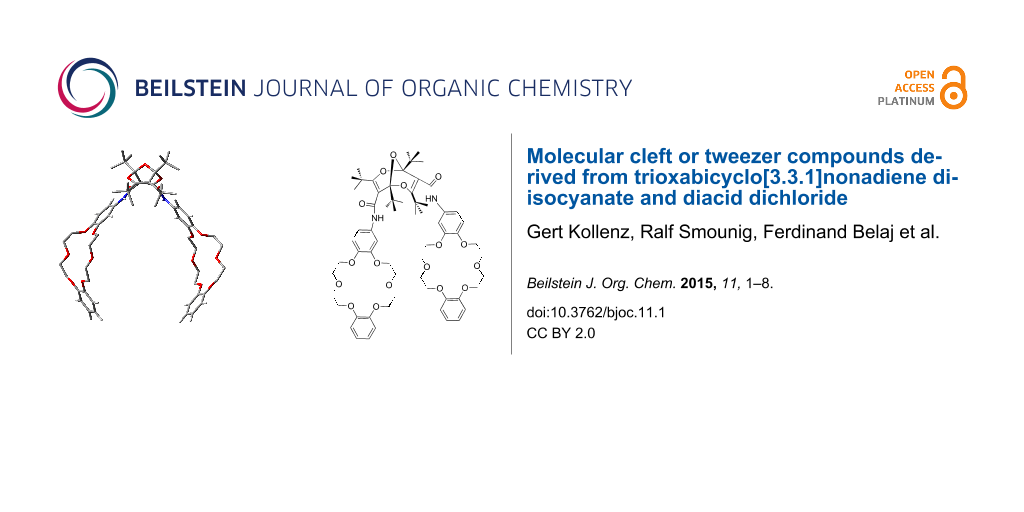
Abstract
The structures of two derivatives of the bisdioxine diisocyanate 1, the bisurea 4 and the biscarbamate 5, are established by X-ray crystallography and DFT calculations. These compounds possess endo,endo structures, in the case of the bisurea 4 with two nearly parallel pendant chains. The X-ray structures are reproduced very well by DFT calculations. Similar endo,endo conformations are calculated for the bisamide crown ether derivatives 7, where two proximate and nearly parallel crown ether units endow the molecules with a claw-like molecular cleft or tweezer structure as evidenced by an enhanced ability to extract some alkali, alkaline earth and rare earth metal ions.

Graphical Abstract
Introduction
The synthesis of the surprisingly stable, monomeric diisocyanate 1 (Figure 1) was reported recently [1]. This and several other derivatives of the unique 2,6,9-trioxabicyclo[3.3.1]nonadiene (“bisdioxine”) system, including the diacid, the diacid dichloride 2 [2] and the diethyl ester 3 [3] are readily synthesized from the stable dimer of dipivaloylketene.
![[1860-5397-11-1-1]](/bjoc/content/figures/1860-5397-11-1-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 2,6,9-Trioxabicyclo[3.3.1]nonadiene (“bisdioxine”) derivatives.
Figure 1: 2,6,9-Trioxabicyclo[3.3.1]nonadiene (“bisdioxine”) derivatives.
DFT calculations at the B3LYP/6-31G** level predict that molecules of this type can exist only in the endo,endo forms shown, i.e., with the functional groups pointing “downwards”, away from the central ether bridge [1]. The calculated endo,endo and endo,exo structures of 1 are shown in Figure 2. However, neither the endo,exo structure, nor the exo,exo isomer (not shown) represent stable energy minima [1]. Optimization of the endo,exo structure leads to ring opening to a new diisocyanate (Scheme 1). Therefore, it is of some importance to ascertain the actual molecular structures of compounds of this type by X-ray crystallography.
![[1860-5397-11-1-2]](/bjoc/content/figures/1860-5397-11-1-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: B3LYP/6-31G**-calculated structures of the stable endo,endo diisocyanate 1 (left) and the unstable endo,exo isomer (right) (hydrogen atoms are omitted for clarity).
Figure 2: B3LYP/6-31G**-calculated structures of the stable endo,endo diisocyanate 1 (left) and the unstable ...
![[1860-5397-11-1-i1]](/bjoc/content/inline/1860-5397-11-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Ring opening taking place on attempted optimization of the calculated, putative endo,exo isomer of 1.
Scheme 1: Ring opening taking place on attempted optimization of the calculated, putative endo,exo isomer of 1...
Here we report the X-ray crystal structures of two derivatives of the diisocyanate 1, viz. the di(hexylurea) 4 and the di(methyl carbamate) 5, as well as the calculated structures of diamide derivatives 7.
Results and Discussion
Diurea and dicarbamate
The di(hexylurea) 4 and the di(methyl carbamate) 5 were synthesized by addition of hexylamine and methanol, respectively, to diisocyanate 1 (Scheme 2) [1]. The crystal structure analysis of 4 confirmed the compound as 1,3,5,7-tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene-4,8-diyl-bis(3-hexylurea). All atoms lie on general positions. The asymmetric unit consists of two molecules, A and B (see Figure 3 and Figure 4) related by a pseudo-inversion center. A refinement with only one molecule in a unit cell with a’ = a/2 – corresponding to the fact that reflections with h odd [Imax 12.74(38)] are far weaker (but still significant) than those with h even [Imax 1000(27)] – resulted in an much higher R value, R1 = 0.0942. In each molecule there is one H atom (H31, H81) bonded to the nitrogen atom, which is cis to the O atom of the urea subunit. These H atoms are not involved in hydrogen bonding. The other H atoms of the same urea subunit (H(N32) and H(N82)) show intramolecular hydrogen bonds to O40 and O90 of 1.970 and 2.006 Å, respectively (dashed lines in Figure 3 and Figure 4). The four other H atoms bonded to N show intermolecular hydrogen bonds forming chains parallel to the [1] direction.
![[1860-5397-11-1-i2]](/bjoc/content/inline/1860-5397-11-1-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the di(hexylurea) and di(methyl carbamate) derivatives 4 and 5.
Scheme 2: Synthesis of the di(hexylurea) and di(methyl carbamate) derivatives 4 and 5.
![[1860-5397-11-1-3]](/bjoc/content/figures/1860-5397-11-1-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Stereoscopic ORTEP plot of molecule A of the di(hexylurea) derivative 4 with atomic numbering scheme. The ellipsoids are drawn at the 50% probability level. The H atoms bonded to N are drawn with arbitrary radii; the other H atoms as well as the disordered atoms with site occupation factors less than 0.5 were omitted for the sake of clarity. The intramolecular hydrogen bond is indicated by a dashed line (H(N32)–O40 = 1.970 Å).
Figure 3: Stereoscopic ORTEP plot of molecule A of the di(hexylurea) derivative 4 with atomic numbering schem...
![[1860-5397-11-1-4]](/bjoc/content/figures/1860-5397-11-1-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Stereoscopic ORTEP plot of molecule B of 4 with atomic numbering scheme. The ellipsoids are drawn at the 50% probability level. The H atoms bonded to N are drawn with arbitrary radii; the other H atoms as well as the disordered atoms with site occupation factors less than 0.5 were omitted for reasons of clarity. The intramolecular hydrogen bond is indicated by a dashed line (H(N82–O90) = 2.006 Å).
Figure 4: Stereoscopic ORTEP plot of molecule B of 4 with atomic numbering scheme. The ellipsoids are drawn a...
The analogous crystal structure analysis of 5 confirmed this compound as dimethyl (1,3,5,7-tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene-4,8-diyl)biscarbamate. The molecules are ordered around two-fold rotation axes through O9 (Figure 5). Each molecule is connected by a donor and an acceptor hydrogen bond to each of two adjacent molecules. Thereby chains parallel to the [101] direction are formed (see Figure S1 in Supporting Information File 1).
![[1860-5397-11-1-5]](/bjoc/content/figures/1860-5397-11-1-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Stereoscopic ORTEP plot of the di(methyl carbamate) 5 showing the atomic numbering scheme. The ellipsoids are drawn at the 50% probability level. The H atoms bonded to N are drawn with arbitrary radii; the H atoms of the methyl groups were omitted for reasons of clarity.
Figure 5: Stereoscopic ORTEP plot of the di(methyl carbamate) 5 showing the atomic numbering scheme. The elli...
It is seen in the crystal structures depicted in Figures 3–5 that compounds 4 and 5 (and therefore also 1) exist in the endo,endo structures, and each functional group is surrounded by three tert-butyl groups, which direct these functional groups away from the ether bridge. This ascertains that the functional groups with their attachments are oriented essentially parallel, i.e., away from the concave bisdioxine backbone. Furthermore, the intramolecular hydrogen bonds depicted in Figure 3 and Figure 4 ascertain that the two pendant hexylamino chains in 4 are held in close proximity.
Calculations
The structures of 4 and 5 were calculated at the B3LYP/6-31G** level, which reproduces the crystal structures very well, as demonstrated in Figure 6 and Figure 7 and in Figure S2 and Figure S3 (Supporting Information File 1).
![[1860-5397-11-1-6]](/bjoc/content/figures/1860-5397-11-1-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The lowest-energy calculated structure of 4 (B3LYP/6-31G**; for other conformers and full details see Supporting Information File 1). Bond lengths are given in Å and angles in degrees. The interchain 2 Å hydrogen bond is indicated by a dashed line.
Figure 6: The lowest-energy calculated structure of 4 (B3LYP/6-31G**; for other conformers and full details s...
![[1860-5397-11-1-7]](/bjoc/content/figures/1860-5397-11-1-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Calculated structure of 5 (B3LYP/6-31G**). Bond lengths are given in Å and angles in degrees. For another view and full details see Supporting Information File 1).
Figure 7: Calculated structure of 5 (B3LYP/6-31G**). Bond lengths are given in Å and angles in degrees. For a...
While there are several possible conformations of 4 and 5 (see Figure S2 and Figure S3, Supporting Information File 1), the lowest-energy conformers shown in Figure 6 and Figure 7 are in very good agreement with the X-ray structures. The nearly parallel dangling chains in the lowest-energy conformer of 4 are particularly noteworthy (Figure 6). As in the crystal structure, the two chains are held in this orientation by 2 Å hydrogen bonds. The calculated structure of the diester 3 (Figure S4, Supporting Information File 1) is also in very good agreement with the previously determined crystal structure [3], but unlike the structures of 4 and 5 described above, the two ester moieties in compound 3 point away from each other with an angle of approximately 120° between them.
Diamides
The endo,endo-structures of compounds 4 and 5 endow them with the character of molecular clefts or tweezers [4-10]. This suggests that other derivatives, such as the bis-crown ether diamides 7 (Scheme 3), would possess similar structures with parallel substituents. Accordingly, we investigated the structures of compound 7a and 7b computationally. Again, there are several possible conformers of each compound, but the lowest-energy conformers shown in Figure 8 and Figure 9 reveal the claw-like character of the dangling chains. This is particularly noteworthy in compound 7b, where a cavity between the two crown-ether moieties is apparent (Figure 9). This suggests that this molecule might be able to form strong complexes with suitable metal ions.
![[1860-5397-11-1-i3]](/bjoc/content/inline/1860-5397-11-1-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of bis-crown ether diamides 7.
Scheme 3: Synthesis of bis-crown ether diamides 7.
![[1860-5397-11-1-8]](/bjoc/content/figures/1860-5397-11-1-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Calculated structure of 7a-a (B3LYP/6-31G**). Bond lengths are given in Å and angles in degrees. For other conformers (7a-b and 7a-c) and full details, see Supporting Information File 1.
Figure 8: Calculated structure of 7a-a (B3LYP/6-31G**). Bond lengths are given in Å and angles in degrees. Fo...
![[1860-5397-11-1-9]](/bjoc/content/figures/1860-5397-11-1-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Calculated structure of 7b-a (B3LYP/6-31G**). For other conformers (7b-b and 7b-c) and full details, see Supporting Information File 1.
Figure 9: Calculated structure of 7b-a (B3LYP/6-31G**). For other conformers (7b-b and 7b-c) and full details...
Cation extraction experiments
The endo,endo structures of bis-crown ether diamides 7 with parallel crown ether moieties would be expected to endow them with enhanced complexation abilities. The extraction of metal ions from aqueous to chloroform solution was examined for the picrates of Na+, K+, Ca2+ and Ce3+ by using a previously described procedure [11]. For comparison, the performance of the crown ether amines 6a and 6b was evaluated with the same extraction procedure. It is seen in Table 1 that there is a modest increase in the extraction ability of 7a (6–13%) over 6a (4–8%), but a more distinct improvement for 7b (11–15%) compared to 6b (3–9%).
Conclusion
X-ray structure determinations confirmed the endo,endo structures of the bisurea 4 and biscarbamate 5, which in turn confirm the endo,endo structure of the diisocyanate 1. Bis-crown ether diamide derivatives 7 also feature endo,endo structures as confirmed by DFT calculations at the B3LYP/6-31G** level. This endows them with claw-like molecular cleft or tweezer properties as manifested in enhanced abilities to extract selected alkali and alkaline earth metal and rare earth ions. The ready availability of diisocyanate 1 and diacid dichloride 2 [1-3,11] paves the way for the synthesis of many other types of compounds with hairpin turns and parallel pendant chains.
Experimental
General
Preparations of the di(hexylurea) 4, the di(ethyl urethane) 5 and the diamides 7 were carried out as previously described [1,11-13].
Crystallography
Crystal structures are represented in ORTEP [14]. Tables of crystal data and bond lengths and angles are presented in Supporting Information File 1. The full data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. CCDC numbers 925965 and 925966.
X-ray diffraction data for 4
All the measurements were performed using graphite-monochromatized MoKα radiation at 100 K: C36H66N4O5, Mr 634.93, monoclinic, space group P21/c, a = 21.0815(7) Å, b = 20.1551(6) Å, c = 17.9547(6) Å, β = 92.203(2)°, V = 7623.3(4) Å3, Z = 8, dcalc = 1.106 g cm−3, μ = 0.073 mm−1. A total of 34296 reflections were collected (Θmax = 25.0°), from which 13358 were unique (Rint = 0.0344), with 8670 having I > 2σ(I). The structure was solved by direct methods (SHELXS-97) [15] and refined by full-matrix least-squares techniques against F2 (SHELXL-97) [15]. Three of the eight tert-butyl groups as well as two of the four hexyl groups in the two molecules of the asymmetric unit are disordered over two orientations. Their site occupation factors were refined to add to unity each and equivalent bonds were restrained to have the same lengths. The same anisotropic displacement parameters were used for equivalent atoms of the disordered groups. The other non-hydrogen atoms were refined with anisotropic displacement parameters without any constraints. The H atoms of the NH groups were refined with N–H distances of 0.88 Å, but without any further positional constraints, and with a common isotropic displacement parameter. The H atoms of the ordered CH2 groups were refined with their isotropic displacement parameters fixed to 1.2 times Ueq of the C atom they are bonded to and idealized geometry with approximately tetrahedral angles and C–H distances of 0.99 Å. The H atoms of the methyl groups were refined with their isotropic displacement parameters fixed to 1.3 times Ueq of the C atom they are bonded to and idealized geometry with tetrahedral angles, staggered conformation, and C–H distances of 0.98 Å. For 902 parameters final R indices of R1 = 0.0670 and wR2 = 0.1708 (GOF = 1.015) were obtained. The largest peak in a difference Fourier map was 0.400 eÅ−3.
X-ray diffraction data for 5
All the measurements were performed using graphite-monochromatized MoKα radiation at 100 K: C26H44N2O7, Mr 496.63, monoclinic, space group C2/c, a = 16.8698(6) Å, b = 10.4411(6) Å, c = 17.1497(7) Å, β = 113.903(3)°, V = 2761.7(2) Å3, Z = 4, dcalc = 1.194 g cm−3, μ = 0.086 mm−1. A total of 11810 reflections were collected (Θmax = 27.5°), from which 3165 were unique (Rint = 0.0254), with 2622 having I > 2σ(I). The structure was solved by direct methods (SHELXS-97) [15] and refined by full-matrix least-squares techniques against F2 (SHELXL-97) [15]. The non-hydrogen atoms were refined with anisotropic displacement parameters without any constraints. The H atom bonded to N4 was refined without any positional constraints with an individual isotropic displacement parameter. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometries with tetrahedral angles, enabling rotation around the X–C bond, and C–H distances of 0.98 Å. For 177 parameters final R indices of R1 = 0.0377 and wR2 = 0.1058 (GOF = 1.045) were obtained. The largest peak in a difference Fourier map was 0.408 eÅ−3.
Extraction experiments
The extractions of ions from H2O to CHCl3 with the aid of bis-crown ether amides 7a and 7c (Table 1) were carried out as described previously [11,16]. Briefly, equimolar amounts of picrate and crown ether in H2O and CHCl3 respectively were shaken vigorously for 10 min. The extent of extraction into CHCl3 was measured by UV spectrophotometry at 354 nm.
Supporting Information
| Supporting Information File 1: Hydrogen bonding pattern in 5, calculated structures of compounds 3, 4, 5, 7a and 7b. X-ray structural data, bond lengths and bond angles for 4 and 5, and preparative and computational details. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024
Return to citation in text: [1] [2] -
Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109
Return to citation in text: [1] [2] [3] -
Chen, C. W.; Whitlock, H. W., Jr. J. Am. Chem. Soc. 1978, 100, 4921–4922. doi:10.1021/ja00483a063
Return to citation in text: [1] -
Rebek, J., Jr.; Marshall, L.; Wolak, R.; McManis, J. J. Am. Chem. Soc. 1984, 106, 1170–1171. doi:10.1021/ja00316a088
Return to citation in text: [1] -
Rebek, J., Jr.; Askew, B.; Killoran, M.; Nemeth, D.; Lin, F. T. J. Am. Chem. Soc. 1987, 109, 2426–2431. doi:10.1021/ja00242a029
Return to citation in text: [1] -
Rebek, J., Jr. Angew. Chem., Int. Ed. Engl. 1990, 29, 245–255. doi:10.1002/anie.199002451
Return to citation in text: [1] -
Zimmerman, S. C. Top. Curr. Chem. 1993, 165, 71–102. doi:10.1007/BFb0111281
Return to citation in text: [1] -
Harmata, M. Acc. Chem. Res. 2004, 37, 862–873. doi:10.1021/ar030164v
Return to citation in text: [1] -
Klärner, F.-G.; Schrader, G. Acc. Chem. Res. 2013, 46, 967–978. doi:10.1021/ar300061c
Return to citation in text: [1] -
Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y
Return to citation in text: [1] [2] [3] [4] -
Smounig, R. Ph.D. Thesis, Karl Franzens Universität Graz, Austria, 1998.
Return to citation in text: [1] -
Heilmayer, W.; Smounig, R.; Gruber, K.; Fabian, W. M. F.; Reidlinger, C.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Tetrahedron 2004, 60, 2857–2867. doi:10.1016/j.tet.2004.01.058
Return to citation in text: [1] -
Johnson, C. K. ORTEP, Report ORNL-3794; Oak Ridge National Laboratory: Tennessee, USA, 1965.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] [2] [3] [4] -
Haberz, P.; Belaj, F.; Gloe, K.; Wentrup, C.; Kollenz, G. Supramol. Chem. 2012, 24, 279–284. doi:10.1080/10610278.2012.658393
Return to citation in text: [1]
| 15. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 11. | Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y |
| 16. | Haberz, P.; Belaj, F.; Gloe, K.; Wentrup, C.; Kollenz, G. Supramol. Chem. 2012, 24, 279–284. doi:10.1080/10610278.2012.658393 |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 15. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 15. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 3. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 14. | Johnson, C. K. ORTEP, Report ORNL-3794; Oak Ridge National Laboratory: Tennessee, USA, 1965. |
| 2. | Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024 |
| 15. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 4. | Chen, C. W.; Whitlock, H. W., Jr. J. Am. Chem. Soc. 1978, 100, 4921–4922. doi:10.1021/ja00483a063 |
| 5. | Rebek, J., Jr.; Marshall, L.; Wolak, R.; McManis, J. J. Am. Chem. Soc. 1984, 106, 1170–1171. doi:10.1021/ja00316a088 |
| 6. | Rebek, J., Jr.; Askew, B.; Killoran, M.; Nemeth, D.; Lin, F. T. J. Am. Chem. Soc. 1987, 109, 2426–2431. doi:10.1021/ja00242a029 |
| 7. | Rebek, J., Jr. Angew. Chem., Int. Ed. Engl. 1990, 29, 245–255. doi:10.1002/anie.199002451 |
| 8. | Zimmerman, S. C. Top. Curr. Chem. 1993, 165, 71–102. doi:10.1007/BFb0111281 |
| 9. | Harmata, M. Acc. Chem. Res. 2004, 37, 862–873. doi:10.1021/ar030164v |
| 10. | Klärner, F.-G.; Schrader, G. Acc. Chem. Res. 2013, 46, 967–978. doi:10.1021/ar300061c |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 2. | Kappe, C. O.; Kollenz, G.; Fabian, W. M. F.; Wentrup, C.; Färber, G. J. Org. Chem. 1993, 58, 3361–3367. doi:10.1021/jo00064a024 |
| 3. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 11. | Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y |
| 3. | Kremsner, J.; Wallfisch, B. C.; Belaj, F.; Uray, G.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Eur. J. Org. Chem. 2008, 3382–3388. doi:10.1002/ejoc.200800109 |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 11. | Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y |
| 12. | Smounig, R. Ph.D. Thesis, Karl Franzens Universität Graz, Austria, 1998. |
| 13. | Heilmayer, W.; Smounig, R.; Gruber, K.; Fabian, W. M. F.; Reidlinger, C.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Tetrahedron 2004, 60, 2857–2867. doi:10.1016/j.tet.2004.01.058 |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 1. | Smounig, R.; Leber, S.; Kvaskoff, D.; Kollenz, G.; Wentrup, C. RSC Adv. 2012, 2, 7743–7746. doi:10.1039/c2ra21073h |
| 11. | Smounig, R.; Kappe, C. O.; Wentrup, C.; Kollenz, G. Monatsh. Chem. 2003, 134, 509–518. doi:10.1007/s00706-002-0547-y |
© 2015 Kollenz et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)