Abstract
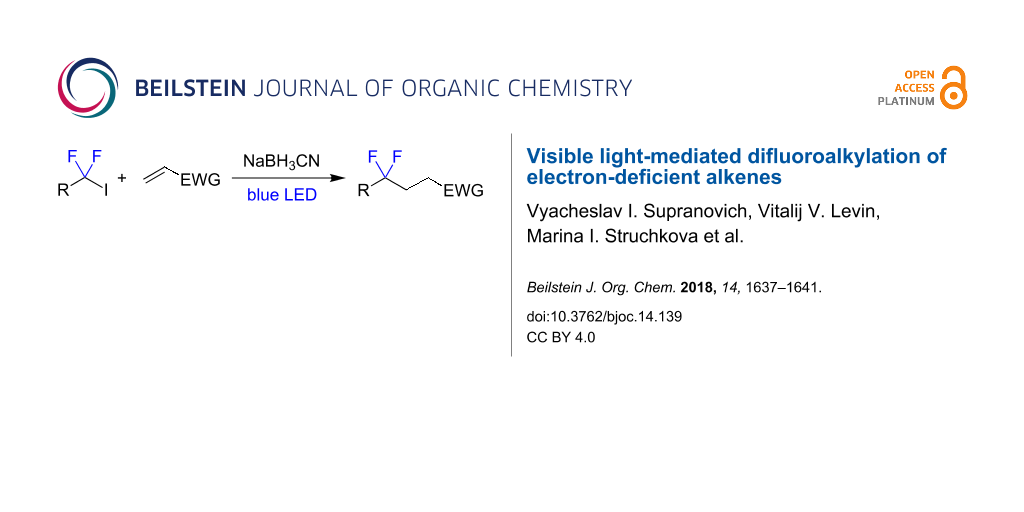
A method for the reductive difluoroalkylation of electron-deficient alkenes using 1,1-difluorinated iodides mediated by irradiation with blue light is described. The reaction involves radical addition of 1,1-difluorinated radicals at the double bond followed by hydrogen atom transfer from sodium cyanoborohydride.

Graphical Abstract
Introduction
Applications of organofluorine compounds in medicinal chemistry and related fields [1,2] have stimulated intensive developments of methods for their synthesis [3,4]. Though the major emphasis has long been placed on trifluoromethylated molecules, compounds bearing the difluoromethylene fragment have attracted increasing attention in recent years [5-8]. Thus, besides the bioisosteric relationship with ethereal oxygen [9-11], the CF2-unit can influence the reactivity of adjacent functional groups [12,13] and can affect conformational properties of chain and cyclic molecules [14,15].
Radical fluoroalkylation of double bonds is a well-established approach for the synthesis of organofluorine compounds [16-24]. While perfluorinated alkyl iodides and bromides are typically employed, reactions with gem-difluorinated iodides are less elaborated, which is primarily associated with availability issues [25-29]. Recently, we developed a general protocol for the synthesis of iodides 1 from organozinc reagents and a source of difluorocarbene [30-32] (Scheme 1). Moreover, it was shown that compounds bearing the CF2I group can be obtained from carbonyl compounds and equivalents of the iododifluoromethyl carbanion [33-36]. We also demonstrated that iodides 1 can alkylate silyl enol ethers [37] under photoredox conditions [38-40]. However, the latter protocol is inapplicable to the addition to electron-deficient alkenes since a radical resulting from the addition step cannot be oxidized by photocatalysts. Herein we report a convenient method for performing hydroperfluoroalkylation of electron-deficient alkenes employing iodides 1 mediated by visible light. The reaction proceeds without the use of a photosensitizer or a photocatalyst.
![[1860-5397-14-139-i1]](/bjoc/content/inline/1860-5397-14-139-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Generation of fluorinated alkyl radicals is typically realized either by single-electron reduction or by radical abstraction of iodine [41-43]. Furthermore, the carbon–iodine bond is prone to homolytic cleavage under UV irradiation [44]. In this regard, we were inspired by the work of Ryu describing the use of energetic UV light (xenon arc lamp) in combination with sodium cyanoborohydride to trigger the reaction of non-fluorinated alkyl iodides [45-47]. Subsequently, we showed that silyldifluoromethyl iodide can generate the corresponding gem-difluorinated radical in combination with an N-heterocyclic carbene/borane complex and 400 nm light [48].
Results and Discussion
Iodide 1a was selected as a model substrate and its reaction with tert-butyl acrylate (2a) was evaluated in the presence of various boron hydrides (Table 1). The reaction was performed in methanol with irradiation by a strip of blue light emitting diodes. The use of sodium cyanoborohydride provided a moderate yield of product 3a and about 60% conversion of 1a (Table 1, entry 1). The addition of pyridine notably increased the yield, presumably, by scavenging boron species with a B–I bond formed in the reaction (Table 1, entry 2). However, some byproducts were observed in amounts of up to 10%, which were tentatively identified as oligomers arising from multiple addition of tert-butyl acrylate to the propagating radical. Fortunately, increase of the amount of borohydride to 2 equiv suppressed the formation of oligomers, and addition product 3a was isolated in 83% yield (Table 1, entry 6). N,N'-Dimethylimidazolidene borane complex (NHC·BH3) was also efficient (Table 1, entry 5), but because of availability and cost issues, sodium cyanoborohydride was used for further studies. We also evaluated the photoredox-mediated hydrofluoroalkylation process using fac-Ir(ppy)3 as a photocatalyst in combination with a suitable donor of hydrogen atom. With triethylamine, no expected product was observed. At the same time, with Hantzsch ester (3 equiv), which can serve as a single-electron reductant and as a source of hydrogen [16], 54% of product 3a was formed. However, difficulties in removing the pyridine byproduct formed from Hantzsch ester and the use of a precious metal photocatalyst make this protocol less practical compared to that with sodium cyanoborohydride.
Table 1: Optimization studies.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-139-i3.svg?max-width=637&scale=1.0)
|
|||
| entry | hydride (equiv) | base (equiv) | yield, %a |
|---|---|---|---|
| 1 | NaBH3CN (1.5) | – | 46 |
| 2 | NaBH3CN (1.5) | pyridine (1.5) | 77b |
| 3 | Py-BH3 (1.5) | pyridine (1.5) | 78b |
| 4 | Py-BH3 (2.0) | pyridine (2.0) | 80b |
| 5 | NHC·BH3 (1.5) | pyridine (1.5) | 76c |
| 6 | NaBH3CN (2.0) | pyridine (1.5) | 83c |
| 7 | NaBH3CN (2.0) | NaOAc (1.5) | 74b |
aDetermined by 19F NMR with internal standard. bOligomeric byproducts (5–11%) were observed. cIsolated yield.
Under the optimized conditions a series of gem-difluorinated iodides 1 were coupled with alkenes 2 (Figure 1). Esters and amides of acrylic acid, acrylonitrile, and vinyl phosphonate used in a small excess (1.2 equiv) were successfully fluoroalkylated. For vinyl(phenyl)sulfone, significant amounts of hydrodeiodination product RfH was formed, likely because of the decreased reactivity of the sulfone double bond toward fluorinated radicals. In this case, two equivalents of the alkene have to be used to achieve good yields of products 3t,u. It should also be pointed out that the reaction tolerates aromatic bromide substituents (products 3b,c,n,o), a boryl fragment (product 3d) and unprotected hydroxy groups (products 3i,j,m). Electron-rich and neutral alkenes were ineffective in this reaction. Indeed, 1-phenyl-(1-trimethylsilyloxy)ethylene and 4-phenylbut-1-ene gave no hydrofluoroalkylation products when combined with iodide 1a under standard conditions.
![[1860-5397-14-139-1]](/bjoc/content/figures/1860-5397-14-139-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Difluoroalkylation of alkenes. Isolated yields are shown. a2 equiv of the alkene were used.
Figure 1: Difluoroalkylation of alkenes. Isolated yields are shown. a2 equiv of the alkene were used.
The proposed mechanism is shown in Scheme 2. First, difluorinated iodide 1 interacts with boron hydride to form a small equilibrium concentration of a halogen-bonded complex [49,50]. This complex is activated by light to effect homolytic cleavage of the carbon–iodine bond with the formation of radical 4 and boron-centered radical anion 5. After the initiation event, the reaction proceeds via a chain mechanism. Thus, radical addition at the double bond gives radical 6, which abstracts a hydrogen atom from cyanoborohydride to generate boryl radical anion 5. The latter species can readily abstract the iodine atom from starting iodide 1 which results in the formation of radical 4 [51,52].
![[1860-5397-14-139-i2]](/bjoc/content/inline/1860-5397-14-139-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, a convenient method for the synthesis of gem-difluorinated compounds via hydrofluoroalkylation of electron-deficient alkenes is described. Readily available boron cyanohydride is used as a source of hydrogen and as a trigger for the generation of free radicals mediated by visible light.
Supporting Information
| Supporting Information File 1: Full experimental details, compound characterization, and copies of NMR spectra. | ||
| Format: PDF | Size: 2.4 MB | Download |
References
-
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Groult, H.; Leroux, F. R.; Tressaud, A., Eds. Modern Synthesis Processes and Reactivity of Fluorinated Compounds; Elsevier: Amsterdam, 2017.
Return to citation in text: [1] -
Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem. – Eur. J. 2015, 21, 12836–12865. doi:10.1002/chem.201501475
Return to citation in text: [1] -
Krishnamoorthy, S.; Prakash, G. K. S. Synthesis 2017, 49, 3394–3406. doi:10.1055/s-0036-1588489
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V. Mendeleev Commun. 2015, 25, 239–244. doi:10.1016/j.mencom.2015.07.001
Return to citation in text: [1] -
Gao, B.; Ni, C.; Hu, J. Chimia 2014, 68, 414–418. doi:10.2533/chimia.2014.414
Return to citation in text: [1] -
Blackburn, G. M.; England, D. A.; Kolkmann, F. J. Chem. Soc., Chem. Commun. 1981, 930–932. doi:10.1039/c39810000930
Return to citation in text: [1] -
Xu, Y.; Aoki, J.; Shimizu, K.; Umezu-Goto, M.; Hama, K.; Takanezawa, Y.; Yu, S.; Mills, G. B.; Arai, H.; Qian, L.; Prestwich, G. D. J. Med. Chem. 2005, 48, 3319–3327. doi:10.1021/jm049186t
Return to citation in text: [1] -
Ivanova, M. V.; Bayle, A.; Besset, T.; Pannecoucke, X.; Poisson, T. Chem. – Eur. J. 2016, 22, 10284–10293. doi:10.1002/chem.201601310
Return to citation in text: [1] -
Baskin, J. M.; Prescher, J. A.; Laughlin, S. T.; Agard, N. J.; Chang, P. V.; Miller, I. A.; Lo, A.; Codelli, J. A.; Bertozzi, C. R. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16793–16797. doi:10.1073/pnas.0707090104
Return to citation in text: [1] -
Codelli, J. A.; Baskin, J. M.; Agard, N. J.; Bertozzi, C. R. J. Am. Chem. Soc. 2008, 130, 11486–11493. doi:10.1021/ja803086r
Return to citation in text: [1] -
O'Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26
Return to citation in text: [1] -
Wang, Y.; Callejo, R.; Slawin, A. M. Z.; O'Hagan, D. Beilstein J. Org. Chem. 2014, 10, 18–25. doi:10.3762/bjoc.10.4
Return to citation in text: [1] -
Sumino, S.; Uno, M.; Fukuyama, T.; Ryu, I.; Matsuura, M.; Yamamoto, A.; Kishikawa, Y. J. Org. Chem. 2017, 82, 5469–5474. doi:10.1021/acs.joc.7b00609
Return to citation in text: [1] [2] -
Zhu, L.; Wang, L.-S.; Li, B.; Fu, B.; Zhang, C.-P.; Li, W. Chem. Commun. 2016, 52, 6371–6374. doi:10.1039/C6CC01944G
Return to citation in text: [1] -
Zhou, S.-H.; Lin, J.-H.; Zhao, G.; Xiao, J.-C.; Cao, W.-G. RSC Adv. 2016, 6, 60080–60083. doi:10.1039/C6RA11557H
Return to citation in text: [1] -
Lin, Q.-Y.; Xu, X.-H.; Qing, F.-L. Org. Biomol. Chem. 2015, 13, 8740–8749. doi:10.1039/C5OB01302J
Return to citation in text: [1] -
Mizuta, S.; Verhoog, S.; Engle, K. M.; Khotavivattana, T.; O'Duill, M.; Wheelhouse, K.; Rassias, G.; Médebielle, M.; Gouverneur, V. J. Am. Chem. Soc. 2013, 135, 2505–2508. doi:10.1021/ja401022x
Return to citation in text: [1] -
Wu, X.; Chu, L.; Qing, F.-L. Angew. Chem., Int. Ed. 2013, 52, 2198–2202. doi:10.1002/anie.201208971
Return to citation in text: [1] -
Wilger, D. J.; Gesmundo, N. J.; Nicewicz, D. A. Chem. Sci. 2013, 4, 3160–3165. doi:10.1039/c3sc51209f
Return to citation in text: [1] -
Erdbrink, H.; Peuser, I.; Gerling, U. I. M.; Lentz, D.; Koksch, B.; Czekelius, C. Org. Biomol. Chem. 2012, 10, 8583–8586. doi:10.1039/c2ob26810h
Return to citation in text: [1] -
Beier, P.; Voltrová, S. Fluorine Notes 2017, 115, 3–4. doi:10.17677/fn20714807.2017.06.02
Return to citation in text: [1] -
Yang, Z.-Y. J. Org. Chem. 2004, 69, 2394–2403. doi:10.1021/jo0354488
Return to citation in text: [1] -
Li, A.-R.; Chen, Q.-Y. Synthesis 1997, 1481–1488. doi:10.1055/s-1997-1378
Return to citation in text: [1] -
Li, A.-R.; Chen, Q.-Y. J. Fluorine Chem. 1997, 82, 151–155. doi:10.1016/S0022-1139(96)03545-2
Return to citation in text: [1] -
Li, A.-R.; Chen, Q.-Y. J. Fluorine Chem. 1997, 81, 99–101. doi:10.1016/S0022-1139(96)03510-5
Return to citation in text: [1] -
Mikami, K.; Tomita, Y.; Itoh, Y. Angew. Chem., Int. Ed. 2010, 49, 3819–3822. doi:10.1002/anie.201000435
Return to citation in text: [1] -
Levin, V. V.; Zemtsov, A. A.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2013, 15, 917–919. doi:10.1021/ol400122k
Return to citation in text: [1] -
Levin, V. V.; Zemtsov, A. A.; Struchkova, M. I.; Dilman, A. D. J. Fluorine Chem. 2015, 171, 97–101. doi:10.1016/j.jfluchem.2014.08.021
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V. Acc. Chem. Res. 2018, 51, 1272–1280. doi:10.1021/acs.accounts.8b00079
Return to citation in text: [1] -
Zhao, Y.; Gao, B.; Hu, J. J. Am. Chem. Soc. 2012, 134, 5790–5793. doi:10.1021/ja301601b
Return to citation in text: [1] -
Miao, W.; Ni, C.; Zhao, Y.; Hu, J. Org. Lett. 2016, 18, 2766–2769. doi:10.1021/acs.orglett.6b01258
Return to citation in text: [1] -
Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2014, 16, 3784–3787. doi:10.1021/ol501674n
Return to citation in text: [1] -
Levin, V. V.; Smirnov, V. O.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 9349–9353. doi:10.1021/acs.joc.5b01590
Return to citation in text: [1] -
Chernov, G. N.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. Adv. Synth. Catal. 2017, 359, 3063–3067. doi:10.1002/adsc.201700423
Return to citation in text: [1] -
Panferova, L. I.; Tsymbal, A. V.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2016, 18, 996–999. doi:10.1021/acs.orglett.6b00117
Return to citation in text: [1] -
Scherbinina, S. I.; Fedorov, O. V.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 12967–12974. doi:10.1021/acs.joc.7b02467
Return to citation in text: [1] -
Panferova, L. I.; Smirnov, V. O.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 745–753. doi:10.1021/acs.joc.6b02344
Return to citation in text: [1] -
Chatterjee, T.; Iqbal, N.; You, Y.; Cho, E. J. Acc. Chem. Res. 2016, 49, 2284–2294. doi:10.1021/acs.accounts.6b00248
Return to citation in text: [1] -
Koike, T.; Akita, M. Top. Catal. 2014, 57, 967–974. doi:10.1007/s11244-014-0259-7
Return to citation in text: [1] -
Baguley, P. A.; Walton, J. C. Angew. Chem., Int. Ed. 1998, 37, 3072–3082. doi:10.1002/(SICI)1521-3773(19981204)37:22<3072::AID-ANIE3072>3.0.CO;2-9
Return to citation in text: [1] -
Uma, S.; Das, P. K. Can. J. Chem. 1994, 72, 865–869. doi:10.1139/v94-113
Return to citation in text: [1] -
Ryu, I.; Uehara, S.; Hirao, H.; Fukuyama, T. Org. Lett. 2008, 10, 1005–1008. doi:10.1021/ol7031043
Return to citation in text: [1] -
Kawamoto, T.; Uehara, S.; Hirao, H.; Fukuyama, T.; Matsubara, H.; Ryu, I. J. Org. Chem. 2014, 79, 3999–4007. doi:10.1021/jo500464q
Return to citation in text: [1] -
Kawamoto, T.; Ryu, I. Org. Biomol. Chem. 2014, 12, 9733–9742. doi:10.1039/C4OB01784F
Return to citation in text: [1] -
Supranovich, V. I.; Levin, V. V.; Struchkova, M. I.; Korlyukov, A. A.; Dilman, A. D. Org. Lett. 2017, 19, 3215–3218. doi:10.1021/acs.orglett.7b01334
Return to citation in text: [1] -
Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484
Return to citation in text: [1] -
Beale, T. M.; Chudzinski, M. G.; Sarwar, M. G.; Taylor, M. S. Chem. Soc. Rev. 2013, 42, 1667–1680. doi:10.1039/C2CS35213C
Return to citation in text: [1] -
Kawamoto, T.; Fukuyama, T.; Ryu, I. J. Am. Chem. Soc. 2012, 134, 875–877. doi:10.1021/ja210585n
Return to citation in text: [1] -
Sheeller, B.; Ingold, K. U. J. Chem. Soc., Perkin Trans. 2 2001, 480–486. doi:10.1039/b009494n
Return to citation in text: [1]
| 51. | Kawamoto, T.; Fukuyama, T.; Ryu, I. J. Am. Chem. Soc. 2012, 134, 875–877. doi:10.1021/ja210585n |
| 52. | Sheeller, B.; Ingold, K. U. J. Chem. Soc., Perkin Trans. 2 2001, 480–486. doi:10.1039/b009494n |
| 16. | Sumino, S.; Uno, M.; Fukuyama, T.; Ryu, I.; Matsuura, M.; Yamamoto, A.; Kishikawa, Y. J. Org. Chem. 2017, 82, 5469–5474. doi:10.1021/acs.joc.7b00609 |
| 49. | Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484 |
| 50. | Beale, T. M.; Chudzinski, M. G.; Sarwar, M. G.; Taylor, M. S. Chem. Soc. Rev. 2013, 42, 1667–1680. doi:10.1039/C2CS35213C |
| 1. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 2. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C |
| 12. | Baskin, J. M.; Prescher, J. A.; Laughlin, S. T.; Agard, N. J.; Chang, P. V.; Miller, I. A.; Lo, A.; Codelli, J. A.; Bertozzi, C. R. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16793–16797. doi:10.1073/pnas.0707090104 |
| 13. | Codelli, J. A.; Baskin, J. M.; Agard, N. J.; Bertozzi, C. R. J. Am. Chem. Soc. 2008, 130, 11486–11493. doi:10.1021/ja803086r |
| 45. | Ryu, I.; Uehara, S.; Hirao, H.; Fukuyama, T. Org. Lett. 2008, 10, 1005–1008. doi:10.1021/ol7031043 |
| 46. | Kawamoto, T.; Uehara, S.; Hirao, H.; Fukuyama, T.; Matsubara, H.; Ryu, I. J. Org. Chem. 2014, 79, 3999–4007. doi:10.1021/jo500464q |
| 47. | Kawamoto, T.; Ryu, I. Org. Biomol. Chem. 2014, 12, 9733–9742. doi:10.1039/C4OB01784F |
| 9. | Blackburn, G. M.; England, D. A.; Kolkmann, F. J. Chem. Soc., Chem. Commun. 1981, 930–932. doi:10.1039/c39810000930 |
| 10. | Xu, Y.; Aoki, J.; Shimizu, K.; Umezu-Goto, M.; Hama, K.; Takanezawa, Y.; Yu, S.; Mills, G. B.; Arai, H.; Qian, L.; Prestwich, G. D. J. Med. Chem. 2005, 48, 3319–3327. doi:10.1021/jm049186t |
| 11. | Ivanova, M. V.; Bayle, A.; Besset, T.; Pannecoucke, X.; Poisson, T. Chem. – Eur. J. 2016, 22, 10284–10293. doi:10.1002/chem.201601310 |
| 48. | Supranovich, V. I.; Levin, V. V.; Struchkova, M. I.; Korlyukov, A. A.; Dilman, A. D. Org. Lett. 2017, 19, 3215–3218. doi:10.1021/acs.orglett.7b01334 |
| 5. | Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem. – Eur. J. 2015, 21, 12836–12865. doi:10.1002/chem.201501475 |
| 6. | Krishnamoorthy, S.; Prakash, G. K. S. Synthesis 2017, 49, 3394–3406. doi:10.1055/s-0036-1588489 |
| 7. | Dilman, A. D.; Levin, V. V. Mendeleev Commun. 2015, 25, 239–244. doi:10.1016/j.mencom.2015.07.001 |
| 8. | Gao, B.; Ni, C.; Hu, J. Chimia 2014, 68, 414–418. doi:10.2533/chimia.2014.414 |
| 41. | Chatterjee, T.; Iqbal, N.; You, Y.; Cho, E. J. Acc. Chem. Res. 2016, 49, 2284–2294. doi:10.1021/acs.accounts.6b00248 |
| 42. | Koike, T.; Akita, M. Top. Catal. 2014, 57, 967–974. doi:10.1007/s11244-014-0259-7 |
| 43. | Baguley, P. A.; Walton, J. C. Angew. Chem., Int. Ed. 1998, 37, 3072–3082. doi:10.1002/(SICI)1521-3773(19981204)37:22<3072::AID-ANIE3072>3.0.CO;2-9 |
| 3. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 4. | Groult, H.; Leroux, F. R.; Tressaud, A., Eds. Modern Synthesis Processes and Reactivity of Fluorinated Compounds; Elsevier: Amsterdam, 2017. |
| 30. | Levin, V. V.; Zemtsov, A. A.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2013, 15, 917–919. doi:10.1021/ol400122k |
| 31. | Levin, V. V.; Zemtsov, A. A.; Struchkova, M. I.; Dilman, A. D. J. Fluorine Chem. 2015, 171, 97–101. doi:10.1016/j.jfluchem.2014.08.021 |
| 32. | Dilman, A. D.; Levin, V. V. Acc. Chem. Res. 2018, 51, 1272–1280. doi:10.1021/acs.accounts.8b00079 |
| 37. | Chernov, G. N.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. Adv. Synth. Catal. 2017, 359, 3063–3067. doi:10.1002/adsc.201700423 |
| 25. | Yang, Z.-Y. J. Org. Chem. 2004, 69, 2394–2403. doi:10.1021/jo0354488 |
| 26. | Li, A.-R.; Chen, Q.-Y. Synthesis 1997, 1481–1488. doi:10.1055/s-1997-1378 |
| 27. | Li, A.-R.; Chen, Q.-Y. J. Fluorine Chem. 1997, 82, 151–155. doi:10.1016/S0022-1139(96)03545-2 |
| 28. | Li, A.-R.; Chen, Q.-Y. J. Fluorine Chem. 1997, 81, 99–101. doi:10.1016/S0022-1139(96)03510-5 |
| 29. | Mikami, K.; Tomita, Y.; Itoh, Y. Angew. Chem., Int. Ed. 2010, 49, 3819–3822. doi:10.1002/anie.201000435 |
| 38. | Panferova, L. I.; Tsymbal, A. V.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2016, 18, 996–999. doi:10.1021/acs.orglett.6b00117 |
| 39. | Scherbinina, S. I.; Fedorov, O. V.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 12967–12974. doi:10.1021/acs.joc.7b02467 |
| 40. | Panferova, L. I.; Smirnov, V. O.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 745–753. doi:10.1021/acs.joc.6b02344 |
| 16. | Sumino, S.; Uno, M.; Fukuyama, T.; Ryu, I.; Matsuura, M.; Yamamoto, A.; Kishikawa, Y. J. Org. Chem. 2017, 82, 5469–5474. doi:10.1021/acs.joc.7b00609 |
| 17. | Zhu, L.; Wang, L.-S.; Li, B.; Fu, B.; Zhang, C.-P.; Li, W. Chem. Commun. 2016, 52, 6371–6374. doi:10.1039/C6CC01944G |
| 18. | Zhou, S.-H.; Lin, J.-H.; Zhao, G.; Xiao, J.-C.; Cao, W.-G. RSC Adv. 2016, 6, 60080–60083. doi:10.1039/C6RA11557H |
| 19. | Lin, Q.-Y.; Xu, X.-H.; Qing, F.-L. Org. Biomol. Chem. 2015, 13, 8740–8749. doi:10.1039/C5OB01302J |
| 20. | Mizuta, S.; Verhoog, S.; Engle, K. M.; Khotavivattana, T.; O'Duill, M.; Wheelhouse, K.; Rassias, G.; Médebielle, M.; Gouverneur, V. J. Am. Chem. Soc. 2013, 135, 2505–2508. doi:10.1021/ja401022x |
| 21. | Wu, X.; Chu, L.; Qing, F.-L. Angew. Chem., Int. Ed. 2013, 52, 2198–2202. doi:10.1002/anie.201208971 |
| 22. | Wilger, D. J.; Gesmundo, N. J.; Nicewicz, D. A. Chem. Sci. 2013, 4, 3160–3165. doi:10.1039/c3sc51209f |
| 23. | Erdbrink, H.; Peuser, I.; Gerling, U. I. M.; Lentz, D.; Koksch, B.; Czekelius, C. Org. Biomol. Chem. 2012, 10, 8583–8586. doi:10.1039/c2ob26810h |
| 24. | Beier, P.; Voltrová, S. Fluorine Notes 2017, 115, 3–4. doi:10.17677/fn20714807.2017.06.02 |
| 14. | O'Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26 |
| 15. | Wang, Y.; Callejo, R.; Slawin, A. M. Z.; O'Hagan, D. Beilstein J. Org. Chem. 2014, 10, 18–25. doi:10.3762/bjoc.10.4 |
| 33. | Zhao, Y.; Gao, B.; Hu, J. J. Am. Chem. Soc. 2012, 134, 5790–5793. doi:10.1021/ja301601b |
| 34. | Miao, W.; Ni, C.; Zhao, Y.; Hu, J. Org. Lett. 2016, 18, 2766–2769. doi:10.1021/acs.orglett.6b01258 |
| 35. | Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2014, 16, 3784–3787. doi:10.1021/ol501674n |
| 36. | Levin, V. V.; Smirnov, V. O.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 9349–9353. doi:10.1021/acs.joc.5b01590 |
© 2018 Supranovich et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)