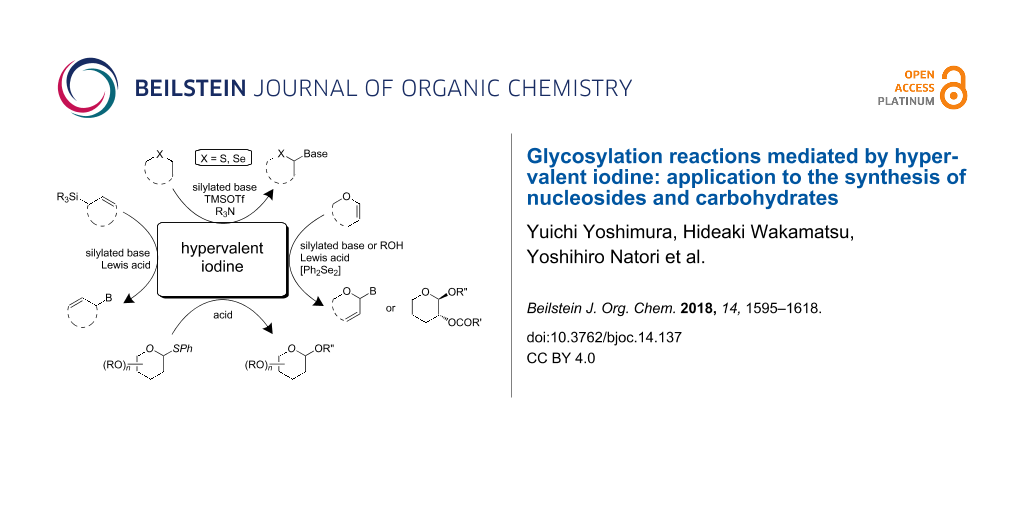
Abstract
To synthesize nucleoside and oligosaccharide derivatives, we often use a glycosylation reaction to form a glycoside bond. Coupling reactions between a nucleobase and a sugar donor in the former case, and the reaction between an acceptor and a sugar donor of in the latter are carried out in the presence of an appropriate activator. As an activator of the glycosylation, a combination of a Lewis acid catalyst and a hypervalent iodine was developed for synthesizing 4’-thionucleosides, which could be applied for the synthesis of 4’-selenonucleosides as well. The extension of hypervalent iodine-mediated glycosylation allowed us to couple a nucleobase with cyclic allylsilanes and glycal derivatives to yield carbocyclic nucleosides and 2’,3’-unsaturated nucleosides, respectively. In addition, the combination of hypervalent iodine and Lewis acid could be used for the glycosylation of glycals and thioglycosides to produce disaccharides. In this paper, we review the use of hypervalent iodine-mediated glycosylation reactions for the synthesis of nucleosides and oligosaccharide derivatives.

Graphical Abstract
Introduction
Nucleic acids and oligosaccharides are both mandatory polymers for the maintenance of life and cell growth. The former exists in nuclei and codes genetic information, which is transformed into proteins through a transcription process known as the “central dogma” (i.e., DNA makes RNA makes proteins). The latter make up the cell walls of microorganisms and also play a role in transmitting information on the cell surface, whose interactions with proteins are a starting point for signal transduction into cells [1]. Since both types of polymers are essential for cell viability, their biological synthesis, including the synthesis of their monomer units, e.g., nucleotides, is highly regulated. Damage to these vital molecules often results in congenital disease with ultimately fatal consequences [2,3]. Accordingly, the study of polymers and their biosynthesis is quite important, and informs the development of new drugs for diseases including cancers and infectious diseases caused by viruses [4-7]. Indeed, many drugs related to nucleic acids and oligosaccharides have been developed and used in clinical fields. Synthetic chemists have contributed to the studies by supplying biological tools for the analyses of these polymers, as well as by synthesizing effective drug candidates for the diseases mentioned above [4,8-14].
To synthesize nucleoside and oligosaccharide derivatives, glycosylation reactions are often used to form a glycoside bond. In the case of nucleoside synthesis, a coupling reaction between a persilylated nucleobase and a sugar donor is typically used [15-17]. On the other hand, the reaction between an acceptor and sugar donor is carried out in the presence of an appropriate activator for oligosaccharide synthesis [18,19]. In both cases, a Lewis acid is generally used as an activator for sugar donors. Our previous review focused on the development of glycosylation reactions and their application to the synthesis of nucleoside derivatives [17]. In this review, we showed our glycosylation reactions under oxidative conditions. These were quite useful and the conceptually similar reactions were widely used for synthesizing nucleoside derivatives. Recently, a combination of a Lewis acid catalyst and hypervalent iodine was developed for synthesizing 4’-thionucleosides, which was based on a Pummerer-type reaction coupled with oxidation. The concept of the oxidative glycosylation reaction was successfully applied to the synthesis of other nucleoside derivatives, including 4’-selenonucleosides and carbocyclic nucleosides. The hypervalent iodine-mediated glycosylation has also been used for oligosaccharide synthesis employing glycals and thioglycosides as sugar donors. In this review, we survey the synthesis of nucleoside and disaccharide derivatives under oxidative conditions mostly based on the hypervalent iodine-mediated glycosylation reactions.
Review
Synthesis of 4’-thionucleosides
Over the last decade, we have steadily pursued the identification of novel antitumor and antiviral nucleoside derivatives [17,20-22]. Matsuda and co-workers reported a 2’-substituted cytidine derivative, DMDC (1), with potent antitumor activity [23,24]. In other reports, Walker [25] and Secrist [26] independently described the potent antiherpesvirus activity of 2’-deoxy-4’-thionucleoside 2, in which sulfur was introduced in place of the sugar ring oxygen of 2’-deoxynucleoside. The results for 2’-substituted nucleosides and 2’-deoxy-4’-thionucleosides strongly suggested that 2’-substituted 4’-thionucleosides would be promising candidates for novel antitumor agents. Thus, we designed a novel 2’-substituted 4’-thiocytidine, 4’-thioDMDC (3), as our target molecule for potential antitumor agents [27,28] (Figure 1).
![[1860-5397-14-137-1]](/bjoc/content/figures/1860-5397-14-137-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Design of potential antineoplastic nucleosides.
Figure 1: Design of potential antineoplastic nucleosides.
At the time we started our project, there had been no reports regarding the synthesis of even 2-substituted 4-thiosugar derivatives. We thus developed the first synthetic route accessing the 4-thiosugar derivative by way of bicyclic intermediate 8 from diacetoneglucose (5). Construction of the bicyclic ring of 8 was achieved by consecutive inter-/intramolecular SN2 reactions of the dimesylate derivative 7 obtained by manipulations of 5. After acetal hydrolysis and the subsequent hydride reduction, 4-thioarabinose derivative 9 was obtained in good yield. Introduction of a TBDPS group at the primary hydroxy group of 9, oxidation and Wittig reaction, followed by deprotection of the benzyl group, gave allyl alcohol 11.
The most popular method to form a glycosyl bond between the sugar moiety and the base of a nucleoside is a Vorbrüggen reaction [15,16], in which a silylated base and sugar donor, e.g., 1-acetoxy sugar, are condensed by a Lewis acid catalyst. It was clear that this reaction could also be used in the synthesis of 4’-thionucleosides as well as normal “4’-oxy” nucleosides. However, for reasons which will be described later, we decided to develop an alternative method to build the glycosyl bond of 4’-thionucleosides by using a direct coupling of a 4-thiosugar sulfoxide and a silylated base under sila-Pummerer conditions [29,30]. We found that treatment of 12, obtained by oxidation of 11, with excess persilylated N4-acetylcytosine in the presence of TMSOTf as a Lewis acid gave an inseparable mixture of α- and β-anomers of 4’-thioDMDC derivatives 15 in good yield. Based on the study of the sila-Pummerer reaction by Kita, it was plausible that the reaction proceeded via the formation of sulfenium ion 14 which was formed by β-elimination of silylated sulfoxide 13. The 4’-thioDMDC derivative 15 was deprotected and the resulting anomeric mixture was separated to furnish 4’-thioDMDC (3) and its α-anomer [27,28] (Scheme 1).
![[1860-5397-14-137-i1]](/bjoc/content/inline/1860-5397-14-137-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
After we reported the synthesis of 4’-thioDMDC using a Pummerer-type glycosylation reaction, Minakawa and Matsuda applied the reaction to the syntheses of 4’-thioribonucleosides. Applying the synthetic scheme of 2’-deoxy-4’-thionucleoside by Walker to a ribo derivative, 2-dimethoxybenzoate 20 was prepared from tribenzylated ribose 16. Introduction of a dimethoxybenzoyl (DMBz) group at the 2-position and diastereoselective formation of sulfoxide 20, favored in Pummerer-type glycosylation reactions and cases where the approach of the nucleophile is restricted, were the key strategies for their synthesis of 4’-thioribonucleosides. Under optimized conditions, the desired 4’-thiouridine derivative 21 was the sole product and it was obtained in excellent yield (Scheme 2). Using the method developed, they succeeded in preparing all four kinds of 4’-thioribonucleosides [31].
![[1860-5397-14-137-i2]](/bjoc/content/inline/1860-5397-14-137-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4’-thioribonucleosides by Minakawa and Matsuda.
Scheme 2: Synthesis of 4’-thioribonucleosides by Minakawa and Matsuda.
We also synthesized 4’-thioribonucleosides constructing the skeleton of the 4-thioribose via a ring-contraction reaction under reductive conditions [32] from 2-mesylate 23, which was obtained from 22. As shown in Scheme 3, the reaction first started to form an episulfonium ion 24 triggered by intramolecular SN2 reaction at the 5-position by sulfur atom. Secondary, ring contraction from thiopyranose to thiofuranose occurred to produce 5-aldehyde 26. Finally, hydride reduction of 26 gave the 4-thiofuranose derivative 27. The Pummerer-type glycosylation reaction of 5-O-silylated sulfoxide 28, by treating with 2,4-bis(trimethylsilyl)uracil (29) and excess diisopropylethylamine (DIPEA) in the presence of TMSOTf, gave 4’-thiouridine derivative 30 in a good yield. The reaction stereoselectively proceeded and resulted the predominant formation of the β-anomer due to steric hindrance of the 2,3-di-O-isopropylidene group.
![[1860-5397-14-137-i3]](/bjoc/content/inline/1860-5397-14-137-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4’-thioribonucleosides by Yoshimura.
Scheme 3: Synthesis of 4’-thioribonucleosides by Yoshimura.
Before our reports regarding the Pummerer-type glycosylation, the synthesis of 4’-thionucleosides was based on the known chemistry: typically, a 1-acetoxy-4-thiosugar or its synthetic equivalent was obtained from natural sugars and subjected to the Vorbrüggen reaction as in the case of 2’-deoxy-4’-thionucleosides [25,26]. When synthesizing 4’-thionucleosides by the way of a sulfide derivative 31, the known chemistry should lead us to use a classical Pummerer reaction to produce 1-acetoxy derivative 33 after converting 31 to the corresponding sulfoxide 32. Even though this scheme should be promising enough, we intended to introduce an additional synthetic idea based on the fact that both of the reaction intermediate of the Vorbrüggen reaction [15,16] of 33 and the sila-Pummerer reaction developed by Kita [29,30] involving sulfoxide 32 would be the same sulfenium ion 34. This new glycosylation reaction was unique and attractive since it was capable of skipping a step. In other words, the reaction could directly access sulfenium ion 34 from sulfoxide 32. Thus we developed the Pummerer-type glycosylation as mentioned above. From these results it can be deduced that the expected sulfenium ion had formed and that the concept of the Pummerer-type glycosylation was actually effective for the formation of the glycosyl bond of 4’-thionucleosides. After we had reported our synthesis of 4’-thioDMDC, the method was widely adopted for the synthesis of 4’-thionucleoside derivatives by other groups and became a standard approach for the glycosylation [33-37]. On the other hand, the conversion from the sulfide to 4’-thionucleoside using the Pummerer-type glycosylation included an oxidation step. If the oxidation of sulfide 31 and the Pummerer-type glycosylation of the sulfoxide 32 could be performed in the same flask, the reaction could bypass two of the reaction steps and would directly produce 4’-thionucleoside 35 from 31. Indeed, the utilization of hypervalent iodine would have enabled this short-cut reaction (Figure 2).
![[1860-5397-14-137-2]](/bjoc/content/figures/1860-5397-14-137-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Concept of the Pummerer-type glycosylation and hypervalent iodine-mediated glycosylation.
Figure 2: Concept of the Pummerer-type glycosylation and hypervalent iodine-mediated glycosylation.
Hypervalent iodine reagents have been widely used in organic synthesis [38]. Although originally used as oxidative agents, their use has spread to coupling reactions, including those for the formation of C–C bonds [39-43]. In the case of C–N bond formation, introduction of an azido group using PhI=O and TMSN3 was reported by Kita and co-workers [44]. Their paper prompted Nishizono et al. to study the glycosylation reaction for 4’-thionucleosides using hypervalent iodine reagents. As a 4-thiosugar donor, 2-p-methoxybenzoate derivative 36 was prepared following Matsuda’s method as shown in Scheme 2, and then was subjected to the Pummerer-type glycosylation mediated by hypervalent iodine. Treatment of 36 with bis(trifluoroacetoxy)iodobenzene (PIFA) and uracil in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) and triethylamine gave a 5:1 mixture of 4’-thiouridine derivative 37 in 55% yield. The reaction of 36 with iodosylbenzene (PhI=O) proceeded stereoselectively and gave only the β-anomer of 37 in 53% yield [45] (Scheme 4).
![[1860-5397-14-137-i4]](/bjoc/content/inline/1860-5397-14-137-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxidative glycosylation of 4-thioribose mediated by hypervalent iodine.
Scheme 4: Oxidative glycosylation of 4-thioribose mediated by hypervalent iodine.
The mechanism of hypervalent iodine-mediated glycosylation can be expressed as shown in Figure 3. The activated hypervalent iodine reagents in the presence of TMSOTf reacted a sulfur atom of 36 to give 38, in which elimination of iodobenzene and HX might subsequently occur to generate a sulfenium ion 40 (path a). The nucleophilic attack of the silylated base to the sulfenium ion 40 favored approaching from the β-face to give only the β-anomer 37 as in the case of Minakawa and Matsuda’s synthesis described above.
![[1860-5397-14-137-3]](/bjoc/content/figures/1860-5397-14-137-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Speculated mechanism of oxidative glycosylation mediated by hypervalent iodine.
Figure 3: Speculated mechanism of oxidative glycosylation mediated by hypervalent iodine.
Nishizono considered that the difference between the stereoselectivities of the coupling reactions in methods A and B was caused by the existence of another reaction path of the sulfonium salt (38 or 39). In path b, the 4-thiosugar 41 was generated and reacted with a nucleobase, giving a mixture of α- and β-anomers since the reaction might occur by the simple SN2 reaction. Thus, the reaction proceeded through both paths a and b in method A, but path a was predominant in the reaction of method B [45] (Figure 3).
Nishizono et al. applied the hypervalent iodine-mediated glycosylation to purine 4’-thionucleosides [46]. However, the reaction of 36 with 6-chloropurine resulted in the formation of a regioisomer reacting at the 4-position without any formation of the desired purine 4’-thionucleoside. The result should relate to the acidity of the α hydrogen adjacent to a sulfur atom, which affects the regioselectivity of the reaction. To study the effects of a protecting group on the reaction, the regioselectivity of the reaction was examined using 42 and 43, which were obtained from 27. When the 5-hydroxy group was protected with a benzoyl group, the coupling reaction of 42 occurred at the 4-position, as in the case mentioned above, to give 46 in 44% yield along with the desired product and its N7 isomer. In contrast, switching the protecting group of 27 at the 5-position to TBS resulted in the exclusive formation of 45 reacted at the 1-position (28%) along with the N7 stereoisomer (10%). These results support the above-mentioned hypothesis. Finally, 4'-thioadenosine (49) was synthesized by treating 45 with TFA followed by methanolic ammonia [46] (Scheme 5).
![[1860-5397-14-137-i5]](/bjoc/content/inline/1860-5397-14-137-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of purine 4’-thioribonucleosides using hypervalent iodine-mediated glycosylation.
Scheme 5: Synthesis of purine 4’-thioribonucleosides using hypervalent iodine-mediated glycosylation.
The same group attempted to apply the oxidative coupling reaction to the synthesis of thietane nucleosides [47]. The substrate of the coupling reaction was prepared as shown in Scheme 6 starting from benzyloxyacetaldehyde (50). When a hypervalent iodine reagent was used for glycosylation with a diastereomeric mixture of sulfide 53, the reaction stereoselectively gave the ring-expanded nucleoside 54 in 30% yield, but did not give the desired thietane nucleoside at all (Scheme 6).
![[1860-5397-14-137-i6]](/bjoc/content/inline/1860-5397-14-137-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Unexpected glycosylation of a thietanose derivative.
Scheme 6: Unexpected glycosylation of a thietanose derivative.
Considered that the ring-expansion occurred in the absence of the hypervalent iodine reagent, the Nishizono and co-workers speculated that the reaction mechanism was as shown in Scheme 7. First, the Lewis acid catalyzed the intramolecular SN2 reaction of sulfur to form the epi-sulfonium ion 55, which proceeded only from the cis-isomer due to the steric requirement. The subsequent nucleophilic attack leaving the benzoate anion resulted in the formation of a ring-expanded product 56, which became a substrate of the hypervalent iodine-mediated glycosylation. As a result, 4’-thiofurano nucleoside 54 was stereoselectively obtained with the assistance of the neighboring benzoyl group as in 58.
![[1860-5397-14-137-i7]](/bjoc/content/inline/1860-5397-14-137-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Speculated mechanism of the ring expansion of a thietanose derivative.
Scheme 7: Speculated mechanism of the ring expansion of a thietanose derivative.
The desired thietanonucleosides 62 and 63 with an anomeric hydroxymethyl group were synthesized by the Pummerer-type glycosylation reaction of trans-cyclobutane sulfoxide 59. The authors concluded that the stereochemistry of the sulfoxide and the nature of the protecting groups had no significant effect on the yield of the Pummerer-type glycosylation [47] (Scheme 8).
![[1860-5397-14-137-i8]](/bjoc/content/inline/1860-5397-14-137-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of thietanonucleosides using the Pummerer-type glycosylation.
Scheme 8: Synthesis of thietanonucleosides using the Pummerer-type glycosylation.
Pummerer-type glycosylation, which was developed by our group, improved the synthesis of 4’-thionucleosides. It greatly contributed to search new biological active nucleoside derivatives. The use of hypervalent iodine reagents helped to further improve their synthesis by saving reaction steps to improve synthetic efficiency.
Synthesis of 4’-selenonucleosides
The unique biological activity of 4’-thionucleosides triggered the synthesis of their chalcogen isosters, 4’-selenonucleosides, the activity of which were reported. The first synthesis of 4’-selenonucleosides was reported by Jeong and co-workers in 2008 [48,49].
As in the case of the 4’-thioribonucleoside described in Scheme 3, Jeong et al. chose a 2,3-di-O-isopropylidene-protected intermediate as a donor of glycosylation, which was synthesized based on their method developed for 4’-thionucleosides. Starting from compound 64, which was obtained from D-gulonic γ-lactone, dimesylate 66 was prepared. The consecutive inter-/intramolecular SN2 reactions of 66 by selenide anion gave a 4-seleno sugar 67 in an excellent yield. After converting 67 to the corresponding selenoxide, the resulting 68 was immediately treated with uracil or N4-benzoylcytosine under the same conditions for Pummerer-type glycosylation to give the desired 4’-selenouridine and 4’-selenocytidine derivatives in moderate yields. Deprotection of the nucleoside derivatives afforded 4’-selenouridine and 4’-selenocytidine, respectively [48] (Scheme 9). In the year in which the first synthesis of 4’-selenonucleoside was reported, Jayakanthan et al. used the same strategy to synthesize 4’-selenonucleosides, including 4’-selenoadenosine [50].
![[1860-5397-14-137-i9]](/bjoc/content/inline/1860-5397-14-137-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: First synthesis of 4’-selenonucleosides.
Scheme 9: First synthesis of 4’-selenonucleosides.
After successful application of the Pummerer-type glycosylation to the synthesis of 4’-selenonucleosides, Jeong’s group reported various 4’-selenonucleoside derivatives by using the same method [51-58]. Minakawa and his group attempted to synthesize 4’-selenonucleosides based on their method described in Scheme 2 [59]. However, the Pummerer-type glycosylation of selenoxide 74 obtained from 73 gave the desired 4’-selenonucleoside in low yield along with the formation of diselenide 76 and deoxygenated 73 (Scheme 10). One of the reasons for the unsatisfactory result was the instability of selenoxide 74. Jeong et al. faced the same problem and suppressed decomposition by the immediate reaction after synthesizing the corresponding selenoxide [48].
![[1860-5397-14-137-i10]](/bjoc/content/inline/1860-5397-14-137-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The Pummerer-type glycosylation of 4-selenoxide 74.
Scheme 10: The Pummerer-type glycosylation of 4-selenoxide 74.
To overcome these problems, Minakawa decided to use hypervalent iodine for the glycosylation reaction [59] as in Nishizono’s synthesis of 4’-thionucleosides [45]. First, they optimized the reaction conditions by examining the reaction of 73 with uracil in the presence of hypervalent iodine reagents. None of the desired pyrimidine nucleoside 75 was formed when the reaction was performed by treatment with iodosylbenzene, TMSOTf and triethylamine in the presence of the silylated uracil (Table 1, entry 1). Instead of trimethylamine, 2,6-lutidine was employed to give 75 in 48% yield together with selenoxide 74 (20%) and starting 73 (8%) (Table 1, entry 2). The use of more reactive hypervalent iodine agents (PIFA and diacetoxyiodobenzene) did not improve the chemical yield of 75 (Table 1, entries 3 and 4). When 73 was treated with iodosylbenzene, TMSOTf, 2,6-lutidine and the silylated uracil in dichloroethane at 50 °C, the reaction gave 75 in 64% yield while suppressing the formation of 74 (Table 1, entry 5).
Table 1: The Pummerer-like glycosylation reaction mediated by hypervalent iodine.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-137-i24.svg?max-width=637&scale=1.0)
|
||||||||
| Conditions | Yield (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Hypervalent iodine (1.2 equiv) | Base (8 equiv) | Solvent | Temp (°C) | Time (h) | 75 | 74 | 73 |
| 1 | PhIO | Et3N | CH2Cl2 | 0 | 4.5 | 0 | 0 | 33 |
| 2 | PhIO | 2,6-lutidine | CH2Cl2 | rt | 17 | 48 | 20 | 8 |
| 3 | PhI(OCOCF3)2 | 2,6-lutidine | CH2Cl2 | rt | 3 | 38 | 0 | 40 |
| 4 | PhI(OAc)2 | 2,6-lutidine | CH2Cl2 | rt | 5 | 25 | 0 | 20 |
| 5 | PhIO | 2,6-lutidine | ClCH2CH2Cl | 50 | 1.5 | 64 | 0 | 13 |
Minakawa’s group attempted to apply the aforementioned reaction to the synthesis of purine derivatives [60]. Based on the reports by Jeong et al., who synthesized 4’-selenoadenosine using the Vorbrüggen reaction [53], they conceived that the hypervalent iodine-mediated reaction of “disarmed” sugar donor 73 bearing an electron-withdrawing group at the 2-position would not readily yield the desired purine derivative. Therefore, they decided to use “armed” seleno sugar 67 as a donor for the hypervalent iodine-mediated glycosylation reaction as in Jeong’s synthesis.
The reaction of 67 was performed by treating with silylated 6-chloropurine, iodosylbenzene, TMSOTf and 2,6-lutidine in dichloroethane at 85 °C for 2.5 h to give the desired N9-isomer 78 in 39% yield along with the formation of the N7-isomer 77 (31%) and the α-isomer (8%, N7/N9 mixture). On the other hand, consumption of 67 required longer times and subsequent isomerization to 78 was insufficient at 50 °C, giving 78 in 31% yield with the predominant formation of 77 (40%). The separated N7 isomer 77 was successfully isomerized to the desired N9 isomer 78 in 53% yield upon treatment with TMSOTf in toluene at 90 °C. Under similar conditions, the hypervalent iodine-mediated glycosylation reaction of 67 in the presence of 2,6-dichloropurine was conducted. The coupling reaction proceeded to give an inseparable mixture of N7-isomer 80 and N9-isomer 81 in 64% yield (80:81 = 1:1). To isomerize the undesired N7-isomer to the desired product as in the case of 2,6-dichloropurine, the subsequent treatment of the resulting mixture with TMSOTf in toluene at 90 °C gave rise to exclusive formation of the desired N9-isomer 81 in 62% yield. Finally, 81 was converted to the desired guanosine derivative 82 [60] (Scheme 11).
![[1860-5397-14-137-i11]](/bjoc/content/inline/1860-5397-14-137-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of purine 4’-selenonucleosides using hypervalent iodine-mediated glycosylation.
Scheme 11: Synthesis of purine 4’-selenonucleosides using hypervalent iodine-mediated glycosylation.
As in the case of 4’-thionucleosides, the use of hypervalent iodine greatly improved the glycosylation reaction with 4-seleosugars by skipping the preparation of unstable selenoxide derivatives.
Synthesis of carbocyclic nucleosides
As described above, in the hypervalent iodine-mediated glycosylation, a thiosugar donor 83 was oxidized to a cationic intermediate 84 with the assistance of a Lewis acid (TMSOTf) and a base and the subsequent nucleophilic attack of silylated base to 84 gave the desired nucleoside 85. The success of the hypervalent iodine-mediated glycosylation led us to apply the reaction to the synthesis of carbocyclic nucleosides. In addition, we were also encouraged by the study of Ochiai, who developed the Friedel–Crafts reaction via umpolung of allylsilanes using hypervalent-iodine reagents [61] and the pioneering work on C–N bond formation using hypervalent iodine by Kita [62]. Thus, we envisioned the use of allylsilanes as a pseudosugar donor for the synthesis of carbocyclic nucleosides. We expected to couple a cyclic allylsilane 86, which could act as a pseudosugar donor for carbocyclic nucleosides 88, with a persilylated nucleobase by using a combination of hypervalent iodine and an appropriate Lewis acid (Figure 4).
![[1860-5397-14-137-4]](/bjoc/content/figures/1860-5397-14-137-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Concept of the oxidative coupling reaction applicable to the synthesis of carbocyclic nucleosides.
Figure 4: Concept of the oxidative coupling reaction applicable to the synthesis of carbocyclic nucleosides.
As shown in Scheme 12, an oxidative coupling reaction was examined using a model reaction [63]. Cycloalkenylsilanes 89a,b and 90a,b were prepared by hydrosilylation of cyclopentadiene and cyclohexadiene. Using TMSOTf as a Lewis acid, the hypervalent iodine-mediated coupling reaction of 89a,b and 90a,b with silylated uracil 29 was examined and the results are summarized in Table 2. Our first attempt to couple triethoxysilanes 89a,b with 29 in the presence of diacetoxyiodobenzene gave cycloalkenyluracil 91a and 91b in 45% and 49% yields respectively (Table 2, entries 1 and 2). On the other hand, the use of trialkylsilanes 90a and 90b successfully improved the chemical yield of 91a and 91b (Table 2, entries 3 and 4). In contrast, the reactions using PIFA, iodosylbenzene, and [hydroxyl(tosyloxy)iodo]benzene (PhI(OH)OTs) resulted in a decrease of the reaction yield (Table 2, entries 5–7).
![[1860-5397-14-137-i12]](/bjoc/content/inline/1860-5397-14-137-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Oxidative coupling reaction mediated by hypervalent iodine.
Scheme 12: Oxidative coupling reaction mediated by hypervalent iodine.
To prove the usefulness of the oxidative coupling reaction mediated by hypervalent iodine, the reaction was applied to the synthesis of a carbocyclic nucleoside derivative designed as a potential anti-HIV agent.
As a target, cyclohexenylcytosine 99 was designed and was planned to synthesize using the oxidative coupling reaction. To prepare the substrate of the coupling reaction, cyclohexenylsilane 96 was synthesized using the Diels–Alder reaction of trimethylsilylbutadiene 92 and dimethyl fumarate (93), which gave cyclohexene diester 94 (1:1 mixture of diastereomers) [64]. Reduction and subsequent separation by silica gel column chromatography gave diols 95a and 95b, the hydroxy groups of which were protected to give di-TBDPS derivatives 96a and 96b. The resulting cyclohexenylsilanes 96a and 96b were subjected to the oxidative coupling reaction with 2,4-bis(trimethylsilyl)uracil (29) using diacetoxyiodobenzene, respectively, and the results are shown in Table 3. The reaction of 96a gave an inseparable mixture containing 4 stereoisomers of 97a–d with a ratio of 6:10:2:1.5, which was determined based on the analysis of its 1H NMR spectrum. The reaction of 96b also gave a similar result. In both reactions, the formation of cyclohexadiene 98 was observed. These results strongly supported that the reaction proceeded through the carbocation intermediate, as expected and depicted in Figure 4, since 98 was considered to be formed by E1 elimination of the allyl cation intermediate. The fact that 96a and 96b showed different reactivities could be explained by the steric interaction between the substituents on the cyclohexene ring and the nucleobase approaching. Compounds 97a–d were converted to the corresponding cytosine analogues [63]. During the course of conversion, all the stereoisomers were separated. Among them, only the cytosine derivative 99 showed weak anti-HIV activity (Scheme 13 and Table 3).
![[1860-5397-14-137-i13]](/bjoc/content/inline/1860-5397-14-137-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of cyclohexenyl nucleosides using an oxidative coupling reaction.
Scheme 13: Synthesis of cyclohexenyl nucleosides using an oxidative coupling reaction.
An oxidative coupling reaction for synthesizing carbocyclic nucleosides mediated by hypervalent iodine was developed. Since the Friedel–Crafts type reaction involved carbocation intermediate, the reaction always gave a mixture of products. Unfortunately, the reaction was not efficient. However, it is worthy that the oxidative coupling reaction contains a novel type of C–N bond formation and would help to synthesize new carbocyclic nucleosides.
Synthesis of dihydropyranonucleosides
The success of the oxidative coupling reaction for constructing a carbocyclic nucleoside skeleton led us to develop a glycosylation reaction applicable to glycal derivatives. Since an electron-rich enol ether unit of glycal could react with oxidative agents, it was expected to form a cationic intermediate as in the case of allylsilanes described above. A direct coupling of glycals with nucleobases is challenging, since it is formally a C–N bond-forming reaction with cleaving of the inactive C–H bond at the γ-position. Actually, the C–N bond-forming reactions using hypervalent iodine agents have attracted much attention [62,65-68]. In the case of the hypervalent iodine-catalyzed coupling reaction with allylsilanes (Figure 5), the reaction involves the following 2 steps: 1) the generation of allyl cation 87 by the oxidation of an allylsilane 86 with PhI(OAc)2 and TMSOTf, and 2) the subsequent nucleophilic attack of the persilylated base to 87 as shown in Figure 5. Therefore, we expected that subjecting the electron-rich glycal 100 to the hypervalent iodine-mediated reaction described above would generate an oxocarbenium ion 101 to serve as an intermediate, giving a nucleoside 102.
![[1860-5397-14-137-5]](/bjoc/content/figures/1860-5397-14-137-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Concept of the oxidative coupling reaction of glycal derivatives.
Figure 5: Concept of the oxidative coupling reaction of glycal derivatives.
First, we attempted model reactions of the oxidative coupling to enol ether using a TMSOTf/PhI(OAc)2 system. After several attempts, we found that the reaction of 3,4-dihydro-2H-pyran (DHP, 103) with PhI(OAc)2 and TMSOTf, starting at −40 °C and then gradually raised to room temperature, gave a dihydropyranyluracil derivative 104 in 31% yield [69]. We also found that when Cu(OTf)2 was used as a catalyst in place of TMSOTf, the reaction gave 104 in 24% yield (Scheme 14).
![[1860-5397-14-137-i14]](/bjoc/content/inline/1860-5397-14-137-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Oxidative coupling reaction of silylated uracil and DHP using hypervalent iodine.
Scheme 14: Oxidative coupling reaction of silylated uracil and DHP using hypervalent iodine.
We speculated that the mechanism of the oxidative coupling reaction was as shown in Scheme 15. DHP (103) was reacted with PhI(OAc)2 to produce an acetoxyiodobenzene derivative 105 with the assistance of TMSOTf. With respect to the pathway from the intermediate 105 to the N1-substituted uracil 104, there were two plausible routes. In path a, a nucleophilic attack of 2,4-bis(trimethylsilyl)uracil (29) occurs prior to an elimination. In path b, on the other hand, an allylic carbocation 110 formed from 108 reacts with 29. From the result that the reaction of 2,3-dihydrofuran gave side products generated from an intermediate resembling 107 (data not shown), it was strongly suggested that the oxidative coupling reaction preferred path a rather than path b (Scheme 15).
![[1860-5397-14-137-i15]](/bjoc/content/inline/1860-5397-14-137-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Proposed mechanism of the oxidative coupling reaction mediated by hypervalent iodine.
Scheme 15: Proposed mechanism of the oxidative coupling reaction mediated by hypervalent iodine.
Because further optimization of the oxidative coupling reaction was not successful, we decided to examine the effect of adding a co-catalyst. The speculated reaction mechanism depicted in Scheme 15 suggested that the instability of the intermediates 105 and 106 might have caused the low yield of the oxidative coupling. Based on this idea, we intended to use (PhSe)2 as a co-catalyst, since it might prevent the formation of unstable 105 and 106 and yield 102 in one step (Figure 6).
![[1860-5397-14-137-6]](/bjoc/content/figures/1860-5397-14-137-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Synthesis of 2’,3’-unsaturated nucleosides using hypervalent iodine and a co-catalyst.
Figure 6: Synthesis of 2’,3’-unsaturated nucleosides using hypervalent iodine and a co-catalyst.
We examined the effect of (PhSe)2 as an additive by the reaction of various glycals and their chemical equivalents [69] and the results are summarized in Table 4. The reaction of 103 and 29 was performed by treatment with PhI(OAc)2 and (PhSe)2 in the presence of catalytic amounts of TMSOTf to selectively yield a trans-isomer of 1-(3-phenylselanyltetrahydropyran-2-yl)uracil (116) in 73% yield (Table 4, entry 1). Although this result was unexpected, it was important, since the reaction appeared to be applicable to access various nucleoside derivatives, including 2’-deoxynucleosides. More importantly, we could avoid the use of unstable reagents such as PhSeBr. In other words, the reaction using hypervalent iodine and stable (PhSe)2 in the presence of a Lewis acid would be expected to yield the same products as the reaction using PhSeBr. The reaction with dihydrofuran (112) furnished 1-(3-phenylselanyltetrahydrofuran-2-yl)uracil (117) in 31% yield (Table 4, entry 2). The reaction of 113 with 29 at −5 °C afforded 118 in 69% yield (Table 4, entry 3). The reaction of 114 gave an anomeric mixture of 119 in 80% yield with the predominant formation of the β-nucleoside (Table 4, entry 4). In contrast, the oxidative glycosylation reaction of D-glucal 115 gave a 1:1 mixture of α-120 and β-120 in 64% yield (Table 4, entry 5). From these data, the oxidative coupling reaction mediated by hypervalent iodine of glycal derivatives can clearly be regarded as a new glycosylation reaction that is applicable to the synthesis of 2’-deoxy- and 2’,3’-dideoxydidehydronucleosides, some of which are known to have anti-HIV activity (Table 4).
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-137-i25.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-137-i26.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-137-i27.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-137-i28.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-137-i29.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-137-i30.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-137-i31.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-137-i32.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-137-i33.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-137-i34.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-137-i35.svg?max-width=637&scale=1.0)
To reveal the scope of this reaction, we designed a new dihydropyranonucleoside as a potential anti-HIV agent and attempted to synthesize it by using the oxidative coupling reaction [70]. First, the PMB-protected epoxide 121 was converted to diene 122. The dihydropyran ring of 123 was constructed by RCM of 122 catalyzed by a Grubbs 1st generation catalyst. The isomerization of the double bond in 123 by treatment with a Wilkinson catalyst under basic conditions afforded glycal 124 (Scheme 16).
![[1860-5397-14-137-i16]](/bjoc/content/inline/1860-5397-14-137-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of dihydropyranonucleoside.
Scheme 16: Synthesis of dihydropyranonucleoside.
The hypervalent iodine-mediated glycosylation of 2,4-bis(trimethylsilyl)uracil (29) with glycal 124 gave an inseparable mixture of α- and β-anomers 125 (α:β = 1:2) in 51% yield as we expected. Compound 125 was then oxidized by treatment with mCPBA, followed by elimination of the resulting selenoxide to give 126. After the separation of anomers, the major β-anomer was converted into a cytosine derivative 127 [70]. However, 127 did not show any activity against HIV whereas its 5’-thio counterpart did show anti-HIV activity (Scheme 16).
The reaction mediated by hypervalent iodine provides an alternative method for constructing glycosidic bonds of nucleoside derivatives by using a glycal as sugar donor. Its usefulness was proved by applying the reaction to synthesize new nucleoside derivatives as mentioned above.
Synthesis of acyclic nucleosides
It is known that the oxidative C–C bond cleavage of glycols, epoxides, and olefins takes place by the action of hypervalent iodine [38,71,72]. For example, Havare and Plattner reported the oxidative cleavage of α-aryl aldehydes using iodosylbenzene to give chain-shortened carbonyl compounds and formaldehyde [71]. In the field of carbohydrate chemistry, similar deformylation by action of hypervalent iodine has also been demonstrated: the β-fragmentation reaction of an anomeric alkoxy radical of carbohydrates was mediated by a hypervalent iodine reagent [73]. The reaction results in the formation of carbohydrates with a reduction of one carbon. From the viewpoint of the synthetic method, the reaction would be useful for dehomologation of aldoses and preparation of chiral synthons deriving from sugars. The reaction procedure involves the initial formation of an alkoxy anomeric radical by a hypervalent iodine reagent in the presence of iodine, which triggers the β-fragmentation of the C1–C2 bond. As a result, a C2 radical is generated and is further oxidized to a carbocation that is reacted with nucleophilic agents to give the desired products.
Boto et al. applied the reaction to the one-pot synthesis of acyclic nucleosides that belong to an important class of nucleosides with antiviral activity [74]. First, they tried to synthesize acyclic nucleosides in a stepwise manner. The substrates 128 and 129 for the fragmentation reaction were synthesized from ribose in a few steps by the conventional method. The oxidative scission of 128 and 129 was carried out by treatment with diacetoxyiodobenzene and iodine under irradiation with visible light to give acetoxy acetals 130 and 131 in good yields with high stereoselectivities. As shown in Scheme 17, the reaction was expected to proceed via the formation of anomeric alkoxyl radicals, which underwent fragmentation to produce radical 132. The radical 132 could be trapped with iodine, giving iodide 133. The oxycarbenium ion 134 generated by the extrusion of iodide from 133 reacted with the acetoxy ion to furnish the resulting acetate derivatives. The acetates 130 and 131 were then treated with silylated thymine or N4-benzoylcytosine in the presence of a Lewis acid to give the desired acyclic nucleosides 135 and 136 in excellent yields. The results revealed that the nucleophilic attack of the nucleobase selectively occurred from the less hindered side of the oxycarbenium ion intermediates, giving 1’,2’-trans isomers as major products (Scheme 17).
![[1860-5397-14-137-i17]](/bjoc/content/inline/1860-5397-14-137-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of acetoxyacetals using hypervalent iodine and addition of silylated base.
Scheme 17: Synthesis of acetoxyacetals using hypervalent iodine and addition of silylated base.
Based on the conditions for the stepwise fragmentation and glycosylation procedure, Boto et al. explored the one-pot version of the reaction [74]. When the β-fragmentation, the first step of oxidative glycosylation, was carried out in CH2Cl2 and then the Lewis acid and the silylated base were added, the acyclic nucleosides were obtained in low yields. Boto and co-workers overcame this problem by replacing the solvent before glycosylation. After the fragmentation reaction was finished, the solvent (CH2Cl2) was removed and replaced with acetonitrile. The resulting mixture was treated with TMSOTf and the silylated base. Under the optimized conditions, the reactions of ribose derivative 128, mannose derivative 137, and rhamnose derivative 138 gave the desired acyclic nucleosides in excellent yields as shown in Scheme 18. It is worth noting that the overall yields for the one-pot process are comparable or superior to those obtained with the two-step procedure (Scheme 18).
![[1860-5397-14-137-i18]](/bjoc/content/inline/1860-5397-14-137-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: One-pot fragmentation-nucleophilic additions mediated by hypervalent iodine.
Scheme 18: One-pot fragmentation-nucleophilic additions mediated by hypervalent iodine.
Synthesis of disaccharides
Classically, carbohydrates have been considered primarily an energy source for life – as in the cases of glucose, fructose and their oligosaccharides, e.g., starch. However, more recently it has been revealed that oligosaccharides and glycoconjugates also play important roles in various biological processes, as mentioned earlier. As a result, the increasing significance of oligosaccharides in biological events has led to a strong demand for synthetic routes towards oligosaccharides, which would also contribute to the identification and development of drug candidates. For example, cancer immunotherapy based on vaccines derived from carbohydrate antigen–adjuvant combinations has received much attention in recent years [75-77]. However, the difficulties associated with the isolation of tumor-associated carbohydrate antigens from natural sources have impeded extensive research. Thus, the most promising approach to the supply of these antigens is to develop a suitable method for their chemical synthesis.
To date, various glycosylation reactions capable of constructing oligosaccharides with high stereoselectivities have been reported [18,19]. Thioglycosides are often used as a sugar donor in these reactions due to their stability under various conditions and specific activation with thiophilic agents. For example, one of the typical conditions used for the construction of oligosaccharides is the combination of Lewis acids and iodine or its chemical equivalents. Fukase and co-workers reported a glycosylation reaction with thioglycoside using hypervalent iodine reagents in the 1990s [78,79]. The outline and postulated mechanism of the reaction are shown in Figure 7. The reaction of iodosylbenzene and electrophiles, e.g., triflic anhydride or Lewis acids, should generate a potent thiophile 143 that reacts with thioglycoside 144 to form an oxocarbenium ion 145. The resulting oxocarbenium ion 145 should in turn react with a sugar acceptor to give the glycosylated product 147 (Figure 7).
![[1860-5397-14-137-7]](/bjoc/content/figures/1860-5397-14-137-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The reaction of thioglycoside with hypervalent iodine in the presence of Lewis acids.
Figure 7: The reaction of thioglycoside with hypervalent iodine in the presence of Lewis acids.
By this reaction, Fukase et al. reported the glycosylation of methyl thioglycoside 148 as a sugar donor to give disaccharides 150 and 152 in high chemical yields as depicted in Scheme 19. As mentioned above, not only triflic anhydride, but various Lewis acids (TMSOTf, Sn(OTf)2, Yb(OTf)3) and a Brønsted acid (TfOH) were proven useful as activators, by which the reaction finished in a short time and gave the products with high stereoselectivity [79].
![[1860-5397-14-137-i19]](/bjoc/content/inline/1860-5397-14-137-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of disaccharides employing thioglycosides under an oxidative coupling reaction mediated by hypervalent iodine.
Scheme 19: Synthesis of disaccharides employing thioglycosides under an oxidative coupling reaction mediated b...
Recently, the reaction was revisited by Kajimoto et al., who sought a glycosylation reaction that could be applied to disarmed thioglycosides using hypervalent iodine reagents [80,81]. One of the reactions they examined was the glycosylation reaction of methyl 2-phthalimidothioglucopyranoside 153 with methyl tribenzylglucopyranoside 149 by PIFA in the presence of various acid catalysts. The results showed that the reaction with PIFA and TfOH afforded the best result, giving disaccharide 154 in 77% yield. On the other hand, the use of bis[cyclohexyl]trifluoromethanesulfonylborane [(cyclo-Hex)2BOTf] and methanesulfonic acid resulted in a poor yield. The synthesis of disaccharides under the optimized conditions was performed using “odorless” thioglycoside 155 and 149 as the donor and the acceptor [81]. Even with the combination of “disarmed” 155 and “armed” 149, the reaction gave rise to the desired disaccharide 157 in 87% yield. The same reaction of the corresponding 3-epimer 156 proceeded smoothly to give the disaccharide 158 in good yield (Scheme 20).
![[1860-5397-14-137-i20]](/bjoc/content/inline/1860-5397-14-137-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of disaccharides using disarmed thioglycosides by hypervalent iodine-mediated glycosylation.
Scheme 20: Synthesis of disaccharides using disarmed thioglycosides by hypervalent iodine-mediated glycosylati...
Randolph and Danishefsky reported a glycal assembly strategy to the synthesis of a branched oligosaccharide [82]. Bennett and co-workers reported that phenyl(trifluoroethyl)iodonium triflimide was a stable promoter for glycosylation reactions using thioglycoside donors [83]. Since the reactions often were unselective in the absence of C2 acetate-directing groups, Bennett et al. investigated the compatibility of the above-mentioned reaction in nitrile solvents documented to have a β-directing effect, with the aim of developing a glycosylation that can be selectively achieved in the absence of directing groups. After preliminary screens, they found that the reaction in the presence of phenyl(trifluoroethyl)iodonium triflimide 160 and the non-nucleophilic base 2,4,6-tri-tert-butylpyrimidine (TTBP) at 0 °C with the solvent combination of 2:1 CH2Cl2/pivalonitrile provided the optimal reaction outcome. However, they also encountered a problem: the reduced solubility of substrate in the solvent system resulted in lower yields. They therefore examined mixed nitrile solvents again, and eventually found that a quaternary solvent mixture composed of 6:1:1:1 CH2Cl2/acetonitrile/isobutyronitrile/pivalonitrile greatly improved both the chemical yields and stereoselectivity, as shown in Scheme 21. The results suggested that both the solvent system and iodonium salt promoter are required for selectivity.
![[1860-5397-14-137-i21]](/bjoc/content/inline/1860-5397-14-137-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Glycosylation using aryl(trifluoroethyl)iodium triflimide.
Scheme 21: Glycosylation using aryl(trifluoroethyl)iodium triflimide.
Even though glycals have a π-electron-rich enol ether unit, reports regarding transformations involving glycal oxidation as well as installation of heteroatom substituents at the C2 position were limited. In 2001, Gin’s group reported the C2-acycloxyglycosylation procedure based on hypervalent iodine chemistry [84]. In this reaction, the use of a combination of hypervalent iodine and Lewis acid was key, as in the reactions described above. In this procedure, a solution of the glycal donor and a (diacyloxyiodo)benzene reagent was first treated with BF3·OEt2. Then, the glycosyl acceptor (R”OH) and a catalytic amount of TfOH were added to the mixture, giving the 1,2-trans disubstituted C2-acyloxylglycoside. A plausible mechanism of the reaction is shown in Figure 8. The first step of the reaction between glycal 161 and (diacyloxyiodo)benzene formed the glycosyl ester intermediate 162 bearing a phenyl iodonium(III) functionality at C2, which was transformed to a diacyloxylated product 163. As evidence in support of this mechanism, they reported that 163 was indeed isolated when the reaction was finished at the first step. In the second step, the resulting diacyloxylated product 163 could effectively glycosylate the appropriate acceptor by the action of TfOH to give the C2-acyloxyglycoside 164 with good selectivity at the anomeric position as a consequence of participation by the neighboring C2 acyloxy group (Figure 8).
![[1860-5397-14-137-8]](/bjoc/content/figures/1860-5397-14-137-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Expected mechanism of hypervalent iodine-mediated glycosylation with glycals.
Figure 8: Expected mechanism of hypervalent iodine-mediated glycosylation with glycals.
They prepared C2-acyloxy glycosides 165, 166, 169, 170 and 172 using hypervalent iodine-mediated coupling reactions with glycals, and the results are shown in Scheme 22. Either (diacetoxyiodo)benzene or (dibenzoyloxyiodo)benzene could serve as an efficient oxidant, and the reactions utilizing them gave the products installing either the acetate or benzoate functionality, respectively, at the C2-position. Both glucal 115 and galactal 167 were amenable to the oxidative glycosylation reaction to stereoselectively give C2-acyloxylated β-glycosides in good yields [84] (Scheme 22).
![[1860-5397-14-137-i22]](/bjoc/content/inline/1860-5397-14-137-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of oligosaccharides by hypervalent iodine-mediated glycosylation with glycals.
Scheme 22: Synthesis of oligosaccharides by hypervalent iodine-mediated glycosylation with glycals.
Hotha and co-workers utilized the reaction of glycals with hypervalent iodine reagents for the stereoselective synthesis of C2 deoxyglycosides and amino acid glycoconjugates [85]. In their work, they also utilized an important chemical attribute of cetylammonium bromide (CTAB) – namely, CTAB forms surfactant-assembled lipophilic nanoreactors stable in organic solvents, which could be used for regioselective functionalization of indenes. Therefore, they investigated the regioselective iodination of glycals by using CTAB and hypervalent iodine reagents for the synthesis of 2-deoxy-2-iodoacetates. In the preliminary experiments, the reaction between per-O-acetylglucal (177) and PhI(OAc)2 in CTAB and KI gave trans-2-iodo α-acetate and its corresponding bromo acetate in a 94:5 ratio. The latter was expected to be formed by halide counter ion exchange between CTAB and KI. Since the reaction occurred as expected, it was applied to the synthesis of amino acid conjugates. Acetyl groups of the (diacetoxyiodo)benzene were exchanged with N- and O-protected amino acids by slow evaporation of a mixture of PhI(OAc)2 and amino acid 173 and 174 in chlorobenzene to give PhI(OCOR)2 compounds 175 and 176. The formation of iodo ester glycosides 178 and 179 from 175 and 176 was achieved in very good yields under the conditions shown in Scheme 23.
![[1860-5397-14-137-i23]](/bjoc/content/inline/1860-5397-14-137-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of 2-deoxy amino acid glycosides.
Scheme 23: Synthesis of 2-deoxy amino acid glycosides.
Notably, the resulting iodo ester glycosides 178 and 179 were considered to have self-assembled structures versatile for the synthesis of serenylated and threonylated glycosides by intramolecular glycosylation. In addition, the access to 2-deoxyglycosides should be easily achievable by subsequent radical deiodination of the products. After several experiments, treatment with a catalytic amount of TMSOTf was found to be suitable for the intramolecular glycosylation, giving the corresponding acid, which was easily converted to the corresponding methyl ester 180 under EDCI/DMAP/MeOH conditions [85]. Similarly, the reaction of the threonine derivative 179 afforded 181 in good yield. Radical deiodination of 180 and 181 using Bu3SnH and AIBN successfully gave 2-deoxy-β-glycosides 182 and 183, which were difficult to synthesize from the corresponding 2-deoxy sugar derivative in a stereoselective manner (Scheme 23).
As mentioned above, the iodo ester glycosides were considered to have self-assembled structures suitable for intramolecular glycosylation. As depicted in Figure 9, treatment of 184 with TMSOTf first cleaved the silyl ether to form 185, which was correctly positioned to undergo intramolecular glycosidation. As a result, the Lewis acid could also facilitate the departure of the anomeric ester and the resulting 185 gave rise to the intramolecular nucleophilic attack to furnish the corresponding acid 186.
![[1860-5397-14-137-9]](/bjoc/content/figures/1860-5397-14-137-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Rationale for the intramolecular migration of the amino acid unit.
Figure 9: Rationale for the intramolecular migration of the amino acid unit.
Glycals and thioglycosides were often used as sugar donors for the glycosylation of oligosaccharides. It is interesting that the hypervalent iodine-mediated oxidative reactions with theses derivatives provide a different method to build glycosidic bonds. Diversity in glycoside bond forming reactions would contribute to improve the oligosaccharide synthesis.
Conclusion
The Pummerer-type glycosylation includes oxidation of a sulfide to the corresponding sulfoxide followed by the TMSOTf-mediated coupling reaction. The reaction utilizing hypervalent iodine reagents could bypass one step of the Pummerer-type glycosylation and directly give 4’-thionucleosides from the corresponding sulfide derivative. The reaction could be efficiently applied to the synthesis of 4’-selenonucleosides as well as 4’-thionucleosides. Based on the concept of hypervalent iodine-mediated glycosylation, a reaction applicable to the synthesis of carbocyclic nucleosides and a coupling reaction between nucleobase and glycal derivatives were developed. The latter reaction was employed to synthesize dihydropyranonucleosides. Oxidative scission is a characteristic reaction mediated by hypervalent iodine reagents and is typically used for dehomologation of sugars. A one-pot glycosylation using this reaction was also developed for the synthesis of acyclic nucleoside derivatives. In addition to nucleoside synthesis, hypervalent iodine-mediated glycosylation could also be applied to the synthesis of oligosaccharides and glycoconjugates when thioglycosides and glycals were used as sugar donors. There is no doubt that the use of hypervalent iodine reagents greatly improved the efficiency of the synthesis of nucleosides and oligosaccharides. The results of these syntheses demonstrate the power of glycoside bond-forming reactions, and should assist in the future identification or synthesis of biologically active nucleoside and glycoconjugate derivatives.
Acknowledgements
Our work described in this review article was supported in part by a Grant-in-Aid for Scientific Research from JSPS and by a grant from the Strategic Research Foundation Grant-aided Project for Private Universities from the Ministry of Education, Culture, Sport, Science, and Technology, Japan (MEXT).
References
-
Varki, A. Glycobiology 2017, 27, 3–49. doi:10.1093/glycob/cww086
Return to citation in text: [1] -
Nyhan, W. L. Mol. Genet. Metab. 2005, 86, 25–33. doi:10.1016/j.ymgme.2005.07.027
Return to citation in text: [1] -
Muntoni, F.; Brockington, M.; Blake, D. J.; Torelli, S.; Brown, S. C. Lancet 2002, 360, 1419–1421. doi:10.1016/s0140-6736(02)11397-3
Return to citation in text: [1] -
Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010
Return to citation in text: [1] [2] -
Schaefer, L.; Tredup, C.; Gubbiotti, M. A.; Iozzo, R. V. FEBS J. 2017, 284, 10–26. doi:10.1111/febs.13963
Return to citation in text: [1] -
Maishi, N.; Hida, K. Cancer Sci. 2017, 108, 1921–1926. doi:10.1111/cas.13336
Return to citation in text: [1] -
Sanderson, R. D.; Elkin, M.; Rapraeger, A. C.; Ilan, N.; Vlodavsky, I. FEBS J. 2017, 284, 42–55. doi:10.1111/febs.13932
Return to citation in text: [1] -
Meadows, D. C.; Gervay-Hague, J. ChemMedChem 2006, 1, 16–29. doi:10.1002/cmdc.200500026
Return to citation in text: [1] -
Stutz, A. E., Ed. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999.
Return to citation in text: [1] -
Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/s0957-4166(00)00113-0
Return to citation in text: [1] -
Saneyoshi, H.; Ito, Y.; Abe, H. J. Am. Chem. Soc. 2013, 135, 13632–13635. doi:10.1021/ja406724k
Return to citation in text: [1] -
Seo, S.; Onizuka, K.; Nishioka, C.; Takahashi, E.; Tsuneda, S.; Abe, H.; Ito, Y. Org. Biomol. Chem. 2015, 13, 4589–4595. doi:10.1039/c5ob00199d
Return to citation in text: [1] -
Kato, A.; Hayashi, E.; Miyauchi, S.; Adachi, I.; Imahori, T.; Natori, Y.; Yoshimura, Y.; Nash, R. J.; Shimaoka, H.; Nakagome, I.; Koseki, J.; Hirono, S.; Takahata, H. J. Med. Chem. 2012, 55, 10347–10362. doi:10.1021/jm301304e
Return to citation in text: [1] -
Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R. J.; Fleet, G. W. J.; Natori, Y.; Watanabe, Y.; Imahori, T.; Yoshimura, Y.; Takahata, H.; Hirono, S. Org. Biomol. Chem. 2016, 14, 1039–1048. doi:10.1039/C5OB02223A
Return to citation in text: [1] -
Niedballa, U.; Vorbrueggen, H. J. Org. Chem. 1974, 39, 3654–3660. doi:10.1021/jo00939a008
Return to citation in text: [1] [2] [3] -
Vorbrüggen, H.; Ruh-Pohlenz, C. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or055.01
Return to citation in text: [1] [2] [3] -
Yoshimura, Y. Heterocycles 2017, 94, 1625–1651. doi:10.3987/REV-17-865
Return to citation in text: [1] [2] [3] -
Hanessian, S.; Lou, B. Chem. Rev. 2000, 100, 4443–4464. doi:10.1021/cr9903454
Return to citation in text: [1] [2] -
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036
Return to citation in text: [1] [2] -
Yoshimura, Y.; Kobayashi, S.; Kaneko, H.; Suzuki, T.; Imamichi, T. Molecules 2015, 20, 4623–4634. doi:10.3390/molecules20034623
Return to citation in text: [1] -
Kiran, Y. B.; Wakamatsu, H.; Natori, Y.; Takahata, H.; Yoshimura, Y. Tetrahedron Lett. 2013, 54, 3949–3952. doi:10.1016/j.tetlet.2013.05.062
Return to citation in text: [1] -
Yoshimura, Y.; Asami, K.; Imamichi, T.; Okuda, T.; Shiraki, K.; Takahata, H. J. Org. Chem. 2010, 75, 4161–4171. doi:10.1021/jo100556u
Return to citation in text: [1] -
Matsuda, A.; Takenuki, K.; Tanaka, M.; Sasaki, T.; Ueda, T. J. Med. Chem. 1991, 34, 812–819. doi:10.1021/jm00106a049
Return to citation in text: [1] -
Yamagami, K.; Fujii, A.; Arita, M.; Okumoto, T.; Sakata, S.; Matsuda, A.; Ueda, T.; Sasaki, T. Cancer Res. 1991, 51, 2319–2323.
Return to citation in text: [1] -
Dyson, M. R.; Coe, P. L.; Walker, R. T. J. Med. Chem. 1991, 34, 2782–2786. doi:10.1021/jm00113a016
Return to citation in text: [1] [2] -
Secrist III, J. A.; Tiwari, K. N.; Riordan, J. M.; Montgomery, J. A. J. Med. Chem. 1991, 34, 2361–2366. doi:10.1021/jm00112a007
Return to citation in text: [1] [2] -
Yoshimura, Y.; Kitano, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1996, 61, 822–823. doi:10.1021/JO9519423
Return to citation in text: [1] [2] -
Yoshimura, Y.; Kitano, K.; Yamada, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1997, 62, 3140–3152. doi:10.1021/JO9700540
Return to citation in text: [1] [2] -
Kita, Y.; Yasuda, H.; Tamura, O.; Itoh, F.; Tamura, Y. Tetrahedron Lett. 1984, 25, 4681–4682. doi:10.1016/s0040-4039(01)91232-8
Return to citation in text: [1] [2] -
Kita, Y.; Tamura, O.; Yasuda, H.; Itoh, F.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 4235–4241. doi:10.1248/cpb.33.4235
Return to citation in text: [1] [2] -
Naka, T.; Minakawa, N.; Abe, H.; Kaga, D.; Matsuda, A. J. Am. Chem. Soc. 2000, 122, 7233–7243. doi:10.1021/ja000541o
Return to citation in text: [1] -
Yoshimura, Y.; Kuze, T.; Ueno, M.; Komiya, F.; Haraguchi, K.; Tanaka, H.; Kano, F.; Yamada, K.; Asami, K.; Kaneko, N.; Takahata, H. Tetrahedron Lett. 2006, 47, 591–594. doi:10.1016/j.tetlet.2005.11.049
Return to citation in text: [1] -
Jeong, L. S.; Moon, H. R.; Choi, Y. J.; Chun, M. W.; Kim, H. O. J. Org. Chem. 1998, 63, 4821–4825. doi:10.1021/JO980196+
Return to citation in text: [1] -
Lim, M. H.; Kim, H. O.; Moon, H. R.; Chun, M. W.; Jeong, L. S. Org. Lett. 2002, 4, 529–531. doi:10.1021/ol017112v
Return to citation in text: [1] -
Jeong, L. S.; Lee, H. W.; Kim, H. O.; Jung, J. Y.; Gao, Z.-G.; Duong, H. T.; Rao, S.; Jacobson, K. A.; Shin, D. H.; Lee, J. A.; Gunaga, P.; Lee, S. K.; Jin, D. Z.; Chun, M. W.; Moon, H. R. Bioorg. Med. Chem. 2006, 14, 4718–4730. doi:10.1016/j.bmc.2006.03.030
Return to citation in text: [1] -
Choi, W. J.; Lee, H. W.; Kim, H. O.; Chinn, M.; Gao, Z.-G.; Patel, A.; Jacobson, K. A.; Moon, H. R.; Jung, Y. H.; Jeong, L. S. Bioorg. Med. Chem. 2009, 17, 8003–8011. doi:10.1016/j.bmc.2009.10.011
Return to citation in text: [1] -
Qu, S.; Mulamoottil, V. A.; Nayak, A.; Ryu, S.; Hou, X.; Song, J.; Yu, J.; Sahu, P. K.; Zhao, L. X.; Choi, S.; Lee, S. K.; Jeong, L. S. Bioorg. Med. Chem. 2016, 24, 3418–3428. doi:10.1016/j.bmc.2016.05.041
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] [2] -
Dohi, T.; Morimoto, K.; Maruyama, A.; Kita, Y. Org. Lett. 2006, 8, 2007–2010. doi:10.1021/ol060333m
Return to citation in text: [1] -
Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668–1669. doi:10.1021/ja808940n
Return to citation in text: [1] -
Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Org. Lett. 2010, 12, 3804–3807. doi:10.1021/ol101498r
Return to citation in text: [1] -
Dohi, T.; Ito, M.; Itani, I.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Org. Lett. 2011, 13, 6208–6211. doi:10.1021/ol202632h
Return to citation in text: [1] -
Ito, M.; Kubo, H.; Itani, I.; Morimoto, K.; Dohi, T.; Kita, Y. J. Am. Chem. Soc. 2013, 135, 14078–14081. doi:10.1021/ja407944p
Return to citation in text: [1] -
Tohma, H. Chem. Commun. 1998, 173–174. doi:10.1039/a707727k
Return to citation in text: [1] -
Nishizono, N.; Baba, R.; Nakamura, C.; Oda, K.; Machida, M. Org. Biomol. Chem. 2003, 1, 3692–3697. doi:10.1039/B305644A
Return to citation in text: [1] [2] [3] -
Nishizono, N.; Soma, K.; Baba, R.; Machida, M.; Oda, K. Heterocycles 2008, 75, 619–634. doi:10.3987/COM-07-11250
Return to citation in text: [1] [2] -
Nishizono, N.; Akama, Y.; Agata, M.; Sugo, M.; Yamaguchi, Y.; Oda, K. Tetrahedron 2011, 67, 358–363. doi:10.1016/j.tet.2010.11.038
Return to citation in text: [1] [2] -
Jeong, L. S.; Tosh, D. K.; Kim, H. O.; Wang, T.; Hou, X.; Yun, H. S.; Kwon, Y.; Lee, S. K.; Choi, J.; Zhao, L. X. Org. Lett. 2008, 10, 209–212. doi:10.1021/ol7025558
Return to citation in text: [1] [2] [3] -
Inagaki, Y.; Minakawa, N.; Matsuda, A. Nucleic Acids Symp. Ser. 2007, 51, 139–140. doi:10.1093/nass/nrm070
Return to citation in text: [1] -
Jayakanthan, K.; Johnston, B. D.; Pinto, B. M. Carbohydr. Res. 2008, 343, 1790–1800. doi:10.1016/j.carres.2008.02.014
Return to citation in text: [1] -
Tosh, D. K.; Choi, W. J.; Kim, H. O.; Lee, Y.; Pal, S.; Hou, X.; Choi, J.; Choi, S.; Jeong, L. S. J. Org. Chem. 2008, 73, 4259–4262. doi:10.1021/jo8003277
Return to citation in text: [1] -
Alexander, V.; Choi, W. J.; Chun, J.; Kim, H. O.; Jeon, J. H.; Tosh, D. K.; Lee, H. W.; Chandra, G.; Choi, J.; Jeong, L. S. Org. Lett. 2010, 12, 2242–2245. doi:10.1021/ol1005906
Return to citation in text: [1] -
Yu, J.; Kim, J.-H.; Lee, H. W.; Alexander, V.; Ahn, H.-C.; Choi, W. J.; Choi, J.; Jeong, L. S. Chem. – Eur. J. 2013, 19, 5528–5532. doi:10.1002/chem.201300741
Return to citation in text: [1] [2] -
Kim, J.-H.; Yu, J.; Alexander, V.; Choi, J. H.; Song, J.; Lee, H. W.; Kim, H. O.; Choi, J.; Lee, S. K.; Jeong, L. S. Eur. J. Med. Chem. 2014, 83, 208–225. doi:10.1016/j.ejmech.2014.06.031
Return to citation in text: [1] -
Sahu, P. K.; Kim, G.; Yu, J.; Ahn, J. Y.; Song, J.; Choi, Y.; Jin, X.; Kim, J.-H.; Lee, S. K.; Park, S.; Jeong, L. S. Org. Lett. 2014, 16, 5796–5799. doi:10.1021/ol502899b
Return to citation in text: [1] -
Alexander, V.; Song, J.; Yu, J.; Choi, J. H.; Kim, J.-H.; Lee, S. K.; Choi, W. J.; Jeong, L. S. Arch. Pharmacal Res. 2015, 38, 966–972. doi:10.1007/s12272-014-0466-6
Return to citation in text: [1] -
Kim, G.; Choi, Y.; Sahu, P. K.; Yu, J.; Qu, S.; Lee, D.; Jeong, L. S. Org. Lett. 2015, 17, 4636–4639. doi:10.1021/acs.orglett.5b02393
Return to citation in text: [1] -
Qu, S.; Kim, G.; Yu, J.; Sahu, P. K.; Choi, Y.; Naik, S. D.; Jeong, L. S. Asian J. Org. Chem. 2016, 5, 735–741. doi:10.1002/ajoc.201600154
Return to citation in text: [1] -
Taniike, H.; Inagaki, Y.; Matsuda, A.; Minakawa, N. Tetrahedron 2011, 67, 7977–7982. doi:10.1016/j.tet.2011.08.020
Return to citation in text: [1] [2] -
Ishii, K.; Saito-Tarashima, N.; Ota, M.; Yamamoto, S.; Okamoto, Y.; Tanaka, Y.; Minakawa, N. Tetrahedron 2016, 72, 6589–6594. doi:10.1016/j.tet.2016.08.071
Return to citation in text: [1] [2] -
Ochiai, M.; Fujita, E.; Arimoto, M.; Yamaguchi, H. Chem. Pharm. Bull. 1985, 33, 41–47. doi:10.1248/cpb.33.41
Return to citation in text: [1] -
Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Chem. Commun. 2007, 1224–1226. doi:10.1039/B616510A
Return to citation in text: [1] [2] -
Yoshimura, Y.; Ohta, M.; Imahori, T.; Imamichi, T.; Takahata, H. Org. Lett. 2008, 10, 3449–3452. doi:10.1021/ol8012155
Return to citation in text: [1] [2] -
Jung, M. E.; Gaede, B. Tetrahedron 1979, 35, 621–625. doi:10.1016/0040-4020(79)87006-4
Return to citation in text: [1] -
Ochiai, M.; Miyamoto, K.; Kaneaki, T.; Hayashi, S.; Nakanishi, W. Science 2011, 332, 448–451. doi:10.1126/science.1201686
Return to citation in text: [1] -
Cho, S. H.; Yoon, J.; Chang, S. J. Am. Chem. Soc. 2011, 133, 5996–6005. doi:10.1021/ja111652v
Return to citation in text: [1] -
Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605–8608. doi:10.1002/anie.201102984
Return to citation in text: [1] -
Souto, J. A.; Zian, D.; Muñiz, K. J. Am. Chem. Soc. 2012, 134, 7242–7245. doi:10.1021/ja3013193
Return to citation in text: [1] -
Yoshimura, Y.; Kan-no, H.; Kiran, Y. B.; Natori, Y.; Saito, Y.; Takahata, H. Synthesis 2012, 44, 1163–1170. doi:10.1055/s-0031-1290749
Return to citation in text: [1] [2] -
Kan-no, H.; Saito, Y.; Omoto, S.; Minato, S.; Wakamatsu, H.; Natori, Y.; Imamichi, T.; Takahata, H.; Yoshimura, Y. Synthesis 2014, 46, 879–886. doi:10.1055/s-0033-1340663
Return to citation in text: [1] [2] -
Havare, N.; Plattner, D. A. Org. Lett. 2012, 14, 5078–5081. doi:10.1021/ol301675v
Return to citation in text: [1] [2] -
Miyamoto, K.; Tada, N.; Ochiai, M. J. Am. Chem. Soc. 2007, 129, 2772–2773. doi:10.1021/ja070179e
Return to citation in text: [1] -
Francisco, C. G.; Freire, R.; González, C. C.; León, E. I.; Riesco-Fagundo, C.; Suárez, E. J. Org. Chem. 2001, 66, 1861–1866. doi:10.1021/jo0057452
Return to citation in text: [1] -
Boto, A.; Hernández, D.; Hernández, R.; Álvarez, E. J. Org. Chem. 2007, 72, 9523–9532. doi:10.1021/jo701608p
Return to citation in text: [1] [2] -
Galonić, D. P.; Gin, D. Y. Nature 2007, 446, 1000–1007. doi:10.1038/nature05813
Return to citation in text: [1] -
Buskas, T.; Thompson, P.; Boons, G. J. Chem. Commun. 2009, 5335–5349. doi:10.1039/b908664c
Return to citation in text: [1] -
Fernández-Tejada, A.; Cañada, F. J.; Jiménez-Barbero, J. Chem. – Eur. J. 2015, 21, 10616–10628. doi:10.1002/chem.201500831
Return to citation in text: [1] -
Fukase, K.; Aoki, Y.; Kinoshita, I.; Suda, Y.; Kurosawa, M.; Zähringer, U.; Rietschel, E. T.; Kusumoto, S. Tetrahedron Lett. 1995, 36, 8645–8648. doi:10.1016/0040-4039(95)01863-D
Return to citation in text: [1] -
Fukase, K.; Kinoshita, I.; Kanoh, T.; Nakai, Y.; Hasuoka, A.; Kusumoto, S. Tetrahedron 1996, 52, 3897–3904. doi:10.1016/S0040-4020(96)00057-9
Return to citation in text: [1] [2] -
Kajimoto, T.; Morimoto, K.; Ogawa, R.; Dohi, T.; Kita, Y. Eur. J. Org. Chem. 2015, 2015, 2138–2142. doi:10.1002/ejoc.201500186
Return to citation in text: [1] -
Kajimoto, T.; Morimoto, K.; Ogawa, R.; Dohi, T.; Kita, Y. Chem. Pharm. Bull. 2016, 64, 838–844. doi:10.1248/cpb.c16-00203
Return to citation in text: [1] [2] -
Randolph, J.; Danishefsky, S. J. J. Am. Chem. Soc. 1993, 115, 8473–8474. doi:10.1021/ja00071a077
Return to citation in text: [1] -
Chu, A.-H. A.; Minciunescu, A.; Bennett, C. S. Org. Lett. 2015, 17, 6262–6265. doi:10.1021/acs.orglett.5b03282
Return to citation in text: [1] -
Shi, L.; Kim, Y.-J.; Gin, D. Y. J. Am. Chem. Soc. 2001, 123, 6939–6940. doi:10.1021/ja015991a
Return to citation in text: [1] [2] -
Islam, M.; Tirukoti, N. D.; Nandi, S.; Hotha, S. J. Org. Chem. 2014, 79, 4470–4476. doi:10.1021/jo500465m
Return to citation in text: [1] [2]
| 46. | Nishizono, N.; Soma, K.; Baba, R.; Machida, M.; Oda, K. Heterocycles 2008, 75, 619–634. doi:10.3987/COM-07-11250 |
| 47. | Nishizono, N.; Akama, Y.; Agata, M.; Sugo, M.; Yamaguchi, Y.; Oda, K. Tetrahedron 2011, 67, 358–363. doi:10.1016/j.tet.2010.11.038 |
| 47. | Nishizono, N.; Akama, Y.; Agata, M.; Sugo, M.; Yamaguchi, Y.; Oda, K. Tetrahedron 2011, 67, 358–363. doi:10.1016/j.tet.2010.11.038 |
| 59. | Taniike, H.; Inagaki, Y.; Matsuda, A.; Minakawa, N. Tetrahedron 2011, 67, 7977–7982. doi:10.1016/j.tet.2011.08.020 |
| 45. | Nishizono, N.; Baba, R.; Nakamura, C.; Oda, K.; Machida, M. Org. Biomol. Chem. 2003, 1, 3692–3697. doi:10.1039/B305644A |
| 59. | Taniike, H.; Inagaki, Y.; Matsuda, A.; Minakawa, N. Tetrahedron 2011, 67, 7977–7982. doi:10.1016/j.tet.2011.08.020 |
| 48. | Jeong, L. S.; Tosh, D. K.; Kim, H. O.; Wang, T.; Hou, X.; Yun, H. S.; Kwon, Y.; Lee, S. K.; Choi, J.; Zhao, L. X. Org. Lett. 2008, 10, 209–212. doi:10.1021/ol7025558 |
| 50. | Jayakanthan, K.; Johnston, B. D.; Pinto, B. M. Carbohydr. Res. 2008, 343, 1790–1800. doi:10.1016/j.carres.2008.02.014 |
| 51. | Tosh, D. K.; Choi, W. J.; Kim, H. O.; Lee, Y.; Pal, S.; Hou, X.; Choi, J.; Choi, S.; Jeong, L. S. J. Org. Chem. 2008, 73, 4259–4262. doi:10.1021/jo8003277 |
| 52. | Alexander, V.; Choi, W. J.; Chun, J.; Kim, H. O.; Jeon, J. H.; Tosh, D. K.; Lee, H. W.; Chandra, G.; Choi, J.; Jeong, L. S. Org. Lett. 2010, 12, 2242–2245. doi:10.1021/ol1005906 |
| 53. | Yu, J.; Kim, J.-H.; Lee, H. W.; Alexander, V.; Ahn, H.-C.; Choi, W. J.; Choi, J.; Jeong, L. S. Chem. – Eur. J. 2013, 19, 5528–5532. doi:10.1002/chem.201300741 |
| 54. | Kim, J.-H.; Yu, J.; Alexander, V.; Choi, J. H.; Song, J.; Lee, H. W.; Kim, H. O.; Choi, J.; Lee, S. K.; Jeong, L. S. Eur. J. Med. Chem. 2014, 83, 208–225. doi:10.1016/j.ejmech.2014.06.031 |
| 55. | Sahu, P. K.; Kim, G.; Yu, J.; Ahn, J. Y.; Song, J.; Choi, Y.; Jin, X.; Kim, J.-H.; Lee, S. K.; Park, S.; Jeong, L. S. Org. Lett. 2014, 16, 5796–5799. doi:10.1021/ol502899b |
| 56. | Alexander, V.; Song, J.; Yu, J.; Choi, J. H.; Kim, J.-H.; Lee, S. K.; Choi, W. J.; Jeong, L. S. Arch. Pharmacal Res. 2015, 38, 966–972. doi:10.1007/s12272-014-0466-6 |
| 57. | Kim, G.; Choi, Y.; Sahu, P. K.; Yu, J.; Qu, S.; Lee, D.; Jeong, L. S. Org. Lett. 2015, 17, 4636–4639. doi:10.1021/acs.orglett.5b02393 |
| 58. | Qu, S.; Kim, G.; Yu, J.; Sahu, P. K.; Choi, Y.; Naik, S. D.; Jeong, L. S. Asian J. Org. Chem. 2016, 5, 735–741. doi:10.1002/ajoc.201600154 |
| 48. | Jeong, L. S.; Tosh, D. K.; Kim, H. O.; Wang, T.; Hou, X.; Yun, H. S.; Kwon, Y.; Lee, S. K.; Choi, J.; Zhao, L. X. Org. Lett. 2008, 10, 209–212. doi:10.1021/ol7025558 |
| 49. | Inagaki, Y.; Minakawa, N.; Matsuda, A. Nucleic Acids Symp. Ser. 2007, 51, 139–140. doi:10.1093/nass/nrm070 |
| 48. | Jeong, L. S.; Tosh, D. K.; Kim, H. O.; Wang, T.; Hou, X.; Yun, H. S.; Kwon, Y.; Lee, S. K.; Choi, J.; Zhao, L. X. Org. Lett. 2008, 10, 209–212. doi:10.1021/ol7025558 |
| 60. | Ishii, K.; Saito-Tarashima, N.; Ota, M.; Yamamoto, S.; Okamoto, Y.; Tanaka, Y.; Minakawa, N. Tetrahedron 2016, 72, 6589–6594. doi:10.1016/j.tet.2016.08.071 |
| 53. | Yu, J.; Kim, J.-H.; Lee, H. W.; Alexander, V.; Ahn, H.-C.; Choi, W. J.; Choi, J.; Jeong, L. S. Chem. – Eur. J. 2013, 19, 5528–5532. doi:10.1002/chem.201300741 |
| 60. | Ishii, K.; Saito-Tarashima, N.; Ota, M.; Yamamoto, S.; Okamoto, Y.; Tanaka, Y.; Minakawa, N. Tetrahedron 2016, 72, 6589–6594. doi:10.1016/j.tet.2016.08.071 |
| 69. | Yoshimura, Y.; Kan-no, H.; Kiran, Y. B.; Natori, Y.; Saito, Y.; Takahata, H. Synthesis 2012, 44, 1163–1170. doi:10.1055/s-0031-1290749 |
| 69. | Yoshimura, Y.; Kan-no, H.; Kiran, Y. B.; Natori, Y.; Saito, Y.; Takahata, H. Synthesis 2012, 44, 1163–1170. doi:10.1055/s-0031-1290749 |
| 63. | Yoshimura, Y.; Ohta, M.; Imahori, T.; Imamichi, T.; Takahata, H. Org. Lett. 2008, 10, 3449–3452. doi:10.1021/ol8012155 |
| 62. | Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Chem. Commun. 2007, 1224–1226. doi:10.1039/B616510A |
| 65. | Ochiai, M.; Miyamoto, K.; Kaneaki, T.; Hayashi, S.; Nakanishi, W. Science 2011, 332, 448–451. doi:10.1126/science.1201686 |
| 66. | Cho, S. H.; Yoon, J.; Chang, S. J. Am. Chem. Soc. 2011, 133, 5996–6005. doi:10.1021/ja111652v |
| 67. | Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605–8608. doi:10.1002/anie.201102984 |
| 68. | Souto, J. A.; Zian, D.; Muñiz, K. J. Am. Chem. Soc. 2012, 134, 7242–7245. doi:10.1021/ja3013193 |
| 63. | Yoshimura, Y.; Ohta, M.; Imahori, T.; Imamichi, T.; Takahata, H. Org. Lett. 2008, 10, 3449–3452. doi:10.1021/ol8012155 |
| 64. | Jung, M. E.; Gaede, B. Tetrahedron 1979, 35, 621–625. doi:10.1016/0040-4020(79)87006-4 |
| 61. | Ochiai, M.; Fujita, E.; Arimoto, M.; Yamaguchi, H. Chem. Pharm. Bull. 1985, 33, 41–47. doi:10.1248/cpb.33.41 |
| 62. | Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Chem. Commun. 2007, 1224–1226. doi:10.1039/B616510A |
| 70. | Kan-no, H.; Saito, Y.; Omoto, S.; Minato, S.; Wakamatsu, H.; Natori, Y.; Imamichi, T.; Takahata, H.; Yoshimura, Y. Synthesis 2014, 46, 879–886. doi:10.1055/s-0033-1340663 |
| 38. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 71. | Havare, N.; Plattner, D. A. Org. Lett. 2012, 14, 5078–5081. doi:10.1021/ol301675v |
| 72. | Miyamoto, K.; Tada, N.; Ochiai, M. J. Am. Chem. Soc. 2007, 129, 2772–2773. doi:10.1021/ja070179e |
| 70. | Kan-no, H.; Saito, Y.; Omoto, S.; Minato, S.; Wakamatsu, H.; Natori, Y.; Imamichi, T.; Takahata, H.; Yoshimura, Y. Synthesis 2014, 46, 879–886. doi:10.1055/s-0033-1340663 |
| 15. | Niedballa, U.; Vorbrueggen, H. J. Org. Chem. 1974, 39, 3654–3660. doi:10.1021/jo00939a008 |
| 16. | Vorbrüggen, H.; Ruh-Pohlenz, C. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or055.01 |
| 17. | Yoshimura, Y. Heterocycles 2017, 94, 1625–1651. doi:10.3987/REV-17-865 |
| 27. | Yoshimura, Y.; Kitano, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1996, 61, 822–823. doi:10.1021/JO9519423 |
| 28. | Yoshimura, Y.; Kitano, K.; Yamada, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1997, 62, 3140–3152. doi:10.1021/JO9700540 |
| 78. | Fukase, K.; Aoki, Y.; Kinoshita, I.; Suda, Y.; Kurosawa, M.; Zähringer, U.; Rietschel, E. T.; Kusumoto, S. Tetrahedron Lett. 1995, 36, 8645–8648. doi:10.1016/0040-4039(95)01863-D |
| 79. | Fukase, K.; Kinoshita, I.; Kanoh, T.; Nakai, Y.; Hasuoka, A.; Kusumoto, S. Tetrahedron 1996, 52, 3897–3904. doi:10.1016/S0040-4020(96)00057-9 |
| 4. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 8. | Meadows, D. C.; Gervay-Hague, J. ChemMedChem 2006, 1, 16–29. doi:10.1002/cmdc.200500026 |
| 9. | Stutz, A. E., Ed. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999. |
| 10. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/s0957-4166(00)00113-0 |
| 11. | Saneyoshi, H.; Ito, Y.; Abe, H. J. Am. Chem. Soc. 2013, 135, 13632–13635. doi:10.1021/ja406724k |
| 12. | Seo, S.; Onizuka, K.; Nishioka, C.; Takahashi, E.; Tsuneda, S.; Abe, H.; Ito, Y. Org. Biomol. Chem. 2015, 13, 4589–4595. doi:10.1039/c5ob00199d |
| 13. | Kato, A.; Hayashi, E.; Miyauchi, S.; Adachi, I.; Imahori, T.; Natori, Y.; Yoshimura, Y.; Nash, R. J.; Shimaoka, H.; Nakagome, I.; Koseki, J.; Hirono, S.; Takahata, H. J. Med. Chem. 2012, 55, 10347–10362. doi:10.1021/jm301304e |
| 14. | Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R. J.; Fleet, G. W. J.; Natori, Y.; Watanabe, Y.; Imahori, T.; Yoshimura, Y.; Takahata, H.; Hirono, S. Org. Biomol. Chem. 2016, 14, 1039–1048. doi:10.1039/C5OB02223A |
| 31. | Naka, T.; Minakawa, N.; Abe, H.; Kaga, D.; Matsuda, A. J. Am. Chem. Soc. 2000, 122, 7233–7243. doi:10.1021/ja000541o |
| 4. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 5. | Schaefer, L.; Tredup, C.; Gubbiotti, M. A.; Iozzo, R. V. FEBS J. 2017, 284, 10–26. doi:10.1111/febs.13963 |
| 6. | Maishi, N.; Hida, K. Cancer Sci. 2017, 108, 1921–1926. doi:10.1111/cas.13336 |
| 7. | Sanderson, R. D.; Elkin, M.; Rapraeger, A. C.; Ilan, N.; Vlodavsky, I. FEBS J. 2017, 284, 42–55. doi:10.1111/febs.13932 |
| 15. | Niedballa, U.; Vorbrueggen, H. J. Org. Chem. 1974, 39, 3654–3660. doi:10.1021/jo00939a008 |
| 16. | Vorbrüggen, H.; Ruh-Pohlenz, C. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or055.01 |
| 75. | Galonić, D. P.; Gin, D. Y. Nature 2007, 446, 1000–1007. doi:10.1038/nature05813 |
| 76. | Buskas, T.; Thompson, P.; Boons, G. J. Chem. Commun. 2009, 5335–5349. doi:10.1039/b908664c |
| 77. | Fernández-Tejada, A.; Cañada, F. J.; Jiménez-Barbero, J. Chem. – Eur. J. 2015, 21, 10616–10628. doi:10.1002/chem.201500831 |
| 2. | Nyhan, W. L. Mol. Genet. Metab. 2005, 86, 25–33. doi:10.1016/j.ymgme.2005.07.027 |
| 3. | Muntoni, F.; Brockington, M.; Blake, D. J.; Torelli, S.; Brown, S. C. Lancet 2002, 360, 1419–1421. doi:10.1016/s0140-6736(02)11397-3 |
| 29. | Kita, Y.; Yasuda, H.; Tamura, O.; Itoh, F.; Tamura, Y. Tetrahedron Lett. 1984, 25, 4681–4682. doi:10.1016/s0040-4039(01)91232-8 |
| 30. | Kita, Y.; Tamura, O.; Yasuda, H.; Itoh, F.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 4235–4241. doi:10.1248/cpb.33.4235 |
| 18. | Hanessian, S.; Lou, B. Chem. Rev. 2000, 100, 4443–4464. doi:10.1021/cr9903454 |
| 19. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 23. | Matsuda, A.; Takenuki, K.; Tanaka, M.; Sasaki, T.; Ueda, T. J. Med. Chem. 1991, 34, 812–819. doi:10.1021/jm00106a049 |
| 24. | Yamagami, K.; Fujii, A.; Arita, M.; Okumoto, T.; Sakata, S.; Matsuda, A.; Ueda, T.; Sasaki, T. Cancer Res. 1991, 51, 2319–2323. |
| 26. | Secrist III, J. A.; Tiwari, K. N.; Riordan, J. M.; Montgomery, J. A. J. Med. Chem. 1991, 34, 2361–2366. doi:10.1021/jm00112a007 |
| 74. | Boto, A.; Hernández, D.; Hernández, R.; Álvarez, E. J. Org. Chem. 2007, 72, 9523–9532. doi:10.1021/jo701608p |
| 17. | Yoshimura, Y. Heterocycles 2017, 94, 1625–1651. doi:10.3987/REV-17-865 |
| 20. | Yoshimura, Y.; Kobayashi, S.; Kaneko, H.; Suzuki, T.; Imamichi, T. Molecules 2015, 20, 4623–4634. doi:10.3390/molecules20034623 |
| 21. | Kiran, Y. B.; Wakamatsu, H.; Natori, Y.; Takahata, H.; Yoshimura, Y. Tetrahedron Lett. 2013, 54, 3949–3952. doi:10.1016/j.tetlet.2013.05.062 |
| 22. | Yoshimura, Y.; Asami, K.; Imamichi, T.; Okuda, T.; Shiraki, K.; Takahata, H. J. Org. Chem. 2010, 75, 4161–4171. doi:10.1021/jo100556u |
| 27. | Yoshimura, Y.; Kitano, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1996, 61, 822–823. doi:10.1021/JO9519423 |
| 28. | Yoshimura, Y.; Kitano, K.; Yamada, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. J. Org. Chem. 1997, 62, 3140–3152. doi:10.1021/JO9700540 |
| 74. | Boto, A.; Hernández, D.; Hernández, R.; Álvarez, E. J. Org. Chem. 2007, 72, 9523–9532. doi:10.1021/jo701608p |
| 71. | Havare, N.; Plattner, D. A. Org. Lett. 2012, 14, 5078–5081. doi:10.1021/ol301675v |
| 18. | Hanessian, S.; Lou, B. Chem. Rev. 2000, 100, 4443–4464. doi:10.1021/cr9903454 |
| 19. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 25. | Dyson, M. R.; Coe, P. L.; Walker, R. T. J. Med. Chem. 1991, 34, 2782–2786. doi:10.1021/jm00113a016 |
| 73. | Francisco, C. G.; Freire, R.; González, C. C.; León, E. I.; Riesco-Fagundo, C.; Suárez, E. J. Org. Chem. 2001, 66, 1861–1866. doi:10.1021/jo0057452 |
| 15. | Niedballa, U.; Vorbrueggen, H. J. Org. Chem. 1974, 39, 3654–3660. doi:10.1021/jo00939a008 |
| 16. | Vorbrüggen, H.; Ruh-Pohlenz, C. Organic Reactions; John Wiley & Sons, Inc., 2004. doi:10.1002/0471264180.or055.01 |
| 32. | Yoshimura, Y.; Kuze, T.; Ueno, M.; Komiya, F.; Haraguchi, K.; Tanaka, H.; Kano, F.; Yamada, K.; Asami, K.; Kaneko, N.; Takahata, H. Tetrahedron Lett. 2006, 47, 591–594. doi:10.1016/j.tetlet.2005.11.049 |
| 25. | Dyson, M. R.; Coe, P. L.; Walker, R. T. J. Med. Chem. 1991, 34, 2782–2786. doi:10.1021/jm00113a016 |
| 26. | Secrist III, J. A.; Tiwari, K. N.; Riordan, J. M.; Montgomery, J. A. J. Med. Chem. 1991, 34, 2361–2366. doi:10.1021/jm00112a007 |
| 81. | Kajimoto, T.; Morimoto, K.; Ogawa, R.; Dohi, T.; Kita, Y. Chem. Pharm. Bull. 2016, 64, 838–844. doi:10.1248/cpb.c16-00203 |
| 82. | Randolph, J.; Danishefsky, S. J. J. Am. Chem. Soc. 1993, 115, 8473–8474. doi:10.1021/ja00071a077 |
| 79. | Fukase, K.; Kinoshita, I.; Kanoh, T.; Nakai, Y.; Hasuoka, A.; Kusumoto, S. Tetrahedron 1996, 52, 3897–3904. doi:10.1016/S0040-4020(96)00057-9 |
| 80. | Kajimoto, T.; Morimoto, K.; Ogawa, R.; Dohi, T.; Kita, Y. Eur. J. Org. Chem. 2015, 2015, 2138–2142. doi:10.1002/ejoc.201500186 |
| 81. | Kajimoto, T.; Morimoto, K.; Ogawa, R.; Dohi, T.; Kita, Y. Chem. Pharm. Bull. 2016, 64, 838–844. doi:10.1248/cpb.c16-00203 |
| 45. | Nishizono, N.; Baba, R.; Nakamura, C.; Oda, K.; Machida, M. Org. Biomol. Chem. 2003, 1, 3692–3697. doi:10.1039/B305644A |
| 46. | Nishizono, N.; Soma, K.; Baba, R.; Machida, M.; Oda, K. Heterocycles 2008, 75, 619–634. doi:10.3987/COM-07-11250 |
| 85. | Islam, M.; Tirukoti, N. D.; Nandi, S.; Hotha, S. J. Org. Chem. 2014, 79, 4470–4476. doi:10.1021/jo500465m |
| 45. | Nishizono, N.; Baba, R.; Nakamura, C.; Oda, K.; Machida, M. Org. Biomol. Chem. 2003, 1, 3692–3697. doi:10.1039/B305644A |
| 38. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 84. | Shi, L.; Kim, Y.-J.; Gin, D. Y. J. Am. Chem. Soc. 2001, 123, 6939–6940. doi:10.1021/ja015991a |
| 39. | Dohi, T.; Morimoto, K.; Maruyama, A.; Kita, Y. Org. Lett. 2006, 8, 2007–2010. doi:10.1021/ol060333m |
| 40. | Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668–1669. doi:10.1021/ja808940n |
| 41. | Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Org. Lett. 2010, 12, 3804–3807. doi:10.1021/ol101498r |
| 42. | Dohi, T.; Ito, M.; Itani, I.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Org. Lett. 2011, 13, 6208–6211. doi:10.1021/ol202632h |
| 43. | Ito, M.; Kubo, H.; Itani, I.; Morimoto, K.; Dohi, T.; Kita, Y. J. Am. Chem. Soc. 2013, 135, 14078–14081. doi:10.1021/ja407944p |
| 85. | Islam, M.; Tirukoti, N. D.; Nandi, S.; Hotha, S. J. Org. Chem. 2014, 79, 4470–4476. doi:10.1021/jo500465m |
| 29. | Kita, Y.; Yasuda, H.; Tamura, O.; Itoh, F.; Tamura, Y. Tetrahedron Lett. 1984, 25, 4681–4682. doi:10.1016/s0040-4039(01)91232-8 |
| 30. | Kita, Y.; Tamura, O.; Yasuda, H.; Itoh, F.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 4235–4241. doi:10.1248/cpb.33.4235 |
| 83. | Chu, A.-H. A.; Minciunescu, A.; Bennett, C. S. Org. Lett. 2015, 17, 6262–6265. doi:10.1021/acs.orglett.5b03282 |
| 33. | Jeong, L. S.; Moon, H. R.; Choi, Y. J.; Chun, M. W.; Kim, H. O. J. Org. Chem. 1998, 63, 4821–4825. doi:10.1021/JO980196+ |
| 34. | Lim, M. H.; Kim, H. O.; Moon, H. R.; Chun, M. W.; Jeong, L. S. Org. Lett. 2002, 4, 529–531. doi:10.1021/ol017112v |
| 35. | Jeong, L. S.; Lee, H. W.; Kim, H. O.; Jung, J. Y.; Gao, Z.-G.; Duong, H. T.; Rao, S.; Jacobson, K. A.; Shin, D. H.; Lee, J. A.; Gunaga, P.; Lee, S. K.; Jin, D. Z.; Chun, M. W.; Moon, H. R. Bioorg. Med. Chem. 2006, 14, 4718–4730. doi:10.1016/j.bmc.2006.03.030 |
| 36. | Choi, W. J.; Lee, H. W.; Kim, H. O.; Chinn, M.; Gao, Z.-G.; Patel, A.; Jacobson, K. A.; Moon, H. R.; Jung, Y. H.; Jeong, L. S. Bioorg. Med. Chem. 2009, 17, 8003–8011. doi:10.1016/j.bmc.2009.10.011 |
| 37. | Qu, S.; Mulamoottil, V. A.; Nayak, A.; Ryu, S.; Hou, X.; Song, J.; Yu, J.; Sahu, P. K.; Zhao, L. X.; Choi, S.; Lee, S. K.; Jeong, L. S. Bioorg. Med. Chem. 2016, 24, 3418–3428. doi:10.1016/j.bmc.2016.05.041 |
| 84. | Shi, L.; Kim, Y.-J.; Gin, D. Y. J. Am. Chem. Soc. 2001, 123, 6939–6940. doi:10.1021/ja015991a |
© 2018 Yoshimura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)