Abstract
Cryptophycins are naturally occurring cytotoxins with great potential for chemotherapy. Since targeted therapy provides new perspectives for treatment of cancer, new potent analogues of cytotoxic agents containing functional groups for conjugation to homing devices are required. We describe the design, synthesis and biological evaluation of three new unit B cryptophycin analogues. The O-methyl group of the unit B D-tyrosine analogue was replaced by an O-(allyloxyethyl) moiety, an O-(hydroxyethyl) group, or an O-(((azidoethoxy)ethoxy)ethoyxethyl) substituent. While the former two maintain cytotoxicity in the subnanomolar range, the attachment of the triethylene glycol spacer with a terminal azide results in a complete loss of activity. Docking studies of the novel cryptophycin analogues to β-tubulin provided a rationale for the observed cytotoxicities.

Graphical Abstract
Introduction
Cryptophycins are natural occurring cyclic depsipeptides that were first isolated from cyanobacteria Nostoc sp. ATCC 53789 in 1990 [1]. Cryptophycins target tubulin, in particular the peptide site of the vinca domain. They block microtubule formation, inhibiting their assembly and, hence, are antimitotic agents [2,3]. Their high cytotoxicity prompted manifold studies that were initially focussed on the total synthesis and structure–activity relationships [4-20]. This work resulted in the identification of cryptophycin-52, a highly biologically active analogue of cryptophycin-1 (Figure 1).
![[1860-5397-14-109-1]](/bjoc/content/figures/1860-5397-14-109-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cryptophycin-1 (1) and -52 (2).
Figure 1: Cryptophycin-1 (1) and -52 (2).
Eli Lilly took cryptophycin-52 into clinical trials. Although almost half of the patients obtained a benefit from the treatment, neurotoxic side effects forced the termination of the clinical trials [21-23]. In order to overcome the side effects of cryptophycin-52 and to better understand the fundamental structure for biological activity, numerous structure–activity relationship studies have been carried out [24-35]. However, like cryptophycin-52, the new analogues were not selective against cancer cells making them not better than its parent.
In recent years the targeted delivery of cytotoxic agents has emerged as a highly promising method to tackle selectivity issues [36-40]. Cryptophycin-52 and many analogues lack an addressable group to conjugate the toxin to a homing device. For this reason, new analogues containing functional groups that would allow the conjugation of a homing device were developed [41-46]. Some of these functionalized analogues have been recently used for the preparation of antibody–drug conjugates (ADCs) and peptide–drug conjugates (PDCs) [46-51]. Nevertheless, there is still a strong need of novel cryptophycin analogues with maintained activity containing a suitable functional group that would allow the conjugation to the homing device. Cryptophycin-1 contains a methoxy group in the aromatic ring of the unit B, which is a chlorinated derivative of D-tyrosine. Different chains for unit B have been investigated, albeit the elongation of the methoxy group is still unknown. Therefore, in the current study, we embarked on the synthesis of novel cryptophycin analogues containing different substituents at the phenolic hydroxy group of the unit B. We intended to investigate whether the high biological activity of the parent compound is retained and thus, construction of ADCs and PDCs would be feasible. This preparation could be done using traceless cleavable linkers that are sensitive to the distinct physiology of the tumour with enhanced level and activity of specific enzymes. The connection between the payload and the linker is of crucial importance since its stability can dramatically change the release and thus, the activity of the compound. For this reason, the included functional groups were designed with the consideration to provide appropriate stability and activity to the future conjugate.
Results and Discussion
Design and synthesis
Previous docking studies have postulated that the methyl group of unit B is not involved in the cryptophycin–tubulin interaction [52]. Moreover, its absence did not produce a dramatic loss of activity [24].
Based on this, we designed cryptophycin analogues modified in the unit B. Instead of the O-methyl group that is present in the natural cryptophycin, we attached a hydroxyethyl group or a triethylene glycol chain terminated with an alcohol or azide, respectively. These functional groups would allow the conjugation of the novel cryptophycin analogues across an appropriate linker to an antibody or peptide. Either a virtually uncleavable triazole (introduced by CuAAC) or scissile ester, carbonate, or carbamate moieties were taken into account.
The synthesis of the modified unit B (Scheme 1) started with the preparation of the two different spacers that were later connected to the phenol. Starting from triethylene glycol (3) or 2-allyloxyethanol (7) tosylations and nucleophilic displacements by azide or iodide substitution provided 6 and 9 in good yields. O-Alkylation of Boc-protected 3-chlorinated D-tyrosine 10 with 6 or 9 gave 11 and 12, again in satisfactory yields (81–85%). Saponification of the ester moiety in 11 and 12 that was formed concomitantly with the O-alkylation in the previous reaction provided Boc-protected modified units B 13 and 14 in 76 and 90% yield, respectively.
![[1860-5397-14-109-i1]](/bjoc/content/inline/1860-5397-14-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of modified unit B (13 and 14). Reagents and conditions: (a) 1) TsCl, DMAP, Et3N, CH2Cl2, rt, 4 h; 2) NaN3, DMF, 70 °C, overnight; (b) TsCl, Et3N, CH2Cl2, rt, overnight; (c) NaI, acetone, reflux, overnight; (d) TsCl, Et3N, CH2Cl2, rt, overnight; (e) NaI, acetone, reflux, overnight; (f) 6 or 9, K2CO3, DMF, 50 °C, overnight; (g) LiOH, H2O/MeOH/THF 1:1:1, rt, 2 h.
Scheme 1: Synthesis of modified unit B (13 and 14). Reagents and conditions: (a) 1) TsCl, DMAP, Et3N, CH2Cl2,...
The synthesis of units C–D and A succeeded as previously described in the literature; unit A (15) and C–D (16) were connected by Yamaguchi esterification to give 17 (Scheme 2) [45]. Then, Fmoc was cleaved from the N-terminus of unit C–D–A (17) using piperidine and the resulting crude amine was coupled to the corresponding modified unit B (13 or 14), affording the according linear cryptophycins 18 and 19 in acceptable yields (51–59%). Compounds 18 and 19 were treated with trifluoroacetic acid for simultaneous Boc and t-Bu removal, which also cleaved the dioxolane ring. Subsequently, macrolactamization was performed under pseudo-high-dilution conditions to afford 20 and 21 as described previously [16]. Then the diol was transformed into the epoxide following a three-step one-pot reaction as extensively used in the synthesis of cryptophycin analogues [46]. Cryptophycin analogues 22 and 23 were obtained in good purity after column chromatography. The allyl ether in 23 was cleaved using Pd(PPh3)4 as Pd(0) source and phenylsilane as scavenger to obtain the cryptophycin analogue 24 in good purity.
![[1860-5397-14-109-i2]](/bjoc/content/inline/1860-5397-14-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of cryptophycin analogues 22, 23 and 24. Reagents and conditions: (a) 4-DMAP, 2,4,6-trichlorobenzoyl chloride, Et3N, THF, 0 °C, 3 h; (b) 1) piperidine, DMF, rt, 2 h; 2) 13 or 14, HOAt, EDC·HCl, Et3N, CH2Cl2, 0 °C → rt, overnight; (c) 1) TFA/CH2Cl2/H2O, rt, 2 h; 2) HATU, HOAt, DIPEA, DMF, rt, slow addition + 2 h; (d) 1) (CH3O)3CH, PPTS, CH2Cl2, rt, 2 h; 2) AcBr, CH2Cl2, rt, 4 h; 3) K2CO3, DME/ethylene glycol (2:1 v/v), rt, 5 min; (e) Pd(PPh3)4, phenylsilane, CH2Cl2, rt, 7 h.
Scheme 2: Synthesis of cryptophycin analogues 22, 23 and 24. Reagents and conditions: (a) 4-DMAP, 2,4,6-trich...
Biological evaluation
The biological activity of the modified unit B analogues was determined in a cell viability assay using the human cervix carcinoma cell line KB-3-1 (Table 1). The cryptophycin analogue 22 showed a dramatic loss of activity compared to cryptophycin-52 (2), while analogues 23 and 24 showed a reduced cytotoxicity although their IC50 values are still in the low nanomolar range. The observed dramatic loss of activity of analogue 22 could be due to its poor internalization or the modification could alter the interaction with tubulin. In order to get an extensive knowledge of the novel analogues, we embarked in docking and modelling studies, herein reported, and internalization studies are ongoing in our research group.
Docking and modelling of cryptophycin derivatives
There is no X-ray analysis of cryptophycin–tubulin complexes available to provide information on the binding site. Based on biochemical evidence, binding close to the vinca-alkaloid binding site of β-tubulin, the so called “peptide-site”, has been proposed [2,52,53]. We performed a docking study to explain the different affinities of the newly synthesized derivatives. The parent compound 2 scored highest with respect to β-tubulin binding (Table 2). Three hydrogen bonds were detected to key residues in the peptide binding pocket of the vinca domain (Lys176, Val177 and Tyr210). Other than previously reported [52], the methoxy group of subunit B forms a hydrogen bond with Lys176 (Figure 2). Another binding mode of 2 with high binding affinity and hydrogen bond formation did not involve any interaction of subunit B, yet it was oriented towards the GDP binding site that might influence GTP hydrolysis.
![[1860-5397-14-109-2]](/bjoc/content/figures/1860-5397-14-109-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Binding mode of 2, showing the interaction to the vinca domain peptide binding pocket (blue). Hydrogen bonds are shown as yellow dots with the interacting amino acid residues in magenta.
Figure 2: Binding mode of 2, showing the interaction to the vinca domain peptide binding pocket (blue). Hydro...
Compound 22 with the triethylene glycol-based substituent prevents correct binding, the binding energy was decreased and mainly nonspecific interactions outside the binding pocket were observed (Figure 3). This was not the case for the other derivatives 23 and 24 (Figure 4).
![[1860-5397-14-109-3]](/bjoc/content/figures/1860-5397-14-109-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Docking of 22 to the vinca domain of β-tubulin. Surface and backbone of β-tubulin are shown in blue, GDP in yellow. No hydrogen bond formation was detected. The orientation of the azidoethoxy-ethoxyethyl substituent prevents the inhibitor from the correct interaction with the protein. The epoxide and benzyl group of subunit A are pointing away from the binding pocket.
Figure 3: Docking of 22 to the vinca domain of β-tubulin. Surface and backbone of β-tubulin are shown in blue...
![[1860-5397-14-109-4]](/bjoc/content/figures/1860-5397-14-109-4.png?scale=2.0&max-width=1024&background=FFFFFF)
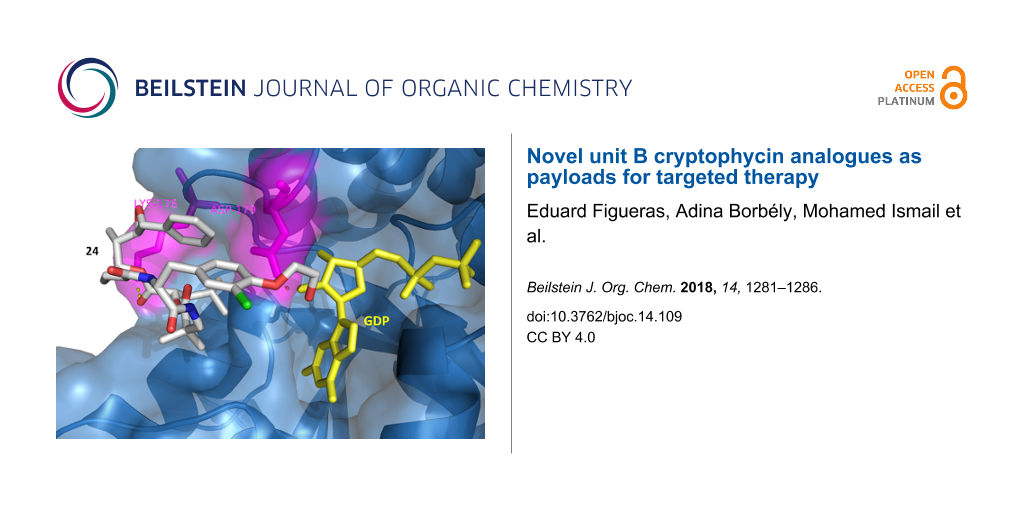
Figure 4: Docking of 24 to β-tubulin. Surface and backbone of β-tubulin are shown in blue, GDP in yellow. H-bonding (yellow dots) was detected with Lys176 and Asp179 in magenta. The benzyl group and the epoxide of subunit A are directed towards the peptide binding pocket, while the hydroxyethyl group is positioned towards the GDP binding pocket forming an H-bond with Asp179.
Figure 4: Docking of 24 to β-tubulin. Surface and backbone of β-tubulin are shown in blue, GDP in yellow. H-b...
Besides hydrogen bond formation and binding affinity of inhibitors 2, 23 and 24, π-interactions and hydrophobic contacts with the binding pocket of the vinca domain were detected that would in turn increase the affinity of the inhibitor and its effect on the protein (Supporting Information File 1, Table S1).
Conclusion
In summary, three new cryptophycin analogues with a modified unit B have been designed and successfully synthesized. The novel analogues were less active than cryptophycin-52 in the KB-3-1 cell line. Analogue 22 showed a dramatic loss of activity whereas analogues 23 and 24 showed a reduced activity but were still very cytotoxic.
Supporting Information
| Supporting Information File 1: Experimental part and analytical data. | ||
| Format: PDF | Size: 3.9 MB | Download |
References
-
Schwartz, R. E.; Hirsch, C. F.; Sesin, D. F.; Flor, J. E.; Chartrain, M.; Fromtling, R. E.; Harris, G. H.; Salvatore, M. J.; Liesch, J. M.; Yudin, K. J. Ind. Microbiol. 1990, 5, 113–123. doi:10.1007/BF01573860
Return to citation in text: [1] -
Bai, R.; Schwartz, R. E.; Kepler, J. A.; Pettit, G. R.; Hamel, E. Cancer Res. 1996, 56, 4398–4406.
Return to citation in text: [1] [2] -
Smith, C. D.; Zhang, X. J. Biol. Chem. 1996, 271, 6192–6198. doi:10.1074/jbc.271.11.6192
Return to citation in text: [1] -
Barrow, R. A.; Hemscheidt, T.; Liang, J.; Paik, S.; Moore, R. E.; Tius, M. A. J. Am. Chem. Soc. 1995, 117, 2479–2490. doi:10.1021/ja00114a011
Return to citation in text: [1] -
Kobayashi, M.; Wang, W.; Ohyabu, N.; Kurosu, M.; Kitagawa, I. Chem. Pharm. Bull. 1995, 43, 1598–1600. doi:10.1248/cpb.43.1598
Return to citation in text: [1] -
de Muys, J.-M.; Rej, R.; Nguyen, D.; Go, B.; Fortin, S.; Lavallée, J.-F. Bioorg. Med. Chem. Lett. 1996, 6, 1111–1116. doi:10.1016/0960-894X(96)00182-5
Return to citation in text: [1] -
Rej, R.; Nguyen, D.; Go, B.; Fortin, S.; Lavallée, J.-F. J. Org. Chem. 1996, 61, 6289–6295. doi:10.1021/jo960816b
Return to citation in text: [1] -
Gardinier, K. M.; Leahy, J. W. J. Org. Chem. 1997, 62, 7098–7099. doi:10.1021/jo971645t
Return to citation in text: [1] -
Norman, B. H.; Hemscheidt, T.; Schultz, R. M.; Andis, S. L. J. Org. Chem. 1998, 63, 5288–5294. doi:10.1021/jo980536r
Return to citation in text: [1] -
Georg, G. I.; Ali, S. M.; Stella, V. J.; Waugh, W. N.; Himmes, R. H. Bioorg. Med. Chem. Lett. 1998, 8, 1959–1962. doi:10.1016/S0960-894X(98)00356-4
Return to citation in text: [1] -
Ghosh, A. K.; Bischoff, A. Org. Lett. 2000, 2, 1573–1575. doi:10.1021/ol000058i
Return to citation in text: [1] -
Eggen, M.; Mossman, C. J.; Buck, S. B.; Nair, S. K.; Bhat, L.; Ali, S. M.; Reiff, E. A.; Boge, T. C.; Georg, G. I. J. Org. Chem. 2000, 65, 7792–7799. doi:10.1021/jo000767+
Return to citation in text: [1] -
Murakami, N.; Wang, W.; Ohyabu, N.; Ito, T.; Tamura, S.; Aoki, S.; Kobayashi, M.; Kitagawa, I. Tetrahedron 2000, 56, 9121–9128. doi:10.1016/S0040-4020(00)00766-3
Return to citation in text: [1] -
Murakami, N.; Tamura, S.; Wang, W.; Takagi, T.; Kobayashi, M. Tetrahedron 2001, 57, 4323–4336. doi:10.1016/S0040-4020(01)00339-8
Return to citation in text: [1] -
Ghosh, A. K.; Swanson, L. J. Org. Chem. 2003, 68, 9823–9826. doi:10.1021/jo035077v
Return to citation in text: [1] -
Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem. – Eur. J. 2005, 11, 4667–4677. doi:10.1002/chem.200500282
Return to citation in text: [1] [2] -
Danner, P.; Bauer, M.; Phukan, P.; Maier, M. E. Eur. J. Org. Chem. 2005, 317–325. doi:10.1002/ejoc.200400558
Return to citation in text: [1] -
Eißler, S.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2008, 273–277. doi:10.1055/s-2007-1000868
Return to citation in text: [1] -
Sammet, B.; Radzey, H.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2009, 417–420. doi:10.1055/s-0028-1087541
Return to citation in text: [1] -
Sammet, B.; Brax, M.; Sewald, N. Beilstein J. Org. Chem. 2011, 7, 243–245. doi:10.3762/bjoc.7.32
Return to citation in text: [1] -
Sessa, C.; Weigang-Köhler, K.; Pagani, O.; Greim, G.; Mor, O.; De Pas, T.; Burgess, M.; Weimer, I.; Johnson, R. Eur. J. Cancer 2002, 38, 2388–2396. doi:10.1016/S0959-8049(02)00489-6
Return to citation in text: [1] -
Stevenson, J. P.; Sun, W.; Gallagher, M.; Johnson, R.; Vaughn, D.; Schuchter, L.; Algazy, K.; Hahn, S.; Enas, N.; Ellis, D.; Thornton, D.; O’Dwyer, P. J. Clin. Cancer Res. 2002, 8, 2524–2529.
Return to citation in text: [1] -
D’Agostino, G.; Del Campo, J.; Mellado, B.; Izquierdo, M. A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J. L.; Scambia, G. Int. J. Gynecol. Cancer 2006, 16, 71–76. doi:10.1111/j.1525-1438.2006.00276.x
Return to citation in text: [1] -
Patel, V. F.; Andis, S. L.; Kennedy, J. H.; Ray, J. E.; Schultz, R. M. J. Med. Chem. 1999, 42, 2588–2603. doi:10.1021/jm980706s
Return to citation in text: [1] [2] -
Varie, D. L.; Shih, C.; Hay, D. A.; Andis, S. L.; Corbett, T. H.; Gossett, L. S.; Janisse, S. K.; Martinelli, M. J.; Moher, E. D.; Schultz, R. M.; Toth, J. E. Bioorg. Med. Chem. Lett. 1999, 9, 369–374. doi:10.1016/S0960-894X(98)00748-3
Return to citation in text: [1] -
Shih, C.; Gossett, L. S.; Gruber, J. M.; Sue Grossman, C.; Andis, S. L.; Schultz, R. M.; Worzalla, J. F.; Corbett, T. H.; Metz, J. T. Bioorg. Med. Chem. Lett. 1999, 9, 69–74. doi:10.1016/S0960-894X(98)00682-9
Return to citation in text: [1] -
Vidya, R.; Eggen, M. J.; Georg, G. I.; Himes, R. H. Bioorg. Med. Chem. Lett. 2003, 13, 757–760. doi:10.1016/S0960-894X(02)01023-5
Return to citation in text: [1] -
Buck, S. B.; Huff, J. K.; Himes, R. H.; Georg, G. I. J. Med. Chem. 2004, 47, 696–702. doi:10.1021/jm030278c
Return to citation in text: [1] -
Buck, S. B.; Huff, J. K.; Himes, R. H.; Georg, G. I. J. Med. Chem. 2004, 47, 3697–3699. doi:10.1021/jm030555f
Return to citation in text: [1] -
Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem. – Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750
Return to citation in text: [1] -
Liu, W. L.; Zhang, J. C.; Jiang, F. Q.; Fu, L. Arch. Pharm. 2009, 342, 577–583. doi:10.1002/ardp.200900067
Return to citation in text: [1] -
Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473
Return to citation in text: [1] -
Weiß, C.; Bogner, T.; Sammet, B.; Sewald, N. Beilstein J. Org. Chem. 2012, 8, 2060–2066. doi:10.3762/bjoc.8.231
Return to citation in text: [1] -
Kumar, A.; Kumar, M.; Sharma, S.; Guru, S. K.; Bhushan, S.; Shah, B. A. Eur. J. Med. Chem. 2015, 93, 55–63. doi:10.1016/j.ejmech.2014.11.068
Return to citation in text: [1] -
Weiss, C.; Figueras, E.; Borbely, A. N.; Sewald, N. J. Pept. Sci. 2017, 23, 514–531. doi:10.1002/psc.3015
Return to citation in text: [1] -
Chari, R. V. J.; Miller, M. L.; Widdison, W. C. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. doi:10.1002/anie.201307628
Return to citation in text: [1] -
Srinivasarao, M.; Low, P. S. Chem. Rev. 2017, 117, 12133–12164. doi:10.1021/acs.chemrev.7b00013
Return to citation in text: [1] -
Krall, N.; Scheuermann, J.; Neri, D. Angew. Chem., Int. Ed. 2013, 52, 1384–1402. doi:10.1002/anie.201204631
Return to citation in text: [1] -
Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. Curr. Top. Med. Chem. 2016, 16, 314–329. doi:10.2174/1568026615666150701114343
Return to citation in text: [1] -
Le Joncour, V.; Laakkonen, P. Bioorg. Med. Chem. 2018, 26, 2797–2806. doi:10.1016/j.bmc.2017.08.052
Return to citation in text: [1] -
Al-awar, R. S.; Ray, J. E.; Schultz, R. M.; Andis, S. L.; Kennedy, J. H.; Moore, R. E.; Liang, J.; Golakoti, T.; Subbaraju, G. V.; Corbett, T. H. J. Med. Chem. 2003, 46, 2985–3007. doi:10.1021/jm0203884
Return to citation in text: [1] -
Al-Awar, R. S.; Corbett, T. H.; Ray, J. E.; Polin, L.; Kennedy, J. H.; Wagner, M. M.; Williams, D. C. Mol. Cancer Ther. 2004, 3, 1061–1067.
Return to citation in text: [1] -
Kotoku, N.; Kato, T.; Narumi, F.; Ohtani, E.; Kamada, S.; Aoki, S.; Okada, N.; Nakagawa, S.; Kobayashi, M. Bioorg. Med. Chem. 2006, 14, 7446–7457. doi:10.1016/j.bmc.2006.07.019
Return to citation in text: [1] -
Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. J. Org. Chem. 2010, 75, 6953–6960. doi:10.1021/jo101563s
Return to citation in text: [1] -
Weiss, C.; Sammet, B.; Sewald, N. Nat. Prod. Rep. 2013, 30, 924–940. doi:10.1039/c3np70022d
Return to citation in text: [1] [2] -
Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2013, 56, 1853–1864. doi:10.1021/jm301346z
Return to citation in text: [1] [2] [3] -
Bouchard, H.; Brun, M.-P.; Commerçon, A.; Zhang, J. Novel conjugates, preparation thereof, and therapeutic use thereof. WO Patent WO2011/001052, Jan 6, 2011.
Return to citation in text: [1] -
Verma, V. A.; Pillow, T. H.; Depalatis, L.; Li, G.; Phillips, G. L.; Polson, A. G.; Raab, H. E.; Spencer, S.; Zheng, B. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. doi:10.1016/j.bmcl.2014.12.070
Return to citation in text: [1] -
Steinkuhler, M. C.; Gallinari, M. P.; Osswald, B.; Sewald, N.; Ritzefeld, M.; Frese, M.; Figueras, E.; Pethö, L. Cryptophycin-based antibody-drug conjugates with novel self-immolative linkers. WO Patent WO2016/146638 A1, Sept 22, 2016.
Return to citation in text: [1] -
Bigot, A.; Bouchard, H.; Brun, M.-P.; Clerc, F.; Zhang, J. Novel cryptophycin compounds and conjugates, their preparation and their therapeutic use. WO Patent WO2017/076998 A1, May 11, 2017.
Return to citation in text: [1] -
Chen, H.; Lin, Z.; Arnst, K. E.; Miller, D. D.; Li, W. Molecules 2017, 22, No. 1281. doi:10.3390/molecules22081281
Return to citation in text: [1] -
Mitra, A.; Sept, D. Biochemistry 2004, 43, 13955–13962. doi:10.1021/bi0487387
Return to citation in text: [1] [2] [3] -
Bai, R.; Pettit, G. R.; Hamel, E. Biochem. Pharmacol. 1990, 39, 1941–1949. doi:10.1016/0006-2952(90)90613-P
Return to citation in text: [1]
| 1. | Schwartz, R. E.; Hirsch, C. F.; Sesin, D. F.; Flor, J. E.; Chartrain, M.; Fromtling, R. E.; Harris, G. H.; Salvatore, M. J.; Liesch, J. M.; Yudin, K. J. Ind. Microbiol. 1990, 5, 113–123. doi:10.1007/BF01573860 |
| 24. | Patel, V. F.; Andis, S. L.; Kennedy, J. H.; Ray, J. E.; Schultz, R. M. J. Med. Chem. 1999, 42, 2588–2603. doi:10.1021/jm980706s |
| 25. | Varie, D. L.; Shih, C.; Hay, D. A.; Andis, S. L.; Corbett, T. H.; Gossett, L. S.; Janisse, S. K.; Martinelli, M. J.; Moher, E. D.; Schultz, R. M.; Toth, J. E. Bioorg. Med. Chem. Lett. 1999, 9, 369–374. doi:10.1016/S0960-894X(98)00748-3 |
| 26. | Shih, C.; Gossett, L. S.; Gruber, J. M.; Sue Grossman, C.; Andis, S. L.; Schultz, R. M.; Worzalla, J. F.; Corbett, T. H.; Metz, J. T. Bioorg. Med. Chem. Lett. 1999, 9, 69–74. doi:10.1016/S0960-894X(98)00682-9 |
| 27. | Vidya, R.; Eggen, M. J.; Georg, G. I.; Himes, R. H. Bioorg. Med. Chem. Lett. 2003, 13, 757–760. doi:10.1016/S0960-894X(02)01023-5 |
| 28. | Buck, S. B.; Huff, J. K.; Himes, R. H.; Georg, G. I. J. Med. Chem. 2004, 47, 696–702. doi:10.1021/jm030278c |
| 29. | Buck, S. B.; Huff, J. K.; Himes, R. H.; Georg, G. I. J. Med. Chem. 2004, 47, 3697–3699. doi:10.1021/jm030555f |
| 30. | Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem. – Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750 |
| 31. | Liu, W. L.; Zhang, J. C.; Jiang, F. Q.; Fu, L. Arch. Pharm. 2009, 342, 577–583. doi:10.1002/ardp.200900067 |
| 32. | Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473 |
| 33. | Weiß, C.; Bogner, T.; Sammet, B.; Sewald, N. Beilstein J. Org. Chem. 2012, 8, 2060–2066. doi:10.3762/bjoc.8.231 |
| 34. | Kumar, A.; Kumar, M.; Sharma, S.; Guru, S. K.; Bhushan, S.; Shah, B. A. Eur. J. Med. Chem. 2015, 93, 55–63. doi:10.1016/j.ejmech.2014.11.068 |
| 35. | Weiss, C.; Figueras, E.; Borbely, A. N.; Sewald, N. J. Pept. Sci. 2017, 23, 514–531. doi:10.1002/psc.3015 |
| 52. | Mitra, A.; Sept, D. Biochemistry 2004, 43, 13955–13962. doi:10.1021/bi0487387 |
| 21. | Sessa, C.; Weigang-Köhler, K.; Pagani, O.; Greim, G.; Mor, O.; De Pas, T.; Burgess, M.; Weimer, I.; Johnson, R. Eur. J. Cancer 2002, 38, 2388–2396. doi:10.1016/S0959-8049(02)00489-6 |
| 22. | Stevenson, J. P.; Sun, W.; Gallagher, M.; Johnson, R.; Vaughn, D.; Schuchter, L.; Algazy, K.; Hahn, S.; Enas, N.; Ellis, D.; Thornton, D.; O’Dwyer, P. J. Clin. Cancer Res. 2002, 8, 2524–2529. |
| 23. | D’Agostino, G.; Del Campo, J.; Mellado, B.; Izquierdo, M. A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J. L.; Scambia, G. Int. J. Gynecol. Cancer 2006, 16, 71–76. doi:10.1111/j.1525-1438.2006.00276.x |
| 4. | Barrow, R. A.; Hemscheidt, T.; Liang, J.; Paik, S.; Moore, R. E.; Tius, M. A. J. Am. Chem. Soc. 1995, 117, 2479–2490. doi:10.1021/ja00114a011 |
| 5. | Kobayashi, M.; Wang, W.; Ohyabu, N.; Kurosu, M.; Kitagawa, I. Chem. Pharm. Bull. 1995, 43, 1598–1600. doi:10.1248/cpb.43.1598 |
| 6. | de Muys, J.-M.; Rej, R.; Nguyen, D.; Go, B.; Fortin, S.; Lavallée, J.-F. Bioorg. Med. Chem. Lett. 1996, 6, 1111–1116. doi:10.1016/0960-894X(96)00182-5 |
| 7. | Rej, R.; Nguyen, D.; Go, B.; Fortin, S.; Lavallée, J.-F. J. Org. Chem. 1996, 61, 6289–6295. doi:10.1021/jo960816b |
| 8. | Gardinier, K. M.; Leahy, J. W. J. Org. Chem. 1997, 62, 7098–7099. doi:10.1021/jo971645t |
| 9. | Norman, B. H.; Hemscheidt, T.; Schultz, R. M.; Andis, S. L. J. Org. Chem. 1998, 63, 5288–5294. doi:10.1021/jo980536r |
| 10. | Georg, G. I.; Ali, S. M.; Stella, V. J.; Waugh, W. N.; Himmes, R. H. Bioorg. Med. Chem. Lett. 1998, 8, 1959–1962. doi:10.1016/S0960-894X(98)00356-4 |
| 11. | Ghosh, A. K.; Bischoff, A. Org. Lett. 2000, 2, 1573–1575. doi:10.1021/ol000058i |
| 12. | Eggen, M.; Mossman, C. J.; Buck, S. B.; Nair, S. K.; Bhat, L.; Ali, S. M.; Reiff, E. A.; Boge, T. C.; Georg, G. I. J. Org. Chem. 2000, 65, 7792–7799. doi:10.1021/jo000767+ |
| 13. | Murakami, N.; Wang, W.; Ohyabu, N.; Ito, T.; Tamura, S.; Aoki, S.; Kobayashi, M.; Kitagawa, I. Tetrahedron 2000, 56, 9121–9128. doi:10.1016/S0040-4020(00)00766-3 |
| 14. | Murakami, N.; Tamura, S.; Wang, W.; Takagi, T.; Kobayashi, M. Tetrahedron 2001, 57, 4323–4336. doi:10.1016/S0040-4020(01)00339-8 |
| 15. | Ghosh, A. K.; Swanson, L. J. Org. Chem. 2003, 68, 9823–9826. doi:10.1021/jo035077v |
| 16. | Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem. – Eur. J. 2005, 11, 4667–4677. doi:10.1002/chem.200500282 |
| 17. | Danner, P.; Bauer, M.; Phukan, P.; Maier, M. E. Eur. J. Org. Chem. 2005, 317–325. doi:10.1002/ejoc.200400558 |
| 18. | Eißler, S.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2008, 273–277. doi:10.1055/s-2007-1000868 |
| 19. | Sammet, B.; Radzey, H.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2009, 417–420. doi:10.1055/s-0028-1087541 |
| 20. | Sammet, B.; Brax, M.; Sewald, N. Beilstein J. Org. Chem. 2011, 7, 243–245. doi:10.3762/bjoc.7.32 |
| 46. | Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2013, 56, 1853–1864. doi:10.1021/jm301346z |
| 2. | Bai, R.; Schwartz, R. E.; Kepler, J. A.; Pettit, G. R.; Hamel, E. Cancer Res. 1996, 56, 4398–4406. |
| 3. | Smith, C. D.; Zhang, X. J. Biol. Chem. 1996, 271, 6192–6198. doi:10.1074/jbc.271.11.6192 |
| 2. | Bai, R.; Schwartz, R. E.; Kepler, J. A.; Pettit, G. R.; Hamel, E. Cancer Res. 1996, 56, 4398–4406. |
| 52. | Mitra, A.; Sept, D. Biochemistry 2004, 43, 13955–13962. doi:10.1021/bi0487387 |
| 53. | Bai, R.; Pettit, G. R.; Hamel, E. Biochem. Pharmacol. 1990, 39, 1941–1949. doi:10.1016/0006-2952(90)90613-P |
| 52. | Mitra, A.; Sept, D. Biochemistry 2004, 43, 13955–13962. doi:10.1021/bi0487387 |
| 45. | Weiss, C.; Sammet, B.; Sewald, N. Nat. Prod. Rep. 2013, 30, 924–940. doi:10.1039/c3np70022d |
| 46. | Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2013, 56, 1853–1864. doi:10.1021/jm301346z |
| 47. | Bouchard, H.; Brun, M.-P.; Commerçon, A.; Zhang, J. Novel conjugates, preparation thereof, and therapeutic use thereof. WO Patent WO2011/001052, Jan 6, 2011. |
| 48. | Verma, V. A.; Pillow, T. H.; Depalatis, L.; Li, G.; Phillips, G. L.; Polson, A. G.; Raab, H. E.; Spencer, S.; Zheng, B. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. doi:10.1016/j.bmcl.2014.12.070 |
| 49. | Steinkuhler, M. C.; Gallinari, M. P.; Osswald, B.; Sewald, N.; Ritzefeld, M.; Frese, M.; Figueras, E.; Pethö, L. Cryptophycin-based antibody-drug conjugates with novel self-immolative linkers. WO Patent WO2016/146638 A1, Sept 22, 2016. |
| 50. | Bigot, A.; Bouchard, H.; Brun, M.-P.; Clerc, F.; Zhang, J. Novel cryptophycin compounds and conjugates, their preparation and their therapeutic use. WO Patent WO2017/076998 A1, May 11, 2017. |
| 51. | Chen, H.; Lin, Z.; Arnst, K. E.; Miller, D. D.; Li, W. Molecules 2017, 22, No. 1281. doi:10.3390/molecules22081281 |
| 16. | Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem. – Eur. J. 2005, 11, 4667–4677. doi:10.1002/chem.200500282 |
| 41. | Al-awar, R. S.; Ray, J. E.; Schultz, R. M.; Andis, S. L.; Kennedy, J. H.; Moore, R. E.; Liang, J.; Golakoti, T.; Subbaraju, G. V.; Corbett, T. H. J. Med. Chem. 2003, 46, 2985–3007. doi:10.1021/jm0203884 |
| 42. | Al-Awar, R. S.; Corbett, T. H.; Ray, J. E.; Polin, L.; Kennedy, J. H.; Wagner, M. M.; Williams, D. C. Mol. Cancer Ther. 2004, 3, 1061–1067. |
| 43. | Kotoku, N.; Kato, T.; Narumi, F.; Ohtani, E.; Kamada, S.; Aoki, S.; Okada, N.; Nakagawa, S.; Kobayashi, M. Bioorg. Med. Chem. 2006, 14, 7446–7457. doi:10.1016/j.bmc.2006.07.019 |
| 44. | Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. J. Org. Chem. 2010, 75, 6953–6960. doi:10.1021/jo101563s |
| 45. | Weiss, C.; Sammet, B.; Sewald, N. Nat. Prod. Rep. 2013, 30, 924–940. doi:10.1039/c3np70022d |
| 46. | Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2013, 56, 1853–1864. doi:10.1021/jm301346z |
| 36. | Chari, R. V. J.; Miller, M. L.; Widdison, W. C. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. doi:10.1002/anie.201307628 |
| 37. | Srinivasarao, M.; Low, P. S. Chem. Rev. 2017, 117, 12133–12164. doi:10.1021/acs.chemrev.7b00013 |
| 38. | Krall, N.; Scheuermann, J.; Neri, D. Angew. Chem., Int. Ed. 2013, 52, 1384–1402. doi:10.1002/anie.201204631 |
| 39. | Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. Curr. Top. Med. Chem. 2016, 16, 314–329. doi:10.2174/1568026615666150701114343 |
| 40. | Le Joncour, V.; Laakkonen, P. Bioorg. Med. Chem. 2018, 26, 2797–2806. doi:10.1016/j.bmc.2017.08.052 |
| 24. | Patel, V. F.; Andis, S. L.; Kennedy, J. H.; Ray, J. E.; Schultz, R. M. J. Med. Chem. 1999, 42, 2588–2603. doi:10.1021/jm980706s |
© 2018 Figueras et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)