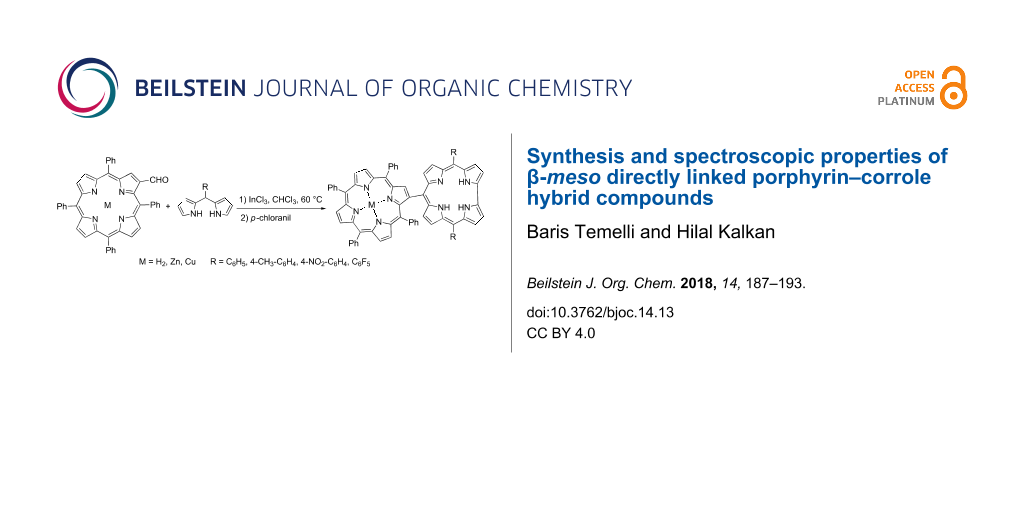
Abstract
The preparation of β-meso directly linked porphyrin–corrole hybrids was realized for the first time via an InCl3-catalyzed condensation reaction of 2-formyl-5,10,15,20-tetraphenylporphyrins with meso-substituted dipyrromethanes. Hybrid compounds have been characterized by 1H NMR, 13C NMR, 2D NMR, UV–vis absorption and fluorescence spectroscopy.

Graphical Abstract
Introduction
Porphyrins and metalloporphyrins play an important role in chemistry, biology, medical and materials sciences because of their presence in biological compounds such as chlorophyll and heme molecules that have very important functions in the metabolism of living organisms [1,2]. In recent years, efforts in porphyrin chemistry have been focused on the synthesis of multichromophore containing compounds and their potential applications in molecular wires, sensors, nonlinear optical devices, photosensitizers and organic conducting materials [3-6]. The first studies on the synthesis of multichromophores were based on obtaining multiporphyrin arrays [7-9] that could stabilize only metal ions in a bivalent state. To overcome this limitation, porphyrin conjugates with different chromophore groups such as fullerene [10-12], BODIPY [13-15], corrole [16-23], phthalocyanine [24-26], subporphyrin [27] and expanded porphyrins [28-30] were prepared and their photophysical and electrochemical properties were characterized. Among these compounds, corroles, contracted porphyrin analogues [31-33], assumed an important place in porphyrin chemistry due to their small cavities, trianionic characters, high fluorescence quantum yields and favorable electronic properties. Porphyrin–corrole conjugates have been successfully used as donor–acceptor systems in photoinduced charge separation processes. It was shown that derivatives of these conjugates could be potentially used in photovoltaic applications [21-23]. In order to achieve rapid energy and electron transfer between macrocycles, the short distance between subunits keeps an important place. Therefore, two important factors affect the physical and electronic properties of porphyrin–corrole conjugates: (i) type of linkers and (ii) position of substitution (meso or β). So far, corrole macrocyles have been integrated into porphyrin conjugates via anthracene, biphenylene, xanthene, dibenzofuran [16-20], amide [21] and triazole [22,23] linkers. Despite the large number of studies on the synthesis of porphyrin–corrole conjugates, systematic studies on directly linked porphyrin–corrole hybrid structures are limited and there is no report on β-porphyrin, meso-corrole linked hybrids of these conjugates. Zheng et al. synthesized meso–meso directly linked porphyrin–corrole dyads by the condensation reaction of meso-formylated porphyrin with meso-substituted dipyrromethanes (Scheme 1A) [34]. Later, Sankar et al. used the same type of condensation reaction to obtain meso–meso linked corrole–porphyrin–corrole (Scheme 1B) [35] and porphyrin–corrrole–porphyrin triads (Scheme 1C) [36]. The only report on the synthesis of meso-porphyrin, β-corrole-linked hybrid structure described a Suzuki–Miyaura cross-coupling reaction between a β-borylated corrole and meso-bromoporphyrins (Scheme 1D) [37].
![[1860-5397-14-13-i1]](/bjoc/content/inline/1860-5397-14-13-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Overview of the synthesis of directly linked porphyrin–corrole hybrid compounds.
Scheme 1: Overview of the synthesis of directly linked porphyrin–corrole hybrid compounds.
Recently, we have successfully synthesized meso–meso and β-meso-linked imine-bridged porphyrin–corrole conjugates and investigated intramolecular energy transfer between macrocycles [38]. As a part of our ongoing research on porphyrin–corrole conjugates, herein we describe a convenient synthesis of a series of novel directly linked porphyrin–corrole hybrid compounds. For this purpose, acid-catalyzed reactions of dipyrromethanes and aldehydes, which have been used frequently for the synthesis of meso-substituted corroles, were carried out [31,39,40]. The key bilane-substituted porphyrin intermediates 3 were obtained by the addition of an excess amount of meso-substituted dipyrromethanes 2 to β-formylated porphyrins 1 and these intermediates were oxidized in the reaction medium to form the desired hybrid structures 4 (Scheme 2).
![[1860-5397-14-13-i2]](/bjoc/content/inline/1860-5397-14-13-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of β-meso directly linked porphyrin–corrole hybrid compounds.
Scheme 2: Synthesis of β-meso directly linked porphyrin–corrole hybrid compounds.
Results and Discussion
Initially, the reaction of 2-formyl-5,10,15,20-tetraphenylporphyrin (1) with 4 equivalents of phenyldipyrromethane (2a) was tested using p-toluenesulfonic acid (TsOH) as catalyst at room temperature but no product was observed in the reaction (Table 1, entry 1). Increasing the reaction temperature affected the production of 4a and the target compound could be obtained with 6% yield (Table 1, entries 2–4). A screen of solvents revealed that chloroform provided the best yield and was superior to 1,2-dichloroethane, toluene and methanol (Table 1, entries 3–6). The ratio of 1a/2a played a little role in improving the yield of product (Table 1, entries 7–9). The yield of 4a increased slightly to 7% when the reaction was carried out with 6 equiv of 2a (Table 1, entry 8). Further increasing the amount of 2a led to the formation of unidentified byproducts in the reaction medium and a reduction of the yield of 4a (Table 1, entry 9). Then, the activities of different catalysts, varying from clay catalysts to Lewis acids, were tested in the model reaction (Table 1, entries 10–19). AgOTf, AlCl3, ZnBr2 and InCl3-catalyzed the reaction in higher yields with an unexpected byproduct, meso–β-substituted directly linked porphyrin–porphyrin dyad 5a (Table 1, entries 16–19). The structure of 5a was confirmed by comparison with literature data [41]. The highest yield of 4a was obtained with InCl3 (19%, Table 1, entry 19). Other catalysts gave no (Table 1, entries 10 and 11) or lower yields of products (Table 1, entries 12–15). When the reaction temperature increased to 80 °C by using 1,2-dichloroethane as solvent in the presence of InCl3 as catalyst, the product yield decreased to 14% (Table 1, entry 20).
Table 1: Optimization of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-13-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | 1a/2a | Solvent | Catalyst | Temp (°C) | 4a (%)a | 5a (%)a |
| 1 | 1:4 | CHCl3 | TsOHb | rt | – | – |
| 2 | 1:4 | CHCl3 | TsOHb | 40 | – | – |
| 3 | 1:4 | CHCl3 | TsOHb | 60 | 6 | – |
| 4 | 1:4 | ClCH2CH2Cl | TsOHb | 60 | 5 | – |
| 5 | 1:4 | MeOH | TsOHb | 60 | 2 | – |
| 6 | 1:4 | toluene | TsOHb | 60 | 4 | – |
| 7 | 1:2 | CHCl3 | TsOHb | 60 | 1 | – |
| 8 | 1:6 | CHCl3 | TsOHb | 60 | 7 | – |
| 9 | 1:10 | CHCl3 | TsOHb | 60 | 4 | – |
| 10 | 1:6 | CHCl3 | Mont. K-10c | 60 | – | – |
| 11 | 1:6 | CHCl3 | BF3·OEt2d | 60 | – | – |
| 12 | 1:6 | CHCl3 | Mont. KSFc | 60 | 2 | – |
| 13 | 1:6 | CHCl3 | Cu(OTf)2b | 60 | 4 | – |
| 14 | 1:6 | CHCl3 | Amberlyst-15c | 60 | 3 | – |
| 15 | 1:6 | CHCl3 | TFAb | 60 | 8 | – |
| 16 | 1:6 | CHCl3 | AgOTfb | 60 | 17 | 3 |
| 17 | 1:6 | CHCl3 | AlCl3d | 60 | 14 | 3 |
| 18 | 1:6 | CHCl3 | ZnBr2b | 60 | 15 | 2 |
| 19 | 1:6 | CHCl3 | InCl3b | 60 | 19 | 4 |
| 20 | 1:6 | ClCH2CH2Cl | InCl3b | 80 | 14 | 3 |
aIsolated yield after column chromatography. Catalyst amount: b10 mol %, c1 mmol aldehyde/1 g of catalyst, d100 mol %.
With the optimized reaction conditions in hand, different meso-substituted dipyrromethanes 2 were subjected to the condensation reactions (Scheme 3). Electronic properties of the substituents on the dipyrromethane generally did not show significant effects on the reactions except for the electron-donating methoxy substituent (Scheme 3, 4d), which resulted in a mixture of unidentified compounds after the reaction. This might be due to an acid-catalyzed rearrangement of the substituents, called scrambling, which is a common problem in porphyrin synthesis in the reaction of dipyrromethanes with aldehydes [42]. When electron-withdrawing 4-nitrophenyl and pentafluorophenyl substituents were used on the dipyrromethane, hybrid compounds 4c and 4e were isolated in 16% and 20% yields, respectively. The synthesis of 4e with InCl3 afforded a very low yield, thus an equimolar amount of AlCl3 was used to obtain this product. In order to expand the scope of the reaction, metal complexes of formylated porphyrins were subjected to the reaction. While Zn-porphyrin containing hybrid compound 4f was isolated in 21%, the Cu(II) complex of β-formylated porphyrin underwent the reaction to produce the hybrid compound 4g in 17% yield (Scheme 3).
![[1860-5397-14-13-i3]](/bjoc/content/inline/1860-5397-14-13-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of porphyrin–corrole hybrid derivatives. *100 mol % of AlCl3 was used as a catalyst.
Scheme 3: Synthesis of porphyrin–corrole hybrid derivatives. *100 mol % of AlCl3 was used as a catalyst.
The structures of the hybrid compounds were identified by using 1H NMR, 13C NMR, 1H,1H-COSY NMR and HRMS techniques (see Supporting Information File 1). The 1H NMR spectrum of 4a is shown in Figure 1. As reported for similar dimeric porphyrin systems, one of the meso-linked phenyl substituents on the porphyrin core was located above the corrole macrocycle and was affected by the ring current. These phenyl protons appear strongly shifted toward higher field. The positions of the protons were assigned by comparing previously reported β–meso-substituted porphyrin arrays [41]. The para-proton (2), the meta-protons (3) and the ortho-protons (4) of the phenyl substituent appeared at 4.05 ppm, 4.64 ppm and 6.62 ppm, respectively. The β-protons of the porphyrin and the corrole macrocycles gave multiplets and doublets between 8.00 and 10.00 ppm as expected. Inner N–H protons of the corrole macrocycle gave a very broad signal and could not be determined due to its high unsymmetrical property and an excess amount of tautomeric structures. The N–H protons of the porphyrin macrocycle resonated at −2.46 ppm (Supporting Information File 1, Figure S1).
![[1860-5397-14-13-1]](/bjoc/content/figures/1860-5397-14-13-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectrum of 4a in CDCl3.
Figure 1: 1H NMR spectrum of 4a in CDCl3.
The absorption spectra of hybrid compounds have been recorded and λmax values are listed with molar extinction coeffients (ε) in Table 2. Split Soret bands were observed for 4b, 4e and metalloporphyrin 4g. These results indicate a strong exciton coupling between the neighbor macrocycles in hybrid structures. The same effect was also observed in the emission spectra. Fluorescence spectra of the compounds gave split emission bands except for 4c. Fluorescence life times and quantum yields were further determined for the hybrid compounds (Table 2). Measured fluorescence life times (4–9 ns), and fluorescence quantum yields are in agreement with the literature data [35,36]. Strong electron-withdrawing pentafluorophenyl-substituted 4e and Zn–porphyrin complex 4f gave higher quantum yields compared to reported literature values for meso–meso-linked porphyrin–corrole dyads [34] and traids [35]. High fluorescence quantum yield properties of the new hybrid compounds are promising for many applications such as organic optoelectronics or biological imaging.
Table 2: Photophysical properties of hybrid compounds.
| Compound | λabs/nm (ε/10−5 M−1·cm−1)a | λem/nmb | Φc | τ/nsd |
| 4a | 421 (5.35), 520 (4.34), 552 (4.08), 595 (4.03), 652 (3.92) | 664, 723 | 0.10 | 7.4 |
| 4b | 421 (5.32), 441 (4.96), 523 (4.18), 559 (4.08), 597 (4.04), 659 (3.53) | 664, 725 | 0.22 | 7.1 |
| 4c | 421 (4.94), 518 (4.08), 596 (3.90), 662 (3.81) | 692 | 0.04 | 8.5 |
| 4e | 420 (5.21), 443 (5.07), 521 (4.32), 554 (4.10), 591 (4.06), 646 (3.86) | 663, 717 | 0.27 | 5.4 |
| 4f | 421 (4.57), 442 (4.46), 524 (3.49), 553 (3.64), 657 (2.98) | 663, 728 | 0.41 | 7.8 |
| 4g | 418 (4.65), 438 (4.47), 542 (3.68), 656 (2.94) | 651, 720 | 0,01 | 4.4 |
aAbsorption spectra were recorded in CHCl3. bFluorescence spectra were recorded in CHCl3 (λex: 420 nm). cBased on TPP in toluene (Φ = 0.11). dExcited at 390 nm.
Conclusion
In summary, we have synthesized the first examples of β-porphyrin, meso-corrole substituted directly linked porphyrin–corrole hybrid compounds via a condensation reaction of meso-substituted dipyrromethanes with β-formylated meso-tetraphenylporphyrins. Spectroscopic and photophysical properties of the new hybrid structures have been determined. Higher quantum yields of some of the synthesized hybrid compounds indicate that these compounds can be considered as good candidates for many important applications ranging from photovoltaics to medicine.
Supporting Information
| Supporting Information File 1: General experimental information, experimental details on the synthesis of products 4 and 5a, and full characterization data of all products. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Kadish, K. M.; Smith, K. M.; Guilard, R. The Porphyrin Handbook; Academic Press: San Diego, 2000; Vol. 1–20.
Return to citation in text: [1] -
Dolphin, D. The Porphyrins; Academic Press: New York, 1979.
Return to citation in text: [1] -
Beletskaya, I.; Tyurin, V. S.; Tsivadze, A. Y.; Guilard, R.; Stern, C. Chem. Rev. 2009, 109, 1659–1713. doi:10.1021/cr800247a
Return to citation in text: [1] -
Wasielewski, M. R. Chem. Rev. 1992, 92, 435–461. doi:10.1021/cr00011a005
Return to citation in text: [1] -
Panda, D. K.; Goodson, F. S.; Ray, S.; Saha, S. Chem. Commun. 2014, 50, 5358–5360. doi:10.1039/C3CC47498D
Return to citation in text: [1] -
Senge, M. O.; Fazekas, M.; Notaras, E. G. A.; Blau, W. J.; Zawadzka, M.; Locos, O. B.; Mhuircheartaigh, E. M. N. Adv. Mater. 2007, 19, 2737–2774. doi:10.1002/adma.200601850
Return to citation in text: [1] -
Aratani, N.; Osuka, A. In Handbook of Porphyrin Science, Synthetic Strategies Toward Multiporphyrinic Architectures; Kadish, K. M.; Smith, K. M.; Guilard, R., Eds.; World Scientific: Singapore, 2010; Vol. 1, pp 1–132.
Return to citation in text: [1] -
Tanaka, T.; Osuka, A. Chem. Soc. Rev. 2015, 44, 943–969. doi:10.1039/C3CS60443H
Return to citation in text: [1] -
Kim, D. Multiporphyrin Arrays: Fundamentals and Applications; Pan Stanford: New York, 2012. doi:10.1201/b11621
Return to citation in text: [1] -
Drovetskaya, T.; Reed, C. A.; Boyd, P. Tetrahedron Lett. 1995, 36, 7971–7974. doi:10.1016/0040-4039(95)01719-X
Return to citation in text: [1] -
Ciammaichella, A.; Dral, P. O.; Clark, T.; Tagliatesta, P.; Sekita, M.; Guldi, D. M. Chem. – Eur. J. 2012, 18, 14008–14016. doi:10.1002/chem.201202245
Return to citation in text: [1] -
Pelado, B.; Abou-Chahine, F.; Calbo, J.; Caballero, R.; de la Cruz, P.; Junquera-Hernández, J. M.; Orti, E.; Tkachenko, N. V.; Langa, F. Chem. – Eur. J. 2015, 21, 5814–5825. doi:10.1002/chem.201406514
Return to citation in text: [1] -
Lee, C. Y.; Hupp, J. T. Langmuir 2010, 26, 3760–3765. doi:10.1021/la9031927
Return to citation in text: [1] -
Kursunlu, A. N. RSC Adv. 2014, 4, 47690–47696. doi:10.1039/C4RA09024A
Return to citation in text: [1] -
Liu, J.-Y.; Ermilov, E. A.; Röder, B.; Ng, D. K. P. J. Porphyrins Phthalocyanines 2013, 17, 831–835. doi:10.1142/S1088424613500533
Return to citation in text: [1] -
Guilard, R.; Jérôme, F.; Gros, C. P.; Barbe, J.-M.; Ou, Z.; Shao, J.; Kadish, K. M. C. R. Acad. Sci., Ser. IIc: Chim. 2001, 4, 245–254. doi:10.1016/S1387-1609(00)01226-3
Return to citation in text: [1] [2] -
Guilard, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Espinosa, E.; Shao, J.; Ou, Z.; Zhan, R.; Kadish, K. M. Inorg. Chem. 2005, 44, 3972–3983. doi:10.1021/ic0501622
Return to citation in text: [1] [2] -
Kadish, K. M.; Shao, J.; Ou, Z.; Frémond, L.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 6744–6754. doi:10.1021/ic050738n
Return to citation in text: [1] [2] -
Kadish, K. M.; Shao, J.; Ou, Z.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 9023–9038. doi:10.1021/ic051073s
Return to citation in text: [1] [2] -
Kadish, K. M.; Frémond, L.; Ou, Z.; Shao, J.; Shi, C.; Anson, F. C.; Burdet, F.; Gros, C. P.; Barbe, J.-M.; Guilard, R. J. Am. Chem. Soc. 2005, 127, 5625–5631. doi:10.1021/ja0501060
Return to citation in text: [1] [2] -
Flamigni, L.; Ventura, B.; Tasior, M.; Gryko, D. T. Inorg. Chim. Acta 2007, 360, 803–813. doi:10.1016/j.ica.2006.03.021
Return to citation in text: [1] [2] [3] -
Nikolaou, V.; Karikis, K.; Farré, Y.; Charalambidis, G.; Odobel, F.; Coutsolelos, A. G. Dalton Trans. 2015, 44, 13473–13479. doi:10.1039/C5DT01730K
Return to citation in text: [1] [2] [3] -
Ngo, T. H.; Zieba, D.; Webre, W. A.; Lim, G. N.; Karr, P. A.; Kord, S.; Jin, S.; Ariga, K.; Galli, M.; Goldup, S.; Hill, J. P.; D’Souza, F. Chem. – Eur. J. 2016, 22, 1301–1312. doi:10.1002/chem.201503490
Return to citation in text: [1] [2] [3] -
Soares, A. R. M.; Martinez-Diaz, V. M.; Bruckner, A.; Pereira, A. M. V. M.; Tomé, J. P. C.; Alonso, C. M. A.; Faustino, M. A. F.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S.; Torres, T.; Guldi, D. M. Org. Lett. 2007, 9, 1557–1560. doi:10.1021/ol0703635
Return to citation in text: [1] -
Pereira, A. M. V. M.; Soares, A. R. M.; Hausmann, A.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S.; Guldi, D. M.; Torres, T. Phys. Chem. Chem. Phys. 2011, 13, 11858–11863. doi:10.1039/c1cp00016k
Return to citation in text: [1] -
KC, C. B.; Lim, G. N.; D’Souza, F. Nanoscale 2015, 7, 6813–6826. doi:10.1039/C5NR00354G
Return to citation in text: [1] -
Inokuma, Y.; Hayashi, S.; Osuka, A. Chem. Lett. 2009, 38, 206–207. doi:10.1246/cl.2009.206
Return to citation in text: [1] -
Tanaka, T.; Aratani, N.; Lim, J. M.; Kim, K. S.; Kim, D.; Osuka, A. Chem. Sci. 2011, 2, 1414–1418. doi:10.1039/c1sc00228g
Return to citation in text: [1] -
Mori, H.; Tanaka, T.; Lee, S.; Lim, J. M.; Kim, D.; Osuka, A. J. Am. Chem. Soc. 2015, 137, 2097–2106. doi:10.1021/ja5130034
Return to citation in text: [1] -
Honjo, K.; Mori, H.; Osuka, A. Asian J. Org. Chem. 2016, 5, 196–200. doi:10.1002/ajoc.201500509
Return to citation in text: [1] -
Orlowski, R.; Gryko, D.; Gryko, D. T. Chem. Rev. 2017, 117, 3102–3137. doi:10.1021/acs.chemrev.6b00434
Return to citation in text: [1] [2] -
Barata, J. F. B.; Neves, M. G. P. M. S.; Faustino, M. A. F.; Tomé, A. C.; Cavaleiro, J. A. S. Chem. Rev. 2017, 117, 3192–3253. doi:10.1021/acs.chemrev.6b00476
Return to citation in text: [1] -
Aviv, I.; Gross, Z. Chem. Commun. 2007, 1987–1999. doi:10.1039/B618482K
Return to citation in text: [1] -
Chen, C.; Zhu, Y.-Z.; Fan, Q.-J.; Song, H.-B.; Zheng, J.-Y. Chem. Lett. 2013, 42, 936–938. doi:10.1246/cl.130324
Return to citation in text: [1] [2] -
Murugavel, M.; Reddy, R. V. R.; Sankar, J. RSC Adv. 2014, 4, 13669–13672. doi:10.1039/C4RA01229A
Return to citation in text: [1] [2] [3] -
Murugavel, M.; Reddy, R. V. R.; Dey, D.; Sankar, J. Chem. – Eur. J. 2015, 21, 14280–14286. doi:10.1002/chem.201501471
Return to citation in text: [1] [2] -
Hiroto, S.; Hisaki, I.; Shinokubo, H.; Osuka, A. Angew. Chem., Int. Ed. 2005, 44, 6763–6766. doi:10.1002/anie.200502335
Return to citation in text: [1] -
Temelli, B.; Ozasik, O.; Yüksel, D. Eur. J. Org. Chem. 2017, 4905–4915. doi:10.1002/ejoc.201700896
Return to citation in text: [1] -
Gryko, D. T. J. Porphyrins Phthalocyanines 2008, 12, 906–917. doi:10.1142/S1088424608000297
Return to citation in text: [1] -
Koszarna, B.; Gryko, D. T. J. Org. Chem. 2006, 71, 3707–3717. doi:10.1021/jo060007k
Return to citation in text: [1] -
Götz, D. C. G.; Bruhn, T.; Senge, M. O.; Bringmann, G. J. Org. Chem. 2009, 74, 8005–8020. doi:10.1021/jo901483q
Return to citation in text: [1] [2] -
Littler, B. J.; Ciringh, Y.; Lindsey, J. S. J. Org. Chem. 1999, 64, 2864–2872. doi:10.1021/jo982452o
Return to citation in text: [1]
| 1. | Kadish, K. M.; Smith, K. M.; Guilard, R. The Porphyrin Handbook; Academic Press: San Diego, 2000; Vol. 1–20. |
| 2. | Dolphin, D. The Porphyrins; Academic Press: New York, 1979. |
| 13. | Lee, C. Y.; Hupp, J. T. Langmuir 2010, 26, 3760–3765. doi:10.1021/la9031927 |
| 14. | Kursunlu, A. N. RSC Adv. 2014, 4, 47690–47696. doi:10.1039/C4RA09024A |
| 15. | Liu, J.-Y.; Ermilov, E. A.; Röder, B.; Ng, D. K. P. J. Porphyrins Phthalocyanines 2013, 17, 831–835. doi:10.1142/S1088424613500533 |
| 34. | Chen, C.; Zhu, Y.-Z.; Fan, Q.-J.; Song, H.-B.; Zheng, J.-Y. Chem. Lett. 2013, 42, 936–938. doi:10.1246/cl.130324 |
| 10. | Drovetskaya, T.; Reed, C. A.; Boyd, P. Tetrahedron Lett. 1995, 36, 7971–7974. doi:10.1016/0040-4039(95)01719-X |
| 11. | Ciammaichella, A.; Dral, P. O.; Clark, T.; Tagliatesta, P.; Sekita, M.; Guldi, D. M. Chem. – Eur. J. 2012, 18, 14008–14016. doi:10.1002/chem.201202245 |
| 12. | Pelado, B.; Abou-Chahine, F.; Calbo, J.; Caballero, R.; de la Cruz, P.; Junquera-Hernández, J. M.; Orti, E.; Tkachenko, N. V.; Langa, F. Chem. – Eur. J. 2015, 21, 5814–5825. doi:10.1002/chem.201406514 |
| 35. | Murugavel, M.; Reddy, R. V. R.; Sankar, J. RSC Adv. 2014, 4, 13669–13672. doi:10.1039/C4RA01229A |
| 7. | Aratani, N.; Osuka, A. In Handbook of Porphyrin Science, Synthetic Strategies Toward Multiporphyrinic Architectures; Kadish, K. M.; Smith, K. M.; Guilard, R., Eds.; World Scientific: Singapore, 2010; Vol. 1, pp 1–132. |
| 8. | Tanaka, T.; Osuka, A. Chem. Soc. Rev. 2015, 44, 943–969. doi:10.1039/C3CS60443H |
| 9. | Kim, D. Multiporphyrin Arrays: Fundamentals and Applications; Pan Stanford: New York, 2012. doi:10.1201/b11621 |
| 21. | Flamigni, L.; Ventura, B.; Tasior, M.; Gryko, D. T. Inorg. Chim. Acta 2007, 360, 803–813. doi:10.1016/j.ica.2006.03.021 |
| 3. | Beletskaya, I.; Tyurin, V. S.; Tsivadze, A. Y.; Guilard, R.; Stern, C. Chem. Rev. 2009, 109, 1659–1713. doi:10.1021/cr800247a |
| 4. | Wasielewski, M. R. Chem. Rev. 1992, 92, 435–461. doi:10.1021/cr00011a005 |
| 5. | Panda, D. K.; Goodson, F. S.; Ray, S.; Saha, S. Chem. Commun. 2014, 50, 5358–5360. doi:10.1039/C3CC47498D |
| 6. | Senge, M. O.; Fazekas, M.; Notaras, E. G. A.; Blau, W. J.; Zawadzka, M.; Locos, O. B.; Mhuircheartaigh, E. M. N. Adv. Mater. 2007, 19, 2737–2774. doi:10.1002/adma.200601850 |
| 22. | Nikolaou, V.; Karikis, K.; Farré, Y.; Charalambidis, G.; Odobel, F.; Coutsolelos, A. G. Dalton Trans. 2015, 44, 13473–13479. doi:10.1039/C5DT01730K |
| 23. | Ngo, T. H.; Zieba, D.; Webre, W. A.; Lim, G. N.; Karr, P. A.; Kord, S.; Jin, S.; Ariga, K.; Galli, M.; Goldup, S.; Hill, J. P.; D’Souza, F. Chem. – Eur. J. 2016, 22, 1301–1312. doi:10.1002/chem.201503490 |
| 28. | Tanaka, T.; Aratani, N.; Lim, J. M.; Kim, K. S.; Kim, D.; Osuka, A. Chem. Sci. 2011, 2, 1414–1418. doi:10.1039/c1sc00228g |
| 29. | Mori, H.; Tanaka, T.; Lee, S.; Lim, J. M.; Kim, D.; Osuka, A. J. Am. Chem. Soc. 2015, 137, 2097–2106. doi:10.1021/ja5130034 |
| 30. | Honjo, K.; Mori, H.; Osuka, A. Asian J. Org. Chem. 2016, 5, 196–200. doi:10.1002/ajoc.201500509 |
| 21. | Flamigni, L.; Ventura, B.; Tasior, M.; Gryko, D. T. Inorg. Chim. Acta 2007, 360, 803–813. doi:10.1016/j.ica.2006.03.021 |
| 22. | Nikolaou, V.; Karikis, K.; Farré, Y.; Charalambidis, G.; Odobel, F.; Coutsolelos, A. G. Dalton Trans. 2015, 44, 13473–13479. doi:10.1039/C5DT01730K |
| 23. | Ngo, T. H.; Zieba, D.; Webre, W. A.; Lim, G. N.; Karr, P. A.; Kord, S.; Jin, S.; Ariga, K.; Galli, M.; Goldup, S.; Hill, J. P.; D’Souza, F. Chem. – Eur. J. 2016, 22, 1301–1312. doi:10.1002/chem.201503490 |
| 27. | Inokuma, Y.; Hayashi, S.; Osuka, A. Chem. Lett. 2009, 38, 206–207. doi:10.1246/cl.2009.206 |
| 16. | Guilard, R.; Jérôme, F.; Gros, C. P.; Barbe, J.-M.; Ou, Z.; Shao, J.; Kadish, K. M. C. R. Acad. Sci., Ser. IIc: Chim. 2001, 4, 245–254. doi:10.1016/S1387-1609(00)01226-3 |
| 17. | Guilard, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Espinosa, E.; Shao, J.; Ou, Z.; Zhan, R.; Kadish, K. M. Inorg. Chem. 2005, 44, 3972–3983. doi:10.1021/ic0501622 |
| 18. | Kadish, K. M.; Shao, J.; Ou, Z.; Frémond, L.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 6744–6754. doi:10.1021/ic050738n |
| 19. | Kadish, K. M.; Shao, J.; Ou, Z.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 9023–9038. doi:10.1021/ic051073s |
| 20. | Kadish, K. M.; Frémond, L.; Ou, Z.; Shao, J.; Shi, C.; Anson, F. C.; Burdet, F.; Gros, C. P.; Barbe, J.-M.; Guilard, R. J. Am. Chem. Soc. 2005, 127, 5625–5631. doi:10.1021/ja0501060 |
| 24. | Soares, A. R. M.; Martinez-Diaz, V. M.; Bruckner, A.; Pereira, A. M. V. M.; Tomé, J. P. C.; Alonso, C. M. A.; Faustino, M. A. F.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S.; Torres, T.; Guldi, D. M. Org. Lett. 2007, 9, 1557–1560. doi:10.1021/ol0703635 |
| 25. | Pereira, A. M. V. M.; Soares, A. R. M.; Hausmann, A.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S.; Guldi, D. M.; Torres, T. Phys. Chem. Chem. Phys. 2011, 13, 11858–11863. doi:10.1039/c1cp00016k |
| 26. | KC, C. B.; Lim, G. N.; D’Souza, F. Nanoscale 2015, 7, 6813–6826. doi:10.1039/C5NR00354G |
| 16. | Guilard, R.; Jérôme, F.; Gros, C. P.; Barbe, J.-M.; Ou, Z.; Shao, J.; Kadish, K. M. C. R. Acad. Sci., Ser. IIc: Chim. 2001, 4, 245–254. doi:10.1016/S1387-1609(00)01226-3 |
| 17. | Guilard, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Espinosa, E.; Shao, J.; Ou, Z.; Zhan, R.; Kadish, K. M. Inorg. Chem. 2005, 44, 3972–3983. doi:10.1021/ic0501622 |
| 18. | Kadish, K. M.; Shao, J.; Ou, Z.; Frémond, L.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 6744–6754. doi:10.1021/ic050738n |
| 19. | Kadish, K. M.; Shao, J.; Ou, Z.; Zhan, R.; Burdet, F.; Barbe, J.-M.; Gros, C. P.; Guilard, R. Inorg. Chem. 2005, 44, 9023–9038. doi:10.1021/ic051073s |
| 20. | Kadish, K. M.; Frémond, L.; Ou, Z.; Shao, J.; Shi, C.; Anson, F. C.; Burdet, F.; Gros, C. P.; Barbe, J.-M.; Guilard, R. J. Am. Chem. Soc. 2005, 127, 5625–5631. doi:10.1021/ja0501060 |
| 21. | Flamigni, L.; Ventura, B.; Tasior, M.; Gryko, D. T. Inorg. Chim. Acta 2007, 360, 803–813. doi:10.1016/j.ica.2006.03.021 |
| 22. | Nikolaou, V.; Karikis, K.; Farré, Y.; Charalambidis, G.; Odobel, F.; Coutsolelos, A. G. Dalton Trans. 2015, 44, 13473–13479. doi:10.1039/C5DT01730K |
| 23. | Ngo, T. H.; Zieba, D.; Webre, W. A.; Lim, G. N.; Karr, P. A.; Kord, S.; Jin, S.; Ariga, K.; Galli, M.; Goldup, S.; Hill, J. P.; D’Souza, F. Chem. – Eur. J. 2016, 22, 1301–1312. doi:10.1002/chem.201503490 |
| 31. | Orlowski, R.; Gryko, D.; Gryko, D. T. Chem. Rev. 2017, 117, 3102–3137. doi:10.1021/acs.chemrev.6b00434 |
| 32. | Barata, J. F. B.; Neves, M. G. P. M. S.; Faustino, M. A. F.; Tomé, A. C.; Cavaleiro, J. A. S. Chem. Rev. 2017, 117, 3192–3253. doi:10.1021/acs.chemrev.6b00476 |
| 33. | Aviv, I.; Gross, Z. Chem. Commun. 2007, 1987–1999. doi:10.1039/B618482K |
| 38. | Temelli, B.; Ozasik, O.; Yüksel, D. Eur. J. Org. Chem. 2017, 4905–4915. doi:10.1002/ejoc.201700896 |
| 36. | Murugavel, M.; Reddy, R. V. R.; Dey, D.; Sankar, J. Chem. – Eur. J. 2015, 21, 14280–14286. doi:10.1002/chem.201501471 |
| 37. | Hiroto, S.; Hisaki, I.; Shinokubo, H.; Osuka, A. Angew. Chem., Int. Ed. 2005, 44, 6763–6766. doi:10.1002/anie.200502335 |
| 35. | Murugavel, M.; Reddy, R. V. R.; Sankar, J. RSC Adv. 2014, 4, 13669–13672. doi:10.1039/C4RA01229A |
| 35. | Murugavel, M.; Reddy, R. V. R.; Sankar, J. RSC Adv. 2014, 4, 13669–13672. doi:10.1039/C4RA01229A |
| 36. | Murugavel, M.; Reddy, R. V. R.; Dey, D.; Sankar, J. Chem. – Eur. J. 2015, 21, 14280–14286. doi:10.1002/chem.201501471 |
| 34. | Chen, C.; Zhu, Y.-Z.; Fan, Q.-J.; Song, H.-B.; Zheng, J.-Y. Chem. Lett. 2013, 42, 936–938. doi:10.1246/cl.130324 |
| 42. | Littler, B. J.; Ciringh, Y.; Lindsey, J. S. J. Org. Chem. 1999, 64, 2864–2872. doi:10.1021/jo982452o |
| 41. | Götz, D. C. G.; Bruhn, T.; Senge, M. O.; Bringmann, G. J. Org. Chem. 2009, 74, 8005–8020. doi:10.1021/jo901483q |
| 31. | Orlowski, R.; Gryko, D.; Gryko, D. T. Chem. Rev. 2017, 117, 3102–3137. doi:10.1021/acs.chemrev.6b00434 |
| 39. | Gryko, D. T. J. Porphyrins Phthalocyanines 2008, 12, 906–917. doi:10.1142/S1088424608000297 |
| 40. | Koszarna, B.; Gryko, D. T. J. Org. Chem. 2006, 71, 3707–3717. doi:10.1021/jo060007k |
| 41. | Götz, D. C. G.; Bruhn, T.; Senge, M. O.; Bringmann, G. J. Org. Chem. 2009, 74, 8005–8020. doi:10.1021/jo901483q |
© 2018 Temelli and Kalkan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)