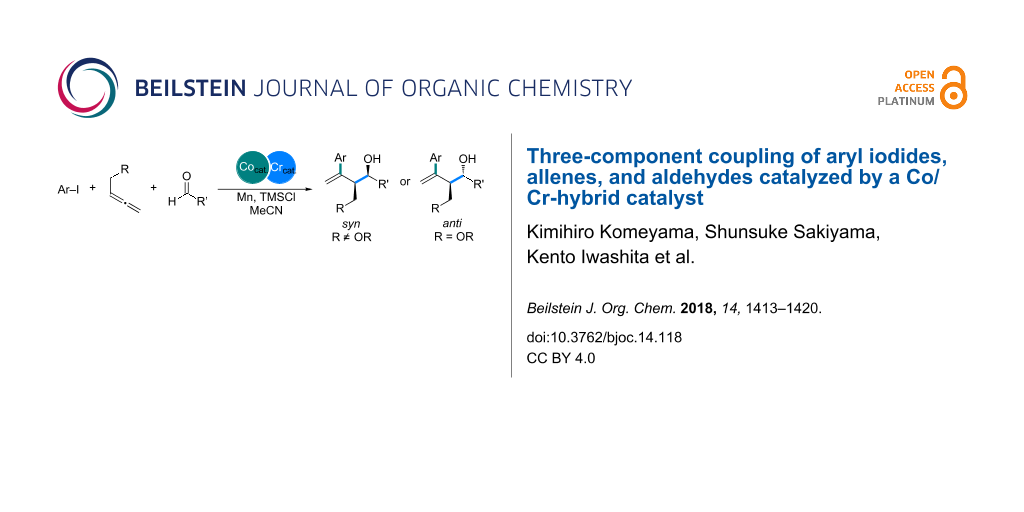
Abstract
The cobalt/chromium-catalyzed three-component coupling of aryl iodides, allenes, and aldehydes has been developed to afford multi-substituted homoallylic alcohols in a diastereoselective manner. Control experiments for understanding the reaction mechanism reveal that the cobalt catalyst is involved in the oxidative addition and carbometalation steps in the reaction, whereas the chromium salt generates highly nucleophilic allylchromium intermediates from allylcobalt species, without the loss of stereochemical information, to allow the addition to aldehydes.

Graphical Abstract
Introduction
Carbon–carbon bond formation is the fundamental and central transformation of synthetic organic chemistry. The elaboration and extension of a carbon framework via a series of carbon–carbon bond-forming reactions are extremely important for medicinal chemistry and agrochemical and natural product synthesis. In these bond formations, organometallics play an essential role because they possess various reactivities depending on the central metal ions that they own. For example, carbon has strong nucleophilicity when bonded to metals with low electronegativity, as demonstrated in the reaction of organolithium, organomagnesium, organozinc, and organochromium A, to facilitate addition reactions of appropriate carbon electrophiles such as aldehydes (Scheme 1, top) [1]. In contrast, π-electrophilic carbon-connected late transition metals B facilitate the carbometalation of carbon–carbon multiple bonds, leading to multi-substituted carbon frameworks (Scheme 1, bottom) [2,3]. The nucleophilic and π-electrophilic organometallic intermediates are used properly in their favorable circumstances.
![[1860-5397-14-118-i1]](/bjoc/content/inline/1860-5397-14-118-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Nucleophilic and π-electrophilic characters of organometallics depending on the central metals.
Scheme 1: Nucleophilic and π-electrophilic characters of organometallics depending on the central metals.
Transmetalation is one of the most vital elemental processes used to drastically change the reactivity of organometallics, involving a wide range of transition metal-catalyzed reactions. For example, transmetalation between an organonickel (or organocobalt) complex and chromium salt results in the formation of a highly nucleophilic organochromium species, which enables efficient addition to aldehydes to give substituted secondary alcohols, as demonstrated in the Nozaki–Hiyama–Kishi (NHK) reaction (Scheme 2) [4-9]. Although the catalyst combination allows the use of organic halides as carbon nucleophiles, a multicomponent coupling reaction using a similar catalyst combination has had limited success [10-12].
![[1860-5397-14-118-i2]](/bjoc/content/inline/1860-5397-14-118-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ni/Cr or Co/Cr-catalyzed NHK reaction.
Scheme 2: Ni/Cr or Co/Cr-catalyzed NHK reaction.
We have recently demonstrated the high π-electron affinity of an organocobalt species that enabled a variety of alkyne functionalization reactions to proceed via carbocobaltation (Scheme 3) [13-15]. Furthermore, a combination of the cobalt and chromium catalyst could be applied to alkynyl iodoarene cyclization/borylation to form cyclized vinylboronic esters, in which transmetalation between the generated vinylcobalt and chromium salt was a critical step (Scheme 4) [16]. As part of our continuing work on the cobalt-catalyzed functionalization of carbon–carbon unsaturated bonds, a three-component coupling method is herein reported for the direct synthesis of highly diastereoselective multi-substituted homoallyl alcohols employing a cobalt/chromium hybrid catalyst (Scheme 5).
![[1860-5397-14-118-i3]](/bjoc/content/inline/1860-5397-14-118-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Functionalization of alkynes via carbocobaltation.
Scheme 3: Functionalization of alkynes via carbocobaltation.
![[1860-5397-14-118-i4]](/bjoc/content/inline/1860-5397-14-118-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclization/borylation of alkynyl iodoarenes using the Co/Cr catalyst.
Scheme 4: Cyclization/borylation of alkynyl iodoarenes using the Co/Cr catalyst.
![[1860-5397-14-118-i5]](/bjoc/content/inline/1860-5397-14-118-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Three-component coupling of aryl iodides, arenes, and aldehydes using Co/Cr catalyst (this work).
Scheme 5: Three-component coupling of aryl iodides, arenes, and aldehydes using Co/Cr catalyst (this work).
Results and Discussion
Initially, suitable reaction conditions were investigated for the three-component coupling reaction between iodobenzene (1a), 5-phenylpenta-1,2-diene (2a), and 4-methylbenzaldehyde (3a) in the presence of CoBr2 (10 mol %), CrCl3 (20 mol %), and manganese powder (2.0 equiv), using trimethylsilyl chloride (TMSCl, 1.2 equiv) as a trapping reagent [7]. These results are summarized in Table 1. The absence of a ligand afforded the homoallyl alcohol 4a in 25% yield as a syn/anti (80:20) mixture of diastereomers, the ratio of which was determined by the coupling constant between the two protons at the C1- and C2-positions of 4a; a coupling value of 3J = ca. 5.0 Hz indicated the syn-form, and a coupling value of 3J = ca. 8.0 Hz indicated the anti-form (Table 1, entry 1) [17]. The addition of PPh3 (20 mol %) resulted in an increase in the product yield to 45% with a similarly diastereomer ratio (Table 1, entry 2). The use of the chromium catalyst, Mn reductant, and TMSCl was crucial for the coupling (Table 1, entries 3–5). Thus, the removal of CrCl3 resulted in the formation of a trace amount of 4a with the complete consumption of allene 2a, whereas most of iodoarene 1a and aldehyde 3a remained unreacted after the reaction was completed (Table 1, entry 3). Using a Zn reductant instead of Mn resulted in a negligible amount of coupling product (Table 1, entry 4), wherein 1a was completely consumed [18]. Reaction conditions without the use of TMSCl produced 4a in a catalytic amount (Table 1, entry 5). Other ligands were also tested (Table 1, entries 5–12), and after the screening of several phosphines and pyridine-type ligands, the latter ligands were found to be the most effective for use in the coupling reaction. Consequently, we found that iminopyridine L3 was the best choice of ligand, and when used, it resulted in the three-component product 4a being obtained in 69% yield with a diastereoselectivity ratio of 92:8 (Table 1, entry 13). Additionally, preformed CoBr2(L3) gave a similar result (Table 1, entry 14). During the transformation, the chromium catalyst ligands inhibited the reaction (Table 1, entries 15–17). Also, the reaction was highly dependent on the solvent used; for example, dimethylformamide (DMF), tetrahydrofuran (THF), 1,4-dioxane, and toluene did not result in any formation of 4a.
Table 1: Screening of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-118-i13.svg?max-width=637&scale=1.0)
|
||||
| entry | ligand (x mol %) | Cr catalyst | NMR yield (%) | isomer ratiob |
| 1 | none | CrCl3 | 25 | 80:20 |
| 2 | PPh3 (20) | CrCl3 | 45 | 85:15 |
| 3 | PPh3 (20) | none | 5 | 82:18 |
| 4c | PPh3 (20) | CrCl3 | trace | – |
| 5d | PPh3 (20) | CrCl3 | 18 | 86:14 |
| 6 | dppe (10) | CrCl3 | 8 | 91:9 |
| 7 | dppb (10) | CrCl3 | trace | – |
| 8 | dppf (10) | CrCl3 | trace | – |
| 9 | xantphos (10) | CrCl3 | trace | – |
| 10 | 2,2′-bpy (10) | CrCl3 | 45 | 92:8 |
| 11 | L1 (10) | CrCl3 | 52 | 93:7 |
| 12 | L2 (10) | CrCl3 | 64 | 91:9 |
| 13 | L3 (10) | CrCl3 | 69 | 92:8 |
| 14e | – | CrCl3 | 65 | 91:9 |
| 15 | L3 (10) | CrCl3(bpy) | 48 | 94:6 |
| 16 | L3 (10) | CrCl3(L3) | 24 | 92:8 |
| 17 | L3 (10) | Cr(salen)Cl | 21 | 91:9 |
aReaction conditions: 1a (0.25 mmol), 2a (0.38 mmol), and 3a (0.25 mmol). bThe ratio (syn:anti) was determined from the 1H NMR of the crude product. cZn was used instead of Mn. dWithout the presence of TMSCl. eCoBr2(L3) was used instead of CoBr2. ![[Graphic 2]](/bjoc/content/inline/1860-5397-14-118-i14.svg?max-width=637&scale=1.18182)
With the optimized conditions in hand, the use of aldehydes in the Co/Cr-catalyzed three-component coupling reaction was explored, as shown in Scheme 6. Electron-rich and electron-deficient aryl aldehydes, as well as 2-furylaldehyde, were well tolerated in the reaction, leading to the formation of the corresponding homoallylic alcohols with similar diastereomer ratios (4a–f). Additionally, alkyl aldehydes were also successfully used in the coupling reaction, albeit resulting in slightly lower yields (4g, 4g’, 4g” and 4h). Next, the generality of the reaction was investigated using aryl iodides (Scheme 7) and allenes (Scheme 8). Although aryl bromides and chlorides did not participate in the coupling, a diverse set of functional groups such as methoxy (4j), halogens (4k and 4l), trifluoromethyl (4m), cyano (4n), and ester (4o) substituents at the para-position of the phenyl ring were successfully used in the reaction, giving rise to the desired coupling products in good yields (47–79%). The homoallylic alcohols 4p–t were formed with a high degree of syn-selectivity because of meta- and ortho-substituted aryl iodides also being well tolerated in the reaction. The protocol was not only limited to the coupling of simple allenes; it was also used with heteroatom-containing functionalized allenes to afford the syn-homoallylic alcohols 4u, 4u’ and 4w in reasonably good yields (Scheme 8).
![[1860-5397-14-118-i6]](/bjoc/content/inline/1860-5397-14-118-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Screening of aldehydes in the Co/Cr-catalyzed three-component coupling reaction. All yields are determined after isolation. The values in brackets indicate the diastereomer ratio of the syn and anti products, determined from 1H NMR spectra.
Scheme 6: Screening of aldehydes in the Co/Cr-catalyzed three-component coupling reaction. All yields are det...
![[1860-5397-14-118-i7]](/bjoc/content/inline/1860-5397-14-118-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Screening of aryl iodides in the Co/Cr-catalyzed three-component coupling reaction. All yields are determined after isolation. The values in brackets indicate the diastereomer ratio of the syn and anti products, determined from 1H NMR spectra.
Scheme 7: Screening of aryl iodides in the Co/Cr-catalyzed three-component coupling reaction. All yields are ...
![[1860-5397-14-118-i8]](/bjoc/content/inline/1860-5397-14-118-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Screening of allenes in the Co/Cr-catalyzed three-component coupling reaction. All yields are determined after isolation. The values in brackets indicate the diastereomer ratio of the syn and anti products, determined from 1H NMR.
Scheme 8: Screening of allenes in the Co/Cr-catalyzed three-component coupling reaction. All yields are deter...
During the investigation of allenes, it was observed that, when oxygen substituents were present at the allenyl position, coupling products with reversed diastereoselectivity were obtained (Scheme 9). Thus, treatment of 4-benzyloxybuta-1,2-diene (5) with 1 and 3a under identical reaction conditions mainly afforded the anti-configured homoallylic alcohols 7–10 in 46–65% yield. A similar reversed diastereoselectivity was observed in the coupling reaction of silyloxy allene 6, leading to 11 in 46% yield (anti/syn = 73:27).
![[1860-5397-14-118-i9]](/bjoc/content/inline/1860-5397-14-118-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reversed diastereoselectivity using allenyl ethers 5 and 6. a4-chlorobenzaldehyde was used instead of 3a.
Scheme 9: Reversed diastereoselectivity using allenyl ethers 5 and 6. a4-chlorobenzaldehyde was used instead ...
A stoichiometric reaction using phenylchromium(II or III) reagents, generated from the reaction of CrCl2 or CrCl3(thf)2 with phenyllithium [19,20], in the presence of allene 2a and aldehyde 3c provided diarylmethanes in 78–85% yields without the visible consumption of allene 2a (Scheme 10, reaction 1). Furthermore, in the absence of the CrCl3 catalyst, allene 2a was utterly consumed, whereas most of iodide 1a and aldehyde 3d were recovered unreacted (Scheme 10, reaction 2). These results indicate that the arylcobalt, rather than the arylchromium intermediate, promoted the allene carbometalation. Additionally, it is thought that the vinylcobalt generated was converted to vinylchromium, which would be inactive in the oligomerization, but is a highly nucleophilic species. According to these findings, the reversed diastereoselectivity in Scheme 9 might be due to stereoelectronic effects [21,22]. Thus, the σ*(C–O) bond stabilizes the forming σ(Co–C) bond in the transition state C of the carbocobaltation step (Scheme 11), facilitating the further selective formation of the branched allylcobalt species D which could be converted into the thermodynamically more stable (E)-allylcobalt species E because of the flexible C(vinyl)–C(allyl) single bond. The generated compound E then undergoes transmetalation with the chromium salt to give the (E)-allylchromium species F. In contrast, the carbometalation of a simple allene produces (Z)-allylcobalt species E′ and the corresponding (Z)-allylchromium product F′ would be provided through a transition state such as C′, in which the cobalt center is connected to a less sterically hindered terminal allene carbon [23].
![[1860-5397-14-118-i10]](/bjoc/content/inline/1860-5397-14-118-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Stoichiometric reaction of phenylchromium(II or III) reagents (reaction 1) and the three-component coupling without CrCl3 catalyst (reaction 2).
Scheme 10: Stoichiometric reaction of phenylchromium(II or III) reagents (reaction 1) and the three-component ...
![[1860-5397-14-118-i11]](/bjoc/content/inline/1860-5397-14-118-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: The origin of the diastereoselectivity in the present three-component coupling.
Scheme 11: The origin of the diastereoselectivity in the present three-component coupling.
On the basis of these results, a plausible catalytic cycle for the cobalt/chromium-catalyzed three-component coupling reaction is shown in Scheme 12. The three-component coupling starts with the oxidative addition of an aryl iodide 1 to a low-valent cobalt species to form an arylcobalt species G that reacts with an allene 2 to stereoselectively generate an allylcobalt H via carbocobaltation. Rapid transmetalation between H and the chromium salt [8,9] triggers the transfer of the cobalt allyl group to the chromium to afford I and the highly nucleophilic allylchromium species J, which retains the same stereochemical information on the olefinic moiety as that of H. The allylchromium species J reacts with the aldehyde 3 at the γ-position of the allyl metal unit via a cyclic six-membered transition state K to give the chromium alkoxide L [24]. Finally, the Cr–O bond is cleaved by TMSCl, generating the active chromium salt for the transmetalation and the silyl ether M, the desilylation of which with a fluoride anion results in the formation of a homoallylic alcohol 4.
![[1860-5397-14-118-i12]](/bjoc/content/inline/1860-5397-14-118-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Plausible reaction mechanism of the three-component coupling.
Scheme 12: Plausible reaction mechanism of the three-component coupling.
Conclusion
The cobalt/chromium-catalyzed three-component coupling reaction of aryl iodides, allenes, and aldehydes to produce highly substituted homoallylic alcohols in a diastereoselective manner has been demonstrated. In the coupling reaction, two catalysts played individual roles; the cobalt catalyst activated aryl iodides to form arylcobalt species, which then performed allene carbocobaltation to form stereo-defined substituted (Z)-allylcobalt intermediates. The chromium catalyst transformed the generated allylcobalt intermediates into highly nucleophilic allylchromium species, without the isomerization of the olefinic moiety, via transmetalation between the generated allylcobalt intermediate and the chromium salt. Moreover, it was found that an oxygen atom present at the allenyl position resulted in a reversed diastereoselectivity of the homoallylic alcohol products; thus, the allene carbocobaltation regioselectivity could be controlled by the stereoelectronic interaction between the forming σ(C–Co) bond and a neighboring σ*(C–O) bond. Further mechanistic studies and expansion of the substrate scope, including synthetic applications of this three-component coupling, are currently in progress.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 4.5 MB | Download |
References
-
Takai, K. Bull. Chem. Soc. Jpn. 2015, 88, 1511. doi:10.1246/bcsj.20150170
Return to citation in text: [1] -
Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698. doi:10.1021/cr050051k
Return to citation in text: [1] -
Tamaru, Y. Modern Organonickel Chemistry; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Okude, Y.; Hirano, S.; Hiyama, T.; Nozaki, H. J. Am. Chem. Soc. 1977, 99, 3179. doi:10.1021/ja00451a061
Return to citation in text: [1] -
Jin, H.; Uenishi, J.; Christ, W. J.; Kishi, Y. J. Am. Chem. Soc. 1986, 108, 5644. doi:10.1021/ja00278a057
Return to citation in text: [1] -
Takai, K.; Tagashira, M.; Kuroda, T.; Oshima, K.; Utimoto, K.; Nozaki, H. J. Am. Chem. Soc. 1986, 108, 6048. doi:10.1021/ja00279a068
Return to citation in text: [1] -
Fürstner, A.; Shi, N. J. Am. Chem. Soc. 1996, 118, 12349. doi:10.1021/ja9625236
Return to citation in text: [1] [2] -
Takai, K.; Nitta, K.; Fujimura, O.; Utimoto, K. J. Org. Chem. 1989, 54, 4732. doi:10.1021/jo00281a004
Return to citation in text: [1] [2] -
Usanov, D. L.; Yamamoto, H. Angew. Chem., Int. Ed. 2010, 49, 8169. doi:10.1002/anie.201002751
Return to citation in text: [1] [2] -
Takai, K.; Matsukawa, N.; Takahashi, A.; Fujii, T. Angew. Chem., Int. Ed. 1998, 37, 152. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<152::AID-ANIE152>3.0.CO;2-8
Return to citation in text: [1] -
Takai, K.; Toratsu, C. J. Org. Chem. 1998, 63, 6450. doi:10.1021/jo9814086
Return to citation in text: [1] -
Xiong, Y.; Zhang, G. J. Am. Chem. Soc. 2018, 140, 2735. doi:10.1021/jacs.7b12760
Return to citation in text: [1] -
Komeyama, K.; Kashihara, T.; Takaki, K. Tetrahedron Lett. 2013, 54, 5659. doi:10.1016/j.tetlet.2013.07.133
Return to citation in text: [1] -
Komeyama, K.; Okamoto, Y.; Takaki, K. Angew. Chem., Int. Ed. 2014, 53, 11325. doi:10.1002/anie.201406807
Return to citation in text: [1] -
Komeyama, K.; Asakura, R.; Fukuoka, H.; Takaki, K. Tetrahedron Lett. 2015, 56, 1735. doi:10.1016/j.tetlet.2015.02.101
Return to citation in text: [1] -
Komeyama, K.; Kiguchi, S.; Takaki, K. Chem. Commun. 2016, 52, 7009. doi:10.1039/c6cc03086f
Return to citation in text: [1] -
Hopkins, C. D.; Malinakova, H. C. Org. Lett. 2004, 6, 2221. doi:10.1021/ol0492795
Return to citation in text: [1] -
Fillon, H.; Gosmini, C.; Périchon, J. J. Am. Chem. Soc. 2003, 125, 3867. doi:10.1021/ja0289494
Return to citation in text: [1] -
Daly, J. J.; Sneeden, R. P. A.; Zeiss, H. H. J. Am. Chem. Soc. 1966, 88, 4287. doi:10.1021/ja00970a049
Return to citation in text: [1] -
Kanno, K.-i.; Liu, Y.; Iesato, A.; Nakajima, K.; Takahashi, T. Org. Lett. 2005, 7, 5453. doi:10.1021/ol052214x
Return to citation in text: [1] -
Ito, H.; Ito, S.; Sasaki, Y.; Matsuura, K.; Sawamura, M. J. Am. Chem. Soc. 2007, 129, 14856. doi:10.1021/ja076634o
Return to citation in text: [1] -
Ohmiya, H.; Makida, Y.; Li, D.; Tanabe, M.; Sawamura, M. J. Am. Chem. Soc. 2010, 132, 879. doi:10.1021/ja9092264
Return to citation in text: [1] -
Yoshida, Y.; Murakami, K.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2010, 132, 8878. doi:10.1021/ja102303s
Return to citation in text: [1] -
Hiyama, T.; Kimura, K.; Nozaki, H. Tetrahedron Lett. 1981, 22, 1037. doi:10.1016/S0040-4039(01)82859-8
Return to citation in text: [1]
| 13. | Komeyama, K.; Kashihara, T.; Takaki, K. Tetrahedron Lett. 2013, 54, 5659. doi:10.1016/j.tetlet.2013.07.133 |
| 14. | Komeyama, K.; Okamoto, Y.; Takaki, K. Angew. Chem., Int. Ed. 2014, 53, 11325. doi:10.1002/anie.201406807 |
| 15. | Komeyama, K.; Asakura, R.; Fukuoka, H.; Takaki, K. Tetrahedron Lett. 2015, 56, 1735. doi:10.1016/j.tetlet.2015.02.101 |
| 10. | Takai, K.; Matsukawa, N.; Takahashi, A.; Fujii, T. Angew. Chem., Int. Ed. 1998, 37, 152. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<152::AID-ANIE152>3.0.CO;2-8 |
| 11. | Takai, K.; Toratsu, C. J. Org. Chem. 1998, 63, 6450. doi:10.1021/jo9814086 |
| 12. | Xiong, Y.; Zhang, G. J. Am. Chem. Soc. 2018, 140, 2735. doi:10.1021/jacs.7b12760 |
| 4. | Okude, Y.; Hirano, S.; Hiyama, T.; Nozaki, H. J. Am. Chem. Soc. 1977, 99, 3179. doi:10.1021/ja00451a061 |
| 5. | Jin, H.; Uenishi, J.; Christ, W. J.; Kishi, Y. J. Am. Chem. Soc. 1986, 108, 5644. doi:10.1021/ja00278a057 |
| 6. | Takai, K.; Tagashira, M.; Kuroda, T.; Oshima, K.; Utimoto, K.; Nozaki, H. J. Am. Chem. Soc. 1986, 108, 6048. doi:10.1021/ja00279a068 |
| 7. | Fürstner, A.; Shi, N. J. Am. Chem. Soc. 1996, 118, 12349. doi:10.1021/ja9625236 |
| 8. | Takai, K.; Nitta, K.; Fujimura, O.; Utimoto, K. J. Org. Chem. 1989, 54, 4732. doi:10.1021/jo00281a004 |
| 9. | Usanov, D. L.; Yamamoto, H. Angew. Chem., Int. Ed. 2010, 49, 8169. doi:10.1002/anie.201002751 |
| 8. | Takai, K.; Nitta, K.; Fujimura, O.; Utimoto, K. J. Org. Chem. 1989, 54, 4732. doi:10.1021/jo00281a004 |
| 9. | Usanov, D. L.; Yamamoto, H. Angew. Chem., Int. Ed. 2010, 49, 8169. doi:10.1002/anie.201002751 |
| 2. | Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698. doi:10.1021/cr050051k |
| 3. | Tamaru, Y. Modern Organonickel Chemistry; Wiley-VCH: Weinheim, Germany, 2005. |
| 24. | Hiyama, T.; Kimura, K.; Nozaki, H. Tetrahedron Lett. 1981, 22, 1037. doi:10.1016/S0040-4039(01)82859-8 |
| 18. | Fillon, H.; Gosmini, C.; Périchon, J. J. Am. Chem. Soc. 2003, 125, 3867. doi:10.1021/ja0289494 |
| 21. | Ito, H.; Ito, S.; Sasaki, Y.; Matsuura, K.; Sawamura, M. J. Am. Chem. Soc. 2007, 129, 14856. doi:10.1021/ja076634o |
| 22. | Ohmiya, H.; Makida, Y.; Li, D.; Tanabe, M.; Sawamura, M. J. Am. Chem. Soc. 2010, 132, 879. doi:10.1021/ja9092264 |
| 17. | Hopkins, C. D.; Malinakova, H. C. Org. Lett. 2004, 6, 2221. doi:10.1021/ol0492795 |
| 23. | Yoshida, Y.; Murakami, K.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2010, 132, 8878. doi:10.1021/ja102303s |
| 7. | Fürstner, A.; Shi, N. J. Am. Chem. Soc. 1996, 118, 12349. doi:10.1021/ja9625236 |
| 16. | Komeyama, K.; Kiguchi, S.; Takaki, K. Chem. Commun. 2016, 52, 7009. doi:10.1039/c6cc03086f |
| 19. | Daly, J. J.; Sneeden, R. P. A.; Zeiss, H. H. J. Am. Chem. Soc. 1966, 88, 4287. doi:10.1021/ja00970a049 |
| 20. | Kanno, K.-i.; Liu, Y.; Iesato, A.; Nakajima, K.; Takahashi, T. Org. Lett. 2005, 7, 5453. doi:10.1021/ol052214x |
© 2018 Komeyama et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)