Abstract
Background: 4-(n-Octyloxy)aniline is a known component in the elaboration of organic materials with mesogenic properties such as N-substituted Schiff bases, perylene bisimide assemblies with a number of 2-amino-4,6-bis[4-(n-octyloxy)phenylamino]-s-triazines, amphiphilic azobenzene-containing linear-dendritic block copolymers and G-0 monomeric or dimeric dendritic liquid crystals with photochromic azobenzene mesogens. The present ab initio study explores a previously unknown use of 4-(n-octyloxy)aniline in the synthesis, structure and supramolecular behaviour of new dendritic melamines.
Results: Starting from 4-(n-octyloxy)aniline, seven G-2 melamine-based dendrimers were obtained in 29–79% overall yields. Their iterative convergent- and chemoselective synthesis consisted of SN2-Ar aminations of cyanuric chloride and final triple N-acylations and Williamson etherifications (→ G-2 covalent trimers) or stoichiometric carboxyl/amino 1:3 neutralisations (→ G-2 ionic trimers). These transformations connected G-1 chloro- and amino-termini dendrons to m-trivalent cores (triazin-2,4,6-triyl and benzene-1,3,5-triyl units) or tripodands (central building blocks), such as N-substituted melamines with 4-hydroxyphenyl or phenyl-4-oxyalkanoic motifs. Owing to the diversity of cores and central building blocks, the structural assortment of the dendritic series was disclosed by solvation effects (affecting reactivity), rotational stereodynamism and self-organisation phenomena (determining a vaulted and/or propeller macromolecular shape in solution). DFT calculations (in solution), (VT) NMR and IR (KBr) spectroscopy supported these assignments. TEM analysis revealed the ability of the title compounds towards self-assembling into homogeneously packed spherical nano-aggregates.
Conclusions: The (non)covalent synthesis and step-by-step structural elucidation of novel G-2 melamine dendrimers based on 4-(n-octyloxy)aniline are reported. Our study demonstrates the crucial influence of the nature (covalent vs ionic) of the dendritic construction in tandem with that of its central building blocks on the aptitude of dendrimers to self-organise in solution and to self-assembly in the solid state.
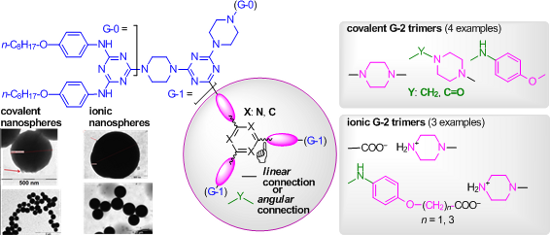
Graphical Abstract
Introduction
N-Substituted melamine (2,4,6-triamino-1,3,5-triazine)-based dendrimers are a class of macromolecules reported as early as 2000 by E. E. Simanek and co-workers [1] and then by K. Takagi and co-workers [2] as part of an innovative development of convergent [1,3,4] and divergent [1,5,6] strategies towards iterative dendritic synthesis. Along with their expansion, both the biological impact of the above arborescent structures, mainly as drug delivery systems [4,7-12], and their utilisation as organic materials have constantly been highlighted [13-17]. In the latter context, dendritic liquid crystals defines a well-established area in the organic materials domain [18], including few examples of s-triazine dendrimers exhibiting mesogenic behaviour. The first cases known so far refer to N-substituted G-0-3 dendritic melamines [19-21] with n-octyl peripheral groups as unconventional columnar liquid crystals and G-0 tris(triazolyl)triazines (available via the “click” reaction between 2,4,6-tris(ethynyl)-s-triazine and various icosanyloxyphenylazides) with liquid crystalline and luminescent properties [22].
On the other hand, mesogenic supramolecular perylene bisimide assemblies with a number of 2-amino-4,6-bis[(4-alkoxy)phenylamino]-s-triazines [23], amphiphilic azobenzene-containing linear-dendritic block copolymers [24] and G-0 monomeric or dimeric dendritic liquid crystals with photochromic azobenzene mesogens [25] called attention on the use of 4-(n-octyloxy)aniline as key building block in the above macromolecules’ elaboration. These recent findings can be seen as well as advances concerning the utilisation of 4-(n-octyloxy)aniline, a “traditional” source for mesogenic N-substituted Schiff bases [26-29].
Following up our contributions in the field of dendritic melamines’ synthesis, structural analysis [30-33] and electrochemistry [34-36], we recently become interested in the inclusion of 4-aminophenol, playing the role of peripheral unit, in G-0-2 dendritic melamines’ preparation, by applying the classic SN2-Ar amination of cyanuric chloride in iterative-convergent strategies. Depending on several factors such as (i) the variable π-deficiency of the s-triazine branch-cells, (ii) basicity and conformational nature of the diaza-six-membered saturated heterocycle as linker and (iii) the global molecular shape, the resulted 4-aminophenol-based melamines displayed relevant redox properties [36] and, in some cases, selective aptitudes to produce MOFs (metal-organic frameworks) [35].
All the above information prompted us towards an ab initio exploration of 4-(n-octyloxy)aniline (seen as the n-octyl ether derivative of 4-aminophenol) as starting material towards new melamine-based dendrimers as synthesis (feasibility and limits), structure and self-assembly propensity. To the best of our knowledge, no similar approach has been reported so far.
Results and Discussion
1 Design
The key elements for the construction and design of the targeted G-2 dendrimers are shown in Figure 1. Cyanuric chloride [1-17], 1,3,5-tris(bromomethyl)benzene [37] and trimesic acid trichloride [38] are commercial chemicals, classically known as reactive m-trivalent (hetero)aryl halo compounds, that are easily convertible into dendritic cores in reactions with various dendrons as N- and O-nucleophiles.
![[1860-5397-14-145-1]](/bjoc/content/figures/1860-5397-14-145-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The key elements for design and construction of the targeted G-2 dendrimers.
Figure 1: The key elements for design and construction of the targeted G-2 dendrimers.
The preparation of the central building block A [1,3,5-tris(piperazinomethyl)benzene] consisted of the amination of 1,3,5-tris(bromomethyl)benzene with commercially available N-Boc-piperazine followed by deprotection, and was achieved according to the literature [39] (97% overall yield in our hands). Similarly, 2,4,6-tris[(4-hydroxy)phenylamino]-s-triazine B, a known starting material for plastics manufacturing [40] and divergent G-1 dendritic synthesis [41], was obtained using 4-aminophenol as amine nucleophile and reacting it with cyanuric chloride by applying a previously patented protocol [40] completed with our own subsequent improvements [36]. With regard to (4-aminophenoxy)alkanoic acid-based tripodal melamines C1 and C3, we recently reported their synthesis in three steps via convergent (starting from N-acetyl-4-aminophenol, also known as paracetamol) and divergent (via A) strategies [42] which are summarised in Scheme 1.
![[1860-5397-14-145-i1]](/bjoc/content/inline/1860-5397-14-145-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Convergent versus divergent three steps (a–c) synthesis of central building blocks C1 and C3.
Scheme 1: Convergent versus divergent three steps (a–c) synthesis of central building blocks C1 and C3.
All central building blocks (A–C) already contain a standard m-trivalent core (benzene-1,3,5-triyl or s-triazin-2,4,6-triyl) connected to 1,4-disubstituted six-membered (hetero)cycle linkers. These linkers were chosen based on their intimate nature: flexible (piperazin-1-yl)methyl (in A), rigid 1,4-phenylene (in B) and rigid 1,4-phenylene with adjustable alkoxy-spacers (n = 1, 3, in C1 and C3, respectively).
G-1 chloro- and piperazine-dendrons, D-Cl and D-N<P>NH, as well as central building blocks B, C1 and C3, had s-triazine rings linked by C(s-triazine)–N(exocyclic) partial double bonds, which exist due to classic LP(N, exocyclic) → π(C=N, s-triazine) delocalisation. A well-documented restricted rotation effect that causes diastereomerism is thus induced [43-45]. In the case of some dendritic melamines, this intrinsic feature can promote specific spatial arrangements at room temperature in solution, for example, asymmetric vs propeller (C3-symmetric) [30,36,45,46], “dendritic choreography” [47], “open-gates ![[Graphic 6]](/bjoc/content/inline/1860-5397-14-145-i10.svg?max-width=637&scale=1.18182) closed-gates” frontier rotamerism [31] and “in out” axial chirality [33].
closed-gates” frontier rotamerism [31] and “in out” axial chirality [33].
In addition, in the targeted G-2 dendrimers (Figure 1), different connections of branches around the core (or central building blocks) were imagined, i.e., linear or angular (covalent vs ionic by carboxyl/amino neutralisation) and were seen as a complementary focus to our work towards structural diversity.
2 Synthesis: feasibilities and failures
2.1 Synthesis of G-1 dendrons
G-1 dendrons D-Cl and D-N<P>NH (Figure 1) were prepared according to the chemistry depicted in Scheme 2. Reaction conditions and quantitative results are listed in Table 1.
![[1860-5397-14-145-i2]](/bjoc/content/inline/1860-5397-14-145-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of G-1 dendrons D-Cl and D-N<P>NH. *As partial conversions of 1 into 2a and 2b based on the effective amounts of these products isolated after column chromatography.
Scheme 2: Synthesis of G-1 dendrons D-Cl and D-N<P>NH. *As partial conversions of 1 into 2a and 2b based on t...
Table 1: Reagents and conditions in the synthesis of compounds 2a, 2b, 3, D-Cl and D-N<P>NH.
| Entry | Reaction | Conditionsa |
|---|---|---|
| 1 | 1 → 2a, 2b | (i) 1.00 equiv n-C8H17-I, 8.00 equiv K2CO3, acetone, −13 °C, 3 h; rt, 12 h; (ii) 1.00 equiv n-C8H17-I, rt, 72 h; 50 °C, 8 h/N2 |
| 2 | 4-OOA → 2ab | (i) 0.50 equiv C3N3Cl3, acetone, 0–5 °C, 2 h; (ii) 1.00 equiv NaHCO3, H2O, 0–5 °C; 45 °C, 3 h; rt, 18 h/N2 |
| 3 | 2a → 3 | 4.00 equiv piperazine, 1.00 equiv K2CO3, THF, rt, 34 h |
| 4 | 3 → D-Cl | 0.50 equiv C3N3Cl3, 1.00 equiv K2CO3, THF, −10 °C, 3 h; rt, 36 h; reflux, 24 h |
| 5 | D-Cl → D-N<P>NH | 4.00 equiv piperazine, 1.00 equiv K2CO3, THF, rt, 40 h; reflux, 15 h |
aFor comments about these conditions, see section 3.1. bThe same double amination of cyanuric chloride with 4-(n-octyloxy)aniline was reported by S. Yagai and co-workers in 2012 [23], with no isolation of 2a, as it was submitted in situ to ammonolysis to give 2-amino-4,6-bis[4-(n-octyloxy)phenylamino]-s-triazine (75% overall yield).
We initially considered using the previously reported 4-hydroxyphenyl N-substituted 2-chloro-4,6-diamino-s-triazine 1 [36,48,49] as starting material. To our disappointment, the attempted O,O’-bis-alkylation of 1 with 1-iodooctane (Williamson etherification, Table 1, entry 1), afforded a multicomponent reaction mixture (TLC monitoring, 100% conversion of 1), from which we succeeded in isolating only G-0 chloro-dendron 2a and the O,O’,N-tris-alkylated side product 2b by column chromatography. Other efforts (not detailed in the present report), such as manipulation under dark conditions or replacing the iodine in the alkylating reagent by bromine, provided even more unsatisfactory results, e.g., the O-mono-alkylated product. By contrast, the double amination of cyanuric chloride with commercial 4-(n-octyloxy)aniline (Table 1, entry 2) gave the desired G-0 chloro-dendron 2a with excellent yield. We note the choice of several authors towards the synthesis of 4-(n-octyloxy)aniline by Williamson etherification of 4-nitrophenol or of N-acetyl-4-aminophenol (paracetamol) and subsequent reduction of the nitro group [23,25,27,29] or acidolysis of the amide sequence [24,26,28]. Our expertise on the use of paracetamol under Williamson etherification conditions [42] (Scheme 1, convergent route) did not impel us in the direction of preliminary 4-(n-octyloxy)aniline preparation; we also took into account its convenient price.
Using G-0 dendron 2a, we synthesised G-1 dendrons in two (→ D-Cl) or three (→ D-N<P>NH) orthogonal transformations, with excellent overall yields (90% and 84% from 2a, respectively). The complete chemoselectivity observed during the installation of the piperazine linkers on 2a (Table 1, entry 3) and on D-Cl (Table 1, entry 5) was ensured by the use of a 300% molar excess of this inexpensive reagent. We note the long-reaction time and large temperature domains (Table 1) required in order to obtain the quantitative results shown in Scheme 2 (to be discussed in section 3.1). Conversely, all G-0 and G-1 dendrons were easily purified by simple crystallisations from boiling methanol or ethanol (Supporting Information File 1, pp. S3–S6; pp. S12–S23, Figures S1–S24).
2.2 Synthesis of G-2 dendrimers
The synthesis of G-2 dendrimers 4–6 (Scheme 3) revealed a moderate reactivity of G-1 dendrons D-N<P>NH and D-Cl with cyanuric chloride and central building blocks A and B. Therefore, in addition to TLC, all reactions were monitored by HRMS as well. They were stopped when no further reaction was observed by TLC and when the expected molecular peak was identified by HRMS (ESI+, ACN + TFA).
![[1860-5397-14-145-i3]](/bjoc/content/inline/1860-5397-14-145-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of G-2 dendrimers 4–6 by m-trimerisations of G-1 dendrons D-Cl and D-N<P>NH.
Scheme 3: Synthesis of G-2 dendrimers 4–6 by m-trimerisations of G-1 dendrons D-Cl and D-N<P>NH.
Under demanding thermal conditions, the complete amination of cyanuric chloride by D-N<P>NH gave the G-2 dendrimer 4 with satisfactory yield. By contrast, treatment of 1,3,5-tris(bromomethyl)benzene with D-N<P>NH (3 equiv, 72 h in refluxing 1,4-dioxane in the presence of K2CO3 as proton scavenger) resulted in no dendrimer formation; decomposition of D-N<P>NH was observed instead (as determined by additional NMR monitoring).
Nevertheless, the desired G-2 dendrimer 5 could be obtained through an alternative route, namely, by amination of G-1 chloro-dendron D-Cl with building block A (Figure 1) under harsh conditions.
To obtain dendrimer 6, we expected to exploit the nucleophilicity of the phenoxide groups in the N,N’,N”-tris(4-hydroxyphenyl)melamine B (Figure 1) based on the previous example of P. Gamez and co-workers [41], which involved reaction with 2-chloro[4,6-di(pyridin-2-ylamino)]-s-triazine in the presence of DIPEA (69% yield after 48 h at 85 °C in pyridine). However, when dendrimer 6 was obtained by reacting B (0.31 equiv) with D-Cl (1.00 equiv), the reaction only proceeded properly when 4.00 equiv K2CO3 per phenolic OH group in B was used as the proton scavenger and deprotonating reagent, i.e., in an identical acid/base molar ratio as that used earlier in the triple Williamson etherification of B (Figure 1, Scheme 1, divergent route) with bromoalkanoic acid ethyl esters [42]. Even so, the yield of 6 was modest, 35%.
Furthermore, we obtained G-2 dendrimers 7a and 7b (Scheme 4) by the triple amidation of trimesic acid trichloride upon treatment with G-1 dendron D-N<P>NH (covalent m-trimerisation →7a) or by simple neutralisation of the latter with trimesic acid (ionic m-trimerisation →7b), the product of which was symbolised as [D-N<P>NH2+]3(−OOC)3C6H3 (Figure 1).
![[1860-5397-14-145-i4]](/bjoc/content/inline/1860-5397-14-145-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of G-2 dendrimers 7–9 by m-trimerisations of G-1 dendron D-N<P>NH.
Scheme 4: Synthesis of G-2 dendrimers 7–9 by m-trimerisations of G-1 dendron D-N<P>NH.
In the synthesis of 7a, unexpectedly difficult conditions for this type of reaction were, once again, required.
Similar amidations run in triplicate using instead the acid chlorides of C1 and C3 provided negative results. Thus, although the protocol for the conversion of these (4-aminophenoxy)alkanoic acid-based tripodal melamines into the corresponding tri acid chlorides (not depicted in Scheme 4) followed typical procedures (in refluxing thionyl chloride with SO2 and HCl generation), a long reaction time (up to 24 h, as determined by HRMS monitoring) was mandatory for this transformation to go to completion. The crude isolated products exhibited very low solubility in solvents usually recommended for standard N-acylations (acetone, THF, 1,4-dioxane, pyridine, etc.). Treatment of these acid chlorides with G-1 dendron D-N<P>NH (3 equiv) under various conditions (microwave-assisted organic reactions included [50]) resulted in no reaction and the starting materials, D-N<P>NH and C1 (or C3) being recovered in good yields after obligatory aqueous work-up. To our surprise, even the trivial derivatisation of the acid chlorides as ethyl esters (precursors of C1 and C3, Scheme 1, divergent route) required at least 24 h in absolute EtOH and only proceeded upon heating. Therefore, we had to limit our efforts to the stoichiometric salts (Figure 1), symbolised as [D-N<P>NH2+]3[C1]3− (8) and [D-N<P>NH2+]3[C3]3− (9). These dendritic constructions by ionic m-trimerisations occurred smoothly and in quantitative yields (Scheme 4) (Supporting Information File 1, pp. S6–S11; pp. S24–S39, Figures S25–S54).
3 Structural assignments
Our research continued with a structural study, implemented by means of DFT calculations, solution (VT) NMR techniques, IR (KBr) spectroscopy and TEM analysis.
3.1 Assignments based on DFT calculations of G-0 and G-1 dendrons: optimal geometry and solvation effects
First, we attempted to explain the generally difficult conditions encountered during synthesis (Schemes 2–4) based on DFT calculations [50-56] (Figure 2 and Table 2).
![[1860-5397-14-145-2]](/bjoc/content/figures/1860-5397-14-145-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The three terms rotamerism of G-0 dendrons 2a and 3 about the C(s-triazine)–N(exocyclic) partial double bonds.
Figure 2: The three terms rotamerism of G-0 dendrons 2a and 3 about the C(s-triazine)–N(exocyclic) partial do...
Table 2: Full geometry optimisation of G-0 dendrons 2a, 3 [51-54], solvation energies of G-0 and G-1 dendrons 2a, 3, D-Cl, D-N<P>NH [54] and lone pair (LP) electron occupation of piperazine Nsp3 nitrogens in dendrons 3 and D-N<P>NH [56,57].
| No. |
Relative conformational total electronic Energy ΔE (kJ mol−1)a
in the gas phase in DMSO |
ΔG of solvation (kJ mol−1) for the (a–a) major rotamera |
LP electron occupation
of piperazine Nsp3 nitrogens (e)b |
||||
|---|---|---|---|---|---|---|---|
| (s–s) | (a–s) | (a–a) | THFc | 1,4-dioxaned | THF | 1,4-dioxane | |
| 2a |
10.464
17.371 |
8.329
15.558 |
0.000
0.000 |
−146.37 | −104.39 | – | – |
| 3 |
4.395
5.923 |
4.730
4.504 |
0.000
0.000 |
−167.54 | −122.51 | 1.92 | 1.92 |
| D-Cl | – | – | – | −378.66 | −284.28 | – | – |
| D-N<P>NH | – | – | – | −401.81 | −301.36 | 1.93 | 1.92 |
aSee Experimental and Supporting Information File 1 (pp. S40 ff) for details of this DFT calculation. bElementary electric charge carried by a single electron. cDielectric constant, ε = 7.4257. dDielectric constant, ε = 2.2099.
As in the case of other amino-s-triazines [30-33,36,45,46], for G-0 dendrons 2a and 3, a topologically idealised model predicted a three terms (syn–syn anti–syn anti–anti) rotational equilibrium about the C(s-triazine)–N(exocyclic) (Figure 2) partial double bonds, at room temperature. Similar to simpler 4-hydroxyphenyl-based amino-s-triazine analogues of 2a and 3 [35,36], the optimised (a–a) geometry was found to be largely dominant, both in the gas phase as well as in solution, e.g., DMSO, which was the most appropriate solvent for (VT) 1H NMR and TEM investigations (see section 3.2).
As expected, the relative ΔE values for the rotational diastereomerism decreased (i) with the decrease of the steric hindrance between the three (hetero)aryl rings in rotamers (s–s) > (a–s) > (a–a), and (ii) with the weakening of the π-deficiency of the s-triazine, 2a > 3. The same optimal geometry (a–a) was found in duplicate, (a–a)/(a–a), in G-1 dimers D-Cl and D-N<P>NH (Figure 2).
Furthermore, for the major (a–a) rotamers, calculations also revealed high solvation energies in the case of all four G-0 and G-1 dendrons. For a single compound, the solvation energy was 33–40% higher in THF vs 1,4-dioxane. Unsurprisingly, piperazine dendrons were more solvated than their chloro precursors, e.g., 3 vs 2a (+14% in THF, +17% in 1,4-dioxane), and D-N<P>NH vs D-Cl (about +6% in both solvents). Taking into account the negligible decrease in LP electron occupation of the piperazine Nsp3 nitrogens in 3 and D-N<P>NH as well, we ascribed the low reactivity of G-1 dendrons D-Cl and D-N<P>NH in our SN2-Ar amination conditions to the increased solvation of their (a–a)/(a–a) rotamers (+140–170%, i.e., more than double) compared to those of the (a–a) rotamers of G-0 3 and 2a.
In addition to all the above, due to the steric accommodation of 12 n-octyloxy chains, the occurrence of a starburst effect [18,58,59] during the covalent m-trimerisations (Scheme 3), was also presumed (see section 3.2.1).
3.2 Assignments by means of NMR and IR spectroscopy
The 1H NMR data deserving of comment are listed in Table 3.
Table 3: Relevant (VT) 1H NMR, temperature gradients and 2D-1H-DOSY NMR data (500 MHz, DMSO-d6) of G-0 dendrons 2a, 3, G-1 dendrons D-Cl, D-N<P>NH and G-2 dendrimers 4–6, 7a, 7b, 8, 9.
| No. | δH (ppm) values of indicative NH protons (see Figure 2) | temperature gradients (TGs) as (ΔδH/ΔΤ) × 103 (ppb K−1)a |
Db
(µm2 s−1) |
dHb
(nm) |
||||
|---|---|---|---|---|---|---|---|---|
|
NH adjacent to
s-triazine T-0 |
NH adjacent
s-triazine T-2 |
NH adjacent to
s-triazine T-0 |
NH adjacent
s-triazine T-2 |
|||||
| 298 K | 363 K | 298 K | 363 K | |||||
| 2ac |
10.03
9.96 9.85 |
9.63 | – | – |
−6.15
−5.08 −3.38 |
198 | 1.10 | |
| 3 | 8.87 | 8.50 | – | – | −5.69 | 191 | 1.14 | |
| D-Cl | 9.08 | 8.70 | – | – | −5.85 | 129 | 1.69 | |
| D-N<P>NH | 8.93 | 8.57 | – | – | −5.54 | 110 | 1.98 | |
| 4 | 9.71 | 8.93 | – | – | −12.00 | 89 | 2.45 | |
| 5 | 8.92 |
8.51
8.53 8.55 8.60 |
– | – |
−6.31
−6.00 −5.69 −4.92 |
88 | 2.48 | |
| 6d | – | 8.55 | – | 9.02 | – | – | – | |
| 7a | 8.94 | 8.55 | – | – | −6.00 | 107 | 2.04 | |
| 7b | 8.93 | 8.53 | – | – | −6.15 |
105
225 |
2.08
0.97 |
|
| 8 | 8.93 | 8.54 | 8.93 | 8.60 | −6.00 | −5.08e | 113 | 1.93 |
| 9 | 8.94 | 8.54 | 8.70 | 8.57 | −6.15 | −2.00f | 115 | 1.90 |
aTGs calculated as [(δH298 K − δHT K)/(298 K – T K)] × 103 < 0 (where T = 363 K). bdH (hydrodynamic diameter) issued from D [diffusion coefficient observed in 2D-1H-DOSY NMR charts as 5.0 mM (2a, 3, D-Cl, D-N<P>NH, 5, 8 and 9) or 2.5 mM (4, 7a and 7b) in DMSO-d6 (η, dynamic viscosity 2.00 × 10−3 kg m−1 s−1) at 298 K] by applying the Stokes–Einstein equation. cMultiple δH and TG values due to more than one (anti–anti major 64% vs anti–syn minor 36%) species found in a frozen rotational equilibrium in agreement with the highest π-deficiency of the s-triazine ring in the analysed series (Figure 2; Supporting Information File 1, p. S12, Figure S1). dDue to the low solubility of compound 6 in DMSO-d6, its convincing NMR spectra could be obtained at 363 K only. eTG as −6.92 ppb K−1 in the starting material C1. fTG as −6.07 ppb K−1 in the starting material C3.
3.2.1 Rotational diastereomerism at dendritic level: As expected, due to it having the highest π-deficient s-triazine group, at room temperature only G-0 chloro dendron 2a exhibited the nearly frozen nature of the anti syn (Figure 2; Supporting Information File 1, pp. S12, Figure S1) rotational equilibrium on a 500 MHz 1H NMR timescale. In contrast, under the same conditions, the rotational diastereomerism of all other compounds (G-0, G-1 and G-2) was identified, as the classically entitled “slow exchange status between unequally populated sites” [60,61]. At room temperature, dendrimers 5, 7a, 8 and 9 displayed unique signals for the topologically equivalent 1H and 13C nuclei of the m-trivalent core and their adjacent angular connections, i.e., consistent with a propeller as local dominant arrangement (Figure 1) [45,46]. Except for G-2 dendrimer 5, upon heating to 90 °C, all compounds became single mediated structures, in states with rapid and free rotation about all the C(s-triazine)–N(exocyclic) bonds. In contrast, in the 1H NMR spectrum of dendrimer 5 recorded at 90 °C, multiple resonances (Table 3 and Figure 3) were displayed by the indicative NH protons, suggesting the occurrence of a significant peripheral steric hindrance, starburst effect [18,58,59], against authentic free rotation upon heating. This fact was not quite surprising because we previously reported a related situation in the case of some G-0 dendrons as N,N’-disubstituted 2-chloro-4,6-diamino-s-triazines with bulky azaspirodecane and propane-1,3-diol ligands and entitled this “abnormal” behaviour “pseudo freely rotating” status [31].
![[1860-5397-14-145-3]](/bjoc/content/figures/1860-5397-14-145-3.svg?scale=1.8&max-width=1024&background=FFFFFF)
Figure 3: Comparative details from 1H NMR spectra of G-2 dendrimer 5 (500 MHz, 5.0 mM in DMSO-d6).
Figure 3: Comparative details from 1H NMR spectra of G-2 dendrimer 5 (500 MHz, 5.0 mM in DMSO-d6).
3.2.2 Dendritic solvation: In line with solvation energy calculations (Table 2), the temperature gradients (TGs) of the indicative NH protons provided additional information on the solvation in the regions surrounding the s-triazine units (branch-cells, T-0 or cores T-2, Scheme 3 and Scheme 4). According to H. Kessler (in 1982, [62]), this parameter is appropriate for describing amide protons solvation in p→π conjugated systems, such as –N(H)–C(=O)– ↔ –N+(H)=C(–O−)– in peptides and proteins, in water. Later on (in 2008), X. K. Moreno and E. E. Simanek [47] demonstrated the validity of the above TGs in the case of amino-s-triazines, –N(H)–C(=N–)– ↔ –N+(H)=C(–N−–)–, as well, in DMSO-d6. Following this new insight, if the TG values of “amidine-like” protons in amino-s-triazines are more negative than −4 ppb K−1 in strong hydrogen bond acceptor solvents, such as DMSO-d6 [45], then the NH groups are exposed to the solvent rather than developing intramolecular hydrogen bonds. Conversely, a TG value less negative than −4 ppb K−1 discloses the NH group preference towards intramolecular hydrogen bonds formation, at room temperature, e.g., with s-triazine LPs N-1, -3, -5. If so, the compounds under investigation (Table 3) exhibited TG values denoting significant (peripheral) >N–H···O=SMe2 hydrogen bond interactions. TGs moderately increased, from −5.55 ppb K−1 in G-1 D-N<P>NH to about −6.00 ppb K−1 in G-2 dendrimers, together with the hydrodynamic diameters, dH, except dendrimers 4 and 5. Dendrimer 4 was by far the most NH/DMSO solvated of the series (TG around −12.00 ppb K−1) in conjunction with one of the highest dH values, of 2.45 nm.
On the other hand, an evaluation of the TG values of the inner (T-2) vs the peripheral (T-0) NH groups in ionic dendrimer 8 showed a comparable aptitude for NH/DMSO H-binding, −5.08 ppb K−1 (−6.92 ppb K−1 in the starting material C1) vs −6.00 ppb K−1, respectively. Furthermore, the replacement of the central methylene spacer (n = 1, in 8) with a trimethylene one (n = 3, in 9) had no influence on the peripheral (T-0) NH groups/DMSO solvation as they maintained almost the same TG (−6.00 ppb K−1 in 8 vs −6.15 ppb K−1 in 9). In contrast, the inner (T-2) NH groups of 9 appeared “protected” against DMSO (as a hydrogen bond acceptor) because their TG was only −2.00 ppb K−1, (much less negative than in the starting material C3, −6.00 ppb K−1), i.e., the three identical intramolecular >N–H···LP(N-1, -3, -5 of s-triazine T-2 core) interactions prevailed, being, most likely, a consequence of the aforementioned (Figure 1) C3 symmetric propeller as local arrangement.
3.2.3 Assignment of ionic interactions in dendrimers 7b, 8 and 9: On the 1H NMR timescale in DMSO-d6, the triple ionic interactions in dendrimers 7b, 8 and 9 (Scheme 4) could only be indirectly assigned, due to rapid proton interchange, symbolised as:
3 D-N<P>NH + (HOOC)3R [D-N<P>NH2+ −OOC]3R
As a consequence of the triple ionisation of the carboxyl groups in tripodal melamines C1 and C3, only a remote upfield shift of their 1H δNH values was observed: from 9.47 ppm (in C1, [42]) to 8.93 ppm (in the tris-anion of 8) and from 8.95 ppm (in C3, [42]) to 8.70 ppm (in the tris-anion of 9) (Table 3). A different diagnosis, based on aliphatic carboxyl group ionisation thus promoting 1H shielding of proximal (α and β) methylene protons, was reported in the case of some G-2 PAMAM ionic dendrimers obtained by –COOH/H2N– neutralisation (NMR solvent, CDCl3) [63].
However, in the case of the cationic counterpart, if a more illustrative series was examined, i.e., D-N<P>NH, 4, 7a, 7b, 8 and 9 (Table 4), δH resonances of the methylene protons α-to the Nsp3 nitrogen of P-1 piperazine linker (in D-N<P>NH vs 7b, 8 and 9) revealed minor fluctuations, between 2.97 and 3.15 ppm. These resonances were found much further upfield with respect to the shifts of the methylene protons α-situated vs the piperazine P-1 Nsp2 nitrogen involved in p→π LP(N)→π(C=N, s-triazine; C=O, amide) conjugation (3.78–3.91 ppm).
Table 4: 1H NMR discrimination of methylene protons in piperazine P-1 linkers of compounds D-N<P>NH, 4, 7a, 7b, 8 and 9.
|
Methylene
locations |
Compound
δH (ppm) (500 MHz, DMSO-d6, 298 K) |
|||||
|---|---|---|---|---|---|---|
| D-N<P>NH | 4 | 7a | 7ba | 8a | 9a | |
| α-to Nsp3 | 3.13 | – | – | 3.09 | 2.97 | 3.15 |
| α-to Nsp2 | 3.91 | 3.80 | 3.78 | 3.78 | 3.78 | 3.78 |
aDownfield shifted resonances (Δδ as +0.25 and +0.33 ppm) of α-CH2 protons to –NH3+ group were reported in the case of some G-2 PAMAM dendrimers obtained by –COOH/H2N– neutralisation (CDCl3) [63].
To conclude, except the well-documented deshielding promoted by the magnetic anisotropy of π-delocalised systems, such as s-triazines and amides [64], no such vicinal effect created by a quaternary EWG of type >Nsp3H2+ [63] was detected in the piperazine P-1 linker of compounds 7b, 8 and 9, even in the case of the strongest proton donor, the trimesic acid (pKa = 3.12, 3.89 and 4.70).
In addition, only in G-1 amino-dendron D-N<P>NH the 1H NMR integration of its termini piperazine P-1 linker disclosed the presence of just 4 methylene protons (instead of 8; Supporting Information File 1, pp. S21, S22, Figures S20 and S21) at room temperature as well as at 90 °C, presumably due to their longer T1 value (longitudinal relaxation time) [60]. No such spectral appearance was observed in the case of G-0 precursor 3 (Supporting Information File 1, p. S17, Figures S11 and S12). Subsequent to the use of D-N<P>NH in covalent (→ 4, 7a, 5) or ionic (→ 7b, 8 and 9) m-trimerisations, the same 1H NMR integration evidenced, throughout, the existence of the expected 8 H/P-1 unit.
On the IR timescale (Figure 4a–d), the comparative spectra of compounds 7a/7b, 7b/trimesic acid, 8/C1 and 9/C3 fully proved the presence of only the tris-carboxylate anions [63,65,66].
(i) The spectrum of dendritic salt 7b was almost the duplicate of that of covalent 7a (Figure 4a), except for the presence of a weak band located at 1702 cm−1 which was attributed to the νC=O stretching absorption of the tris-carboxylate anion in 7b [65,66].
(ii) Indeed (Figure 4b), the shift to a lower field and weaker intensity of the strong νC=O band (from 1725 cm−1 for the COOH groups of trimesic acid to 1702 cm−1 for the COO− groups of 7b), together with the disappearance of the νOH stretching band (3090 cm−1) of the carboxyl groups (strongly H-associated in trimesic acid) confirmed the existence of the trimesic tris-carboxylate anion in 7b. Nevertheless, the stretching (νNH, 3000–2700 cm−1) or deformation (δNH, 1620–1560 cm−1) bands [66,67] of the protonated >NH2+ group of the P-1 piperazine linker could not be definitely allocated due to the overlapping in the above regions between the absorptions of the piperazine methylene groups (νCH2), carbonyl bonds (νC=O) in the COO− groups, aryl bonds (νC=C) and s-triazine bonds (νC=N).
(iii) Similar attributions were applied in the case of ionic dendrimers 8 (Figure 4c) and 9 (Figure 4d). The vanishing of the carboxyl νOH bands from C2 and C3 in the dendritic environment of 8 and 9 established the ionic nature of the latter. Due to their overall lower symmetry in comparison with trimesic acid, tripodal aminophenoxy-acids-based N-substituted melamines C2 and C3 exhibited multiple (3–4) carboxyl νC=O stretching absorptions, in agreement with their different degrees of H-bond association, i.e., dimers (1742 and 1709 cm−1), polymers (1679, 1625, 1622 and, presumably, even 1600 cm−1). Succeeding the ionic trimerisations, all these bands were absent in the IR spectra of 8 and 9, with the remaining carboxylate νC=O stretching band being located most likely at approximately 1600 cm−1.
![[1860-5397-14-145-4]](/bjoc/content/figures/1860-5397-14-145-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparative IR spectra (KBr) of compounds 7a vs 7b (a), 7b vs trimesic acid (b), 8 vs C1 (c) and 9 vs C3 (d).
Figure 4: Comparative IR spectra (KBr) of compounds 7a vs 7b (a), 7b vs trimesic acid (b), 8 vs C1 (c) and 9 ...
In conclusion, the exact protonated site in ionic dendrimers 7b, 8 and 9 could not be attributed accurately either in solution (1H NMR), or in the solid state (IR). Consequently, their proposed cationic structures shown in Figure 1 and Scheme 4 (linker P-1 as piperazin-1-ium moiety) should be seen intuitively, a proton sponge-like comportment of G-1 piperazine dendron D-N<P>NH being not entirely ruled out [68]. In line with this hypothesis, we recently described [35] the aptitude of the non-O-n-octylated analogue of G-2 dendrimer 4 (Scheme 3) to deprotonate Hemin completely (1:5 molar ratio), thus generating a new MOF for H2O2 amperometric detection. More insights on this debate were obtained by means of DFT calculations only (see section 3.3).
3.3 Assignments by means of 2D-1H-DOSY NMR in tandem with DFT calculations at dendritic level
Except for dendrimers 4 (dH 2.45 nm) and 5 (dH 2.48 nm) (Table 3), in the series of trimers 7–9 the expected increase in the hydrodynamic diameters (dH) with respect to their G-1 monomeric precursor, D-N<P>NH (1.98 nm), was negligible (1.90–2.04 nm).
2D-1H-DOSY NMR charts of G-2 dendrimers 7a (covalent) vs ionic 7b, 8 and 9 (Figure 5) displayed unique structures, with a partial dissociation of 7b, presumably due to the sterically induced imperfect accommodation of the three G-1 cationic dendrons D-N<P>NH2+ around the smallest central tris-anion of the series, trimesic tris-carboxylate.
![[1860-5397-14-145-5]](/bjoc/content/figures/1860-5397-14-145-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 2D-1H-DOSY NMR charts (DMSO-d6, 500 MHz, 298 K) of compounds 7a, 7b (2.5 mM), 8 and 9 (5.0 mM).
Figure 5: 2D-1H-DOSY NMR charts (DMSO-d6, 500 MHz, 298 K) of compounds 7a, 7b (2.5 mM), 8 and 9 (5.0 mM).
In other words, the 2D-1H-DOSY NMR charts were consistent with the envisaged 3(D-N<P>NH):1 (trimesic acid, C1 or C3) stoichiometric assembly of dendrimers 7b, 8 and 9. That is, we had to assume that in series D-N<P>NH, 7–9, the correlation between D values (by means of hydrodynamic diameters dH, Table 3) and the macromolecular size was not direct, i.e., dendritic structures with different molecular weight displaced related volumes of solvent. The above encountered situation was comparable to that of some PAMAM dendrimers exhibiting D values around 100 μm2 s−1 [69].
Therefore, an additional effort to estimate the above situation in the case of dendrimers 7–9, by means of DFT calculations in solution (DMSO) (Figures 6–8), was straightforward.
First, the optimisation of the geometry of G-2 dendrimer 7a (Figure 6) identified its global form to be almost vaulted although the degree of freedom of the six G-1 dendritic units appeared restricted. Indeed, one of them (marked by the red arrow) had an opposite direction to the other five, caused, probably, by the propeller orientation (Figure 1) of the three covalent junctions around the benzene core.
![[1860-5397-14-145-6]](/bjoc/content/figures/1860-5397-14-145-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The DFT optimised geometry at M062X/def2-TZVP level of theory of G-2 dendrimer 7a in DMSO (hydrogen atoms were omitted for reasons of simplicity).
Figure 6: The DFT optimised geometry at M062X/def2-TZVP level of theory of G-2 dendrimer 7a in DMSO (hydrogen...
In contrast, the energetic minimum of the ionic analogue 7b could be accessed in a gradual approach only. Thus, we had to start from a preliminary DFT calculation of the trimesic tris-carboxylate anion (Figure 7a) because its identical partial double bond lengths (dC![[Graphic 1]](/bjoc/content/inline/1860-5397-14-145-i5.svg?max-width=637&scale=1.18182) Oδ−) and oxygen atoms negative charges values (δ−, as Natural Population (no) according to the NBO analysis) were then taken as references with regard to those of the same tris-anion installed in a model G-1 ionic environment, 10 (Figure 7b). We considered dendrimer 10 as a simplified version of 7b from which the 4,6-bis[4-(n-octyloxy)phenylamino]-s-triazin-2-yl G-0 branches were removed. Oxygen atoms negative charges in 10 (Figure 7b), δ(a)– and δ(b)–, did not differ significantly vs those of the initial trimesic tris-carboxylate anion (δ–, Figure 7a). Besides the electrostatic attraction between the tris-carboxylate anion and the P-1 piperazin-1-ium melamine G-1 cations, the occurrence of important H-bonding three interactions (>C
Oδ−) and oxygen atoms negative charges values (δ−, as Natural Population (no) according to the NBO analysis) were then taken as references with regard to those of the same tris-anion installed in a model G-1 ionic environment, 10 (Figure 7b). We considered dendrimer 10 as a simplified version of 7b from which the 4,6-bis[4-(n-octyloxy)phenylamino]-s-triazin-2-yl G-0 branches were removed. Oxygen atoms negative charges in 10 (Figure 7b), δ(a)– and δ(b)–, did not differ significantly vs those of the initial trimesic tris-carboxylate anion (δ–, Figure 7a). Besides the electrostatic attraction between the tris-carboxylate anion and the P-1 piperazin-1-ium melamine G-1 cations, the occurrence of important H-bonding three interactions (>C![[Graphic 2]](/bjoc/content/inline/1860-5397-14-145-i6.svg?max-width=637&scale=1.18182) Oδ(b)–···H(ax or eq)-NH+<) were identified, being supported by the bond lengths decreasing order as 1.265 Å (d(C
Oδ(b)–···H(ax or eq)-NH+<) were identified, being supported by the bond lengths decreasing order as 1.265 Å (d(C![[Graphic 3]](/bjoc/content/inline/1860-5397-14-145-i7.svg?max-width=637&scale=1.18182) Oδ(b)–) in 10) > 1.252 Å (d(C
Oδ(b)–) in 10) > 1.252 Å (d(C![[Graphic 4]](/bjoc/content/inline/1860-5397-14-145-i8.svg?max-width=637&scale=1.18182) Oδ–) in trimesic tris-carboxylate anion) > 1.244 Å (d(C
Oδ–) in trimesic tris-carboxylate anion) > 1.244 Å (d(C![[Graphic 5]](/bjoc/content/inline/1860-5397-14-145-i9.svg?max-width=637&scale=1.18182) Oδ(a)–) in 10) together with Σrcov (O, H): 1.40 Å < d(Oδ(b)–···H): 1.60 Å < ΣrwdW (O, H): 2.60 Å [70]. One must observe that, contrarily to our planned design of 7b, as [D-N<P>NH2+]3(–OOC)3C6H3 (Figure 1, linear connection, as a six-membered chelate, to the m-trivalent core), the H-bonding network in model 10 determined simpler angular connections of the P-1 piperazin-1-ium melamine G-1 cations around the trimesic tris-carboxylate anion core in a statistically favoured asymmetric manner (Figure 1). In spite of this local “irregularity”, entirely re-found in the optimised geometry of 7b (Figure 7), its global profile was of type propeller due to the adopted vault shape of its three G-1 branches (D, Figure 1), i.e., concavity vs convexity.
Oδ(a)–) in 10) together with Σrcov (O, H): 1.40 Å < d(Oδ(b)–···H): 1.60 Å < ΣrwdW (O, H): 2.60 Å [70]. One must observe that, contrarily to our planned design of 7b, as [D-N<P>NH2+]3(–OOC)3C6H3 (Figure 1, linear connection, as a six-membered chelate, to the m-trivalent core), the H-bonding network in model 10 determined simpler angular connections of the P-1 piperazin-1-ium melamine G-1 cations around the trimesic tris-carboxylate anion core in a statistically favoured asymmetric manner (Figure 1). In spite of this local “irregularity”, entirely re-found in the optimised geometry of 7b (Figure 7), its global profile was of type propeller due to the adopted vault shape of its three G-1 branches (D, Figure 1), i.e., concavity vs convexity.
![[1860-5397-14-145-7]](/bjoc/content/figures/1860-5397-14-145-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The DFT optimised geometry at M062X/def2-TZVP level of theory of trimesic tris-carboxylate anion (a), of model G-1 dendrimer 10 (b) and of G-2 dendrimer 7b in DMSO.
Figure 7: The DFT optimised geometry at M062X/def2-TZVP level of theory of trimesic tris-carboxylate anion (a...
Extrapolation of the ionic relationships found for model 10 to compounds with higher mass 8 and 9 (Figure 8) provided their energetic minima as to correspond to (i) a local propeller orientation of the G-1 branches around the s-triazin-2,4,6-triyl core (initial design, Figure 1) and (ii) a global vaulted form (“candelabrum” allure) promoted, as in the case of 7b, by the same arched G-1 branches.
![[1860-5397-14-145-8]](/bjoc/content/figures/1860-5397-14-145-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The DFT optimised geometry at M062X/def2-TZVP level of theory of G-2 dendrimers 8 and 9 in DMSO.
Figure 8: The DFT optimised geometry at M062X/def2-TZVP level of theory of G-2 dendrimers 8 and 9 in DMSO.
3.4 Assignments by means of TEM analyses
We also considered of interest to corroborate our ab initio study with an introductory exploration by means of TEM (transmission electronic microscopy). In this purpose, the sampling was made by dissolving compounds D-N<P>NH and 4–9 (ca. 1 mg) in DMSO (1 mL, see section 3.1) under sonication. One drop of solution was deposited on a Formvar/Carbon coated copper grid (300 mesh) and let to free evaporate to dryness at room temperature (24 h). Except for compound 4, agglomerations of homogeneously packed spherical nano-aggregates (Figure 9 and Figure 10) were thus obtained. They were comparable with those of the recently reported G-2 PAMAM ionic (–COO−/H3N+–) dendrimers, already mentioned [63]. Each sample preparation was repeated three times and the hereafter discussed numerical data were mediated.
![[1860-5397-14-145-9]](/bjoc/content/figures/1860-5397-14-145-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: TEM images of homogeneously packed spherical nano-aggregates (a) and their agglomerations (b) in the case of G-1 amino-dendron D-N<P>NH and of covalent G-2 dendrimers 5–7a. D is the average diameter of nanospheres; the red arrow shows the nano-particles that aggregate.
Figure 9: TEM images of homogeneously packed spherical nano-aggregates (a) and their agglomerations (b) in th...
![[1860-5397-14-145-10]](/bjoc/content/figures/1860-5397-14-145-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: TEM images of homogeneously packed spherical nano-aggregates (a) and their agglomerations (b) in the case of G-2 ionic dendrimers 7b, 8 and 9; the red arrow indicates the nano-particles that aggregate. D is the average diameter of nanospheres.
Figure 10: TEM images of homogeneously packed spherical nano-aggregates (a) and their agglomerations (b) in th...
The size of nanospheres (expressed by their average diameters, D) ranged in a large domain (234–1454 nm) with a quite wide distribution of D values in all situations (Supporting Information File 1, p. S41, Figure S55). With the exception of compound 6, all other G-2 dendrimers were, in fact, trimers of the same G-1 dendron, D-N<P>NH. Therefore, for the present discussion, we will limit our assignments to the structural diversity impact of the central building blocks (or cores) together with that of the dendritic elaboration (covalent vs ionic), seen both accountable for the observed different propensity for self-assembly as nano-aggregates. Indeed, covalent dendrimers 5–7a generated nanospheres with much smaller average diameter (259–732 nm, Figure 9) than the ionic 7b–9 (992–1454 nm, Figure 10). Moreover, according to the literature [71], the last ones appeared to be among the greatest previously reported nanosystems as polymeric nanoparticles. In covalent series, G-1 dendron D-N<P>NH itself (as unexpected reference) self-assembled into nanospheres with a high D value (694 nm). We ascribed this packing ability to three basic π stacking interactions [72] associating stratified parallel-displaced dendritic scaffolds (Figure 11):
(i) (π–π) between the electron-poor N-atoms, adjacent to s-triazine rings, as π-acceptors, from one hand, and the π-enriched (hetero)cycles, s-triazines and 4-(n-octyloxy)phenylamino peripheral units, on the other hand, as π-donors.
(ii) (π–H) of type (s-triazine) adjacent >Nsp2–H···π-enriched (hetero)cycles.
![[1860-5397-14-145-11]](/bjoc/content/figures/1860-5397-14-145-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Proposed π-stacking interactions in compounds D-N<P>NH and 5–7a.
Figure 11: Proposed π-stacking interactions in compounds D-N<P>NH and 5–7a.
Surprisingly, single trimer 4 ([D-N<P>N]3C3N3, Figure 1), containing s-triazine as linearly linked core, gave no distinct spherical aggregation (Supporting Information File 1, p. S41, Figure S56). In contrast, by replacing the P-1 piperazine linker in 4 with a 4-oxyphenylamino unit, the resulting dendrimer 6 ([D3]B, Figure 1), still encompassing s-triazine as core but propeller angularly coupled, evidenced a total opposite behaviour, i.e., aggregation under the form of nanospheres with the highest (732 nm) D value.
Trimers of D-N<P>NH angularly-propeller attached to 1,3,5-trisubstituted benzene cores, as π-electron-enhanced (1,3,5-tris(methylene)benzene, [D]3A (Figure 1), compound 5, D = 234 nm) or π-poor (1,3,5-tris(formyl)benzene, [D-N<P>N]3(OC)3C6H3 (Figure 1), compound 7a, D = 378 nm) self-assembled into the smallest spherical nano-aggregates.
By far, due to their electrostatic interactions (attractions vs repulsions) G-2 ionic dendrimers 7b, 8 and 9 (Figure 10) produced the most developed spherical nano-aggregates. The ionic relationships masked those deduced in covalent series, namely the π–π and π–H stacking and angular-propeller connectivity of the G-1 dendritic branches to the m-trivalent core.
Conclusion
Starting from 4-(n-octyloxy)aniline, the five-step orthogonal convergent synthesis of a new seven terms series of G-2 melamine based-dendrimers was achieved in overall yields ranging between 29 and 79%. By means of DFT calculation in solution, the reaction conditions were found mandatory to the high solvation of the G-0 and G-1 dendrons exhibiting a major (anti–anti) (parallel) rotamerism of the peripheral 4-(n-octyloxy)phenyl units about the C(s-triazine)–N(exocyclic) partial double bonds. The final iterative synthetic step was realised by covalent and/or (carboxyl/amino) ionic trimerisations, to recommend two G-1 N-substituted melamine dendrons with piperzine-1,4-diyl (linkers) and 4-(n-octyloxyphenyl)amino (peripheral units), D-Cl and D-N<P>NH, as promising scaffolds for future dendritic elaborations.
Tandem DFT-(VT) NMR investigations revealed (i) the regular shape in solution of the terminal 4-(n-octyloxy)phenyl units (“parallel”, anti–anti), (ii) the propeller arrangement, in the case of the angular connections of G-1 dendrons around the m-trivalent core, (iii) the vaulted shapes of G-2 dendrimers and (iv) in one case, the occurrence of a starburst effect. The “salt”-like nature of the G-2 dendrimers (obtained by a carboxyl/amino 1:3 stoichiometric trimerisation) could be unambiguously assigned by means of 1H NMR (in solution) and IR (solid state), confirming the existence of the only tris-carboxylate anions. TEM analysis indicated the aptitude of our G-2 vaulted melamines for π–π and π–H stacking self-assembly into homogeneously packed spherical nano-aggregates. Their size was tailored primarily by the covalent vs ionic nature of dendrimers, i.e., the last ones producing nanospheres with more than 1000 nm averaged D values. The structural variety of the covalent dendritic elaboration around the m-trivalent cores (1,4-phenylene over piperazin-1,4-diyl adjacent linkers, propeller over asymmetric rotamerism of the angular over linear connectivity) modulated the degree of nano-aggregation.
Experimental
General
All reagents and solvents were of commercial quality and required no purification prior to use. Melting points were carried out on an ELECTROTHERMAL instrument and were not corrected. Microanalyses were performed on a Carlo Erba CHNOS 1160 apparatus.
TLC monitoring was performed by using aluminium sheets with silica gel 60 F254 (Merck) (visualisation under UV at λ = 254 nm).
Column chromatography was conducted on silica gel Si 60 (0.063–0.200 mm, Merck).
IR spectra were recorded on a JASCO FTIR 6100 Spectrometer. Only relevant absorption maxima (in cm−1) are listed throughout as being s (strong), m (medium) or w (weak).
NMR spectra were recorded on a Bruker AM 500 instrument operating at 500 or 125 MHz for 1H and 13C nuclei, respectively. All chemical shifts (δ values) are given in parts per million (ppm); all homocoupling patterns (nJH,H values) are given in Hertz. In the NMR descriptions, some specific abbreviations were used: “br s” (broad singlet), “br t” (broad triplet), “br m” (broad multiplet), “dd” (doublet of doublets), “tt app. q” (triplet of triplets app. as a quartet), “tt app. qi” (triplet of triplets app. as a quintet), “tt app. s” (triplet of triplets app. as a sextet), “tt app. sp” (triplet of triplets app. as a septet), “ddd app. td” (doublet of doublets of doublets app. as a triplet of doublets), T-0, T-1 and T-2 (s-triazine branch cell according to generations 0, 1 and 2), P-0 and P-1 (piperazine as linker according to generations 0 and 1).
Data for 2D-1H-DOSY NMR spectra of compounds D-N<P>NH, 4, 5, 7–9 (2.5 or 5 mM in DMSO-d6 at 298 K on 500 MHz timescale) were acquired by means of the ledbpgp2s pulse sequence. The diffusion time (Δ) ranged from 150 to 220 ms and the gradient pulse length (δ) ranged from 1.7 to 1.9 ms. The size of the raw data set was 32 × 16 k. The gradient intensity values were equally spaced from 2% to 95%. The DOSY spectrum was calculated using the Bruker TOPSPIN Software. The inverse Laplace transformation in the indirectly detected dimension was carried out by means of the MaxEnt algorithm. Log (D) was calculated with D expressed in µm2 s−1: D-N<P>NH (5 mM, δ = 1.7 ms, Δ = 220 ms), 4 (2.5 mM, δ = 1.8 ms, Δ = 150 ms), 5 (5 mM, δ = 1.9 ms, Δ = 190 ms), 7a (2.5 mM, δ = 1.7 ms, Δ = 150 ms), 7b (2.5 mM, δ = 1.8 ms, Δ = 150 ms), 8 (5 mM, δ = 1.9 ms, Δ = 150 ms), 9 (5 mM, δ = 1.7 ms, Δ = 150 ms).
Mass spectra were obtained on an LTQ ORBITRAP XL (Thermo Scientific) instrument which was externally calibrated using the manufacturer’s ESI(+) calibration mix. The samples were introduced into the spectrometer by direct infusion.
DFT calculations
The full geometry optimisation of compounds 2a, 3, 7a and 7b were carried out using DFT level of theory considering the M06-2X exchange-correlation functional together with the def2-TZVP basis set in the presence of a solvent environment implemented in the Gaussian 09 program package. The solvent effects were taken into account via the Polarisable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM) and considering DMSO (ε = 46.826) as the solvent environment; the total electronic energies of the anti–anti conformations were taken as the reference value.
Free energies of solvation (ΔG) of compounds 2a, 3, D-Cl and D-N<P>NH were calculated at the M06-2X/def2-TZVP level of theory (solvent model: THF and 1,4-dioxane) through the SMD solvation model of Truhlar.
The LP electron occupation of piperazine Nsp3 nitrogen in compounds 3 and D-N<P>NH was obtained by considering the NBO electron population analysis at the M06-2X/def2-TZVP level of theory, where e is the elementary electric charge carried by a single electron.
The molecular graphics (figures) were created by using GaussView software.
TEM analysis
TEM images were obtained with a Hitachi H-7650 transmission electron microscope operating at 80 keV.
Supporting Information
| Supporting Information File 1: All procedures for the synthesis of compounds 2a, 2b, 3, D-Cl, D-N<P>NH, 4-9 together with their full analytical data, (VT) NMR and MS spectra. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
The financial support from the Grant provided by the Ministry of Education and Scientific Research, UEFISCDI and ANCSI, Romania is gratefully acknowledged: Project PN-II-ID-PCE-2011-3-0128. A.B. thanks the Data Center of NIRDIMT Cluj-Napoca for providing the computational infrastructure and technical assistance.
References
-
Zhang, W.; Simanek, E. E. Org. Lett. 2000, 2, 843–845. doi:10.1021/ol005585g
Return to citation in text: [1] [2] [3] [4] -
Takagi, K.; Hattori, T.; Kunisada, H.; Yuki, Y. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 4385–4395. doi:10.1002/1099-0518(20001215)38:24<4385::AID-POLA70>3.0.CO;2-Y
Return to citation in text: [1] [2] -
Zhang, W.; Nowlan, D. T.; Thomson, L. M.; Lackowski, M. W.; Simanek, E. E. J. Am. Chem. Soc. 2001, 123, 8914–8922. doi:10.1021/ja0041369
Return to citation in text: [1] [2] -
Steffensen, M. B.; Simanek, E. E. Angew. Chem., Int. Ed. 2004, 43, 5178–5180. doi:10.1002/anie.200460031
Angew. Chem. 2004, 39, 5290–5292. doi:10.1002/ange.200460031
Return to citation in text: [1] [2] [3] -
Hollink, E.; Simanek, E. E. Org. Lett. 2006, 8, 2293–2295. doi:10.1021/ol060559p
Return to citation in text: [1] [2] -
Lim, J.; Mintzer, M. A.; Perez, L. M.; Simanek, E. E. Org. Lett. 2010, 12, 1148–1151. doi:10.1021/ol902669g
Return to citation in text: [1] [2] -
Chen, H.-T.; Neerman, M. F.; Parrish, A. R.; Simanek, E. E. J. Am. Chem. Soc. 2004, 126, 10044–10048. doi:10.1021/ja048548j
Return to citation in text: [1] [2] -
Neerman, M. F.; Zhang, W.; Parrish, A. R.; Simanek, E. E. Int. J. Pharm. 2004, 281, 129–132. doi:10.1016/j.ijpharm.2004.04.023
Return to citation in text: [1] [2] -
Lim, J.; Simanek, E. E. Mol. Pharmaceutics 2005, 2, 273–277. doi:10.1021/mp050030e
Return to citation in text: [1] [2] -
Merkel, M. O.; Mintzer, M. A.; Sitterberg, J.; Bakowsky, U.; Simanek, E. E.; Kissel, T. Bioconjugate Chem. 2009, 20, 1799–1806. doi:10.1021/bc900243r
Return to citation in text: [1] [2] -
Lim, J.; Simanek, E. E. Adv. Drug Delivery Rev. 2012, 64, 826–835. doi:10.1016/j.addr.2012.03.008
Return to citation in text: [1] [2] -
Simanek, E. E.; Enciso, A. E.; Pavan, G. M. Expert Opin. Drug Discovery 2013, 8, 1057–1069. doi:10.1517/17460441.2013.813479
Return to citation in text: [1] [2] -
Zhang, W.; Gonzalez, S. O.; Simanek, E. E. Macromolecules 2002, 35, 9015–9021. doi:10.1021/ma021101f
Return to citation in text: [1] [2] -
Acosta, E. J.; Deng, Y.; White, G. N.; Dixon, J. B.; McInnes, K. J.; Senseman, S. A.; Frantzen, A. S.; Simanek, E. E. Chem. Mater. 2003, 15, 2903–2909. doi:10.1021/cm0302328
Return to citation in text: [1] [2] -
Acosta, E. J.; Gonzalez, S. O.; Simanek, E. E. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 168–177. doi:10.1002/pola.20493
Return to citation in text: [1] [2] -
Yoo, Y.; Lunn, D. J.; Gonzalez, S.; Ristich, J. A.; Simanek, E. E.; Shantz, F. D. Chem. Mater. 2006, 18, 2935–2942. doi:10.1021/cm0520564
Return to citation in text: [1] [2] -
Lalwani, S.; Venditto, V. J.; Chouai, A.; Rivera, E. G.; Shaunak, S.; Simanek, E. E. Macromolecules 2009, 42, 3152–3161. doi:10.1021/ma802250c
Return to citation in text: [1] [2] -
Vögtle, F.; Richardt, G.; Werner, N. Dendrimer Chemistry: Concepts, Syntheses, Properties, Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2009; 3, 14, 299–300. doi:10.1002/9783527626953
Return to citation in text: [1] [2] [3] -
Lai, L.-L.; Wang, L.-Y.; Lee, C.-H.; Lin, Y.-C.; Cheng, K.-L. Org. Lett. 2006, 8, 1541–1544. doi:10.1021/ol0531076
Return to citation in text: [1] -
Lai, L.-L.; Lee, C.-H.; Wang, L.-Y.; Cheng, K.-L.; Hsu, H.-F. J. Org. Chem. 2008, 73, 485–490. doi:10.1021/jo701990w
Return to citation in text: [1] -
Lai, L.-L.; Wang, S.-W.; Cheng, K.-L.; Lee, J.-J.; Wang, T.-H.; Hsu, H.-F. Chem. – Eur. J. 2012, 18, 15361–15367. doi:10.1002/chem.201200933
Return to citation in text: [1] -
Beltrán, E.; Serrano, J. L.; Sierra, T.; Giménez, R. Org. Lett. 2010, 12, 1404–1407. doi:10.1021/ol902900y
Return to citation in text: [1] -
Yagai, S.; Usui, M.; Seki, T.; Murayama, H.; Kikkawa, Y.; Uemura, S.; Karatsu, T.; Kitamura, A.; Asano, A.; Seki, S. J. Am. Chem. Soc. 2012, 134, 7983–7994. doi:10.1021/ja302574b
Return to citation in text: [1] [2] [3] -
Shi, Z.; Lu, H.; Chen, Z.; Cheng, R.; Chen, D. Polymer 2012, 53, 359–369. doi:10.1016/j.polymer.2011.11.047
Return to citation in text: [1] [2] -
Kim, D.-Y.; Lee, S.-A.; Choi, Y.-J.; Hwang, S.-H.; Kuo, S.-W.; Nah, C.; Lee, M.-H.; Jeong, K.-U. Chem. – Eur. J. 2014, 20, 5689–5695. doi:10.1002/chem.201303924
Return to citation in text: [1] [2] -
Okuno, T.; Izuoka, A.; Ito, T.; Kubo, S.; Sugawara, T.; Sato, N.; Sugawara, Y. J. Chem. Soc., Perkin Trans. 2 1998, 889–896. doi:10.1039/a707737h
Return to citation in text: [1] [2] -
López de Murillas, D.; Piñol, R.; Ros, M. B.; Serrano, J. L.; Sierra, T.; de la Fuente, M. R. J. Mater. Chem. 2004, 14, 1117–1127. doi:10.1039/B310955K
Return to citation in text: [1] [2] -
Li, W.; Bu, W.; Li, H.; Wu, L.; Lib, M. Chem. Commun. 2005, 3785–3787. doi:10.1039/b503550c
Return to citation in text: [1] [2] -
Jeong, M. J.; Park, J. H.; Lee, C.; Chang, J. Y. Org. Lett. 2006, 8, 2221–2224. doi:10.1021/ol060479l
Return to citation in text: [1] [2] -
Pintea, M.; Fazekas, M.; Lameiras, P.; Cadis, I.; Berghian, C.; Silaghi-Dumitrescu, I.; Popa, F.; Bele, C.; Plé, N.; Darabantu, M. Tetrahedron 2008, 64, 8851–8870. doi:10.1016/j.tet.2008.06.071
Return to citation in text: [1] [2] [3] -
Popa, F.; Lameiras, P.; Moldovan, O.; Tomoaia-Cotisel, M.; Hénon, E.; Martinez, A.; Sacalis, C.; Mocanu, A.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2012, 68, 8945–8967. doi:10.1016/j.tet.2012.07.096
Return to citation in text: [1] [2] [3] [4] -
Moldovan, O.; Lameiras, P.; Nagy, I.; Opruta, T.; Popa, F.; Antheaume, C.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2013, 69, 2199–2213. doi:10.1016/j.tet.2012.12.059
Return to citation in text: [1] [2] -
Moldovan, O.; Nagy, I.; Lameiras, P.; Antheaume, C.; Sacalis, C.; Darabantu, M. Tetrahedron: Asymmetry 2015, 26, 683–701. doi:10.1016/j.tetasy.2015.05.003
Return to citation in text: [1] [2] [3] -
Lates, V.; Gligor, D.; Darabantu, M.; Muresan, L. M. J. Appl. Electrochem. 2007, 37, 631–636. doi:10.1007/s10800-007-9293-5
Return to citation in text: [1] -
Deac, A. R.; Morar, C.; Turdean, G. L.; Darabantu, M.; Gál, E.; Bende, A.; Muresan, L. M. J. Solid State Electrochem. 2016, 20, 3071–3081. doi:10.1007/s10008-016-3298-0
Return to citation in text: [1] [2] [3] [4] -
Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V. K. J. Org. Chem. 1985, 50, 2003–2004. doi:10.1021/jo00211a052
Return to citation in text: [1] -
Hernandez, B. A.; Chang, V.; Villanueva, I.; Heagy, M. D. J. Org. Chem. 1999, 64, 6905–6906. doi:10.1021/jo990560k
Return to citation in text: [1] -
Ricken, S.; Osinski, P. W.; Eilbracht, P.; Haag, R. J. Mol. Catal. A: Chem. 2006, 257, 78–88. doi:10.1016/j.molcata.2006.04.037
Return to citation in text: [1] -
Negoro, M.; Kawata, K. Process for Producing 2,4,6-Tris(hydroxyphenylamino)-1,3,5-triazines and 2,4,6-Tris(substituted phenylamino)-1,3,5-triazines. U.S. Patent 2005/0209453 A1, Sept 22, 2005.
Return to citation in text: [1] [2] -
de Hoog, P.; Gamez, P.; Driessen, W. L.; Reedijk, J. Tetrahedron Lett. 2002, 43, 6783–6786. doi:10.1016/S0040-4039(02)01498-3
Return to citation in text: [1] [2] -
Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048
Return to citation in text: [1] [2] [3] [4] [5] -
Katritzky, A. R.; Oniciu, D. C.; Ghiviriga, I.; Barkock, A. J. Chem. Soc., Perkin Trans. 2 1995, 785–792. doi:10.1039/P29950000785
Return to citation in text: [1] -
Katritzky, A. R.; Ghiviriga, I.; Steel, P. G.; Oniciu, D. C. J. Chem. Soc., Perkin Trans. 2 1996, 443–447. doi:10.1039/p29960000443
Return to citation in text: [1] -
Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G
Return to citation in text: [1] [2] [3] [4] [5] -
Fazekas, M.; Pintea, M.; Lameiras, P.; Lesur, A.; Berghian, C.; Silaghi-Dumitrescu, I.; Plé, N.; Darabantu, M. Eur. J. Org. Chem. 2008, 2473–2494. doi:10.1002/ejoc.200700847
Return to citation in text: [1] [2] [3] -
Moreno, X. K.; Simanek, E. E. Macromolecules 2008, 41, 4108–4114. doi:10.1021/ma702143f
Return to citation in text: [1] [2] -
Bhat, H. R.; Singh, U. P.; Gahtori, P.; Ghosh, S. K.; Gogoi, K.; Prakash, A.; Singh, R. K. New J. Chem. 2013, 37, 2654–2662. doi:10.1039/c3nj00317e
Return to citation in text: [1] -
Bhat, H. R.; Singh, U. P.; Gahtori, P.; Ghosh, S. K.; Gogoi, K.; Prakash, A.; Singh, R. K. RSC Adv. 2013, 3, 2942–2952. doi:10.1039/c2ra21915h
Return to citation in text: [1] -
Enciso, A. E.; Ramirez-Crescencio, F.; Zeiser, M.; Redón, R.; Simanek, E. E. Polym. Chem. 2015, 6, 5219–5224. doi:10.1039/C5PY00899A
Return to citation in text: [1] [2] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] [2] -
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a
Return to citation in text: [1] [2] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] [2] -
Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009
Return to citation in text: [1] [2] [3] -
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n
Return to citation in text: [1] -
Foster, J. P.; Weinhold, F. J. Am. Chem. Soc. 1980, 102, 7211–7218. doi:10.1021/ja00544a007
Return to citation in text: [1] [2] -
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735–746. doi:10.1063/1.449486
Return to citation in text: [1] -
de Gennes, P. G.; Hervet, H. J. Phys., Lett. 1983, 44, 351–360. doi:10.1051/jphyslet:01983004409035100
Return to citation in text: [1] [2] -
Tomalia, D. A. Aldrichimica Acta 2004, 37, 39–57.
Return to citation in text: [1] [2] -
Friebolin, H. Basic One- and Two Dimensional NMR Spectroscopy; VCH Verlagsgesellschaft: Weinheim, New York, 1991; 147–165, 263–291.
Return to citation in text: [1] [2] -
Eliel, E. L.; Wilen, H. S. Stereochemistry of the Organic Compounds; John Wiley & Sons: New York, 1994; pp 642 ff.
Return to citation in text: [1] -
Kessler, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 512–523. doi:10.1002/anie.198205121
Angew. Chem. 1982, 94, 509–520. doi:10.1002/ange.19820940705
Return to citation in text: [1] -
Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A
Return to citation in text: [1] [2] [3] [4] [5] -
Stewart, W. E.; Siddall, T. H. Chem. Rev. 1970, 70, 517–551. doi:10.1021/cr60267a001
Return to citation in text: [1] -
Parker, F. S. Applications of Infrared Spectroscopy in Biochemistry, Biology and Medicine; Plenum Press: New York, 1971; pp 35 ff. doi:10.1007/978-1-4684-1872-9
Return to citation in text: [1] [2] -
Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. Identification Spectrométrique de Composés Organiques; De Boeck & Larcier s.a., 2007; pp 95–96, 101.
Return to citation in text: [1] [2] [3] -
Heacock, R. A.; Marion, L. Can. J. Chem. 1956, 34, 1782–1795. doi:10.1139/v56-231
Return to citation in text: [1] -
Simanek, E. E.; Enciso, A. E. CHAPTER 10 Cationic Triazine Dendrimers: Synthesis, Characterization, and Biological Applications. Cationic Polymers in Regenerative Medicine; The Royal Society of Chemistry, 2015; pp 249–267. doi:10.1039/9781782620105-00249
Return to citation in text: [1] -
van Dongen, M. A.; Orr, B. G.; Banaszak Holl, M. M. J. Phys. Chem. B 2014, 118, 7195–7202. doi:10.1021/jp504059p
Return to citation in text: [1] -
Alvarez, S. Dalton Trans. 2013, 42, 8617–8636. doi:10.1039/c3dt50599e
Return to citation in text: [1] -
Bhatia, S. Nanoparticles types, classification, characterization, fabrication methods and drug delivery applications. Natural Polymer Drug Delivery Systems; Springer International Publishing: Switzerland , 2016; pp 33–93. doi:10.1007/978-3-319-41129-3_2
Return to citation in text: [1] -
Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525–5534. doi:10.1021/ja00170a016
Return to citation in text: [1]
| 30. | Pintea, M.; Fazekas, M.; Lameiras, P.; Cadis, I.; Berghian, C.; Silaghi-Dumitrescu, I.; Popa, F.; Bele, C.; Plé, N.; Darabantu, M. Tetrahedron 2008, 64, 8851–8870. doi:10.1016/j.tet.2008.06.071 |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 45. | Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G |
| 46. | Fazekas, M.; Pintea, M.; Lameiras, P.; Lesur, A.; Berghian, C.; Silaghi-Dumitrescu, I.; Plé, N.; Darabantu, M. Eur. J. Org. Chem. 2008, 2473–2494. doi:10.1002/ejoc.200700847 |
| 47. | Moreno, X. K.; Simanek, E. E. Macromolecules 2008, 41, 4108–4114. doi:10.1021/ma702143f |
| 31. | Popa, F.; Lameiras, P.; Moldovan, O.; Tomoaia-Cotisel, M.; Hénon, E.; Martinez, A.; Sacalis, C.; Mocanu, A.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2012, 68, 8945–8967. doi:10.1016/j.tet.2012.07.096 |
| 72. | Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525–5534. doi:10.1021/ja00170a016 |
| 41. | de Hoog, P.; Gamez, P.; Driessen, W. L.; Reedijk, J. Tetrahedron Lett. 2002, 43, 6783–6786. doi:10.1016/S0040-4039(02)01498-3 |
| 42. | Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048 |
| 24. | Shi, Z.; Lu, H.; Chen, Z.; Cheng, R.; Chen, D. Polymer 2012, 53, 359–369. doi:10.1016/j.polymer.2011.11.047 |
| 26. | Okuno, T.; Izuoka, A.; Ito, T.; Kubo, S.; Sugawara, T.; Sato, N.; Sugawara, Y. J. Chem. Soc., Perkin Trans. 2 1998, 889–896. doi:10.1039/a707737h |
| 28. | Li, W.; Bu, W.; Li, H.; Wu, L.; Lib, M. Chem. Commun. 2005, 3785–3787. doi:10.1039/b503550c |
| 42. | Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048 |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 48. | Bhat, H. R.; Singh, U. P.; Gahtori, P.; Ghosh, S. K.; Gogoi, K.; Prakash, A.; Singh, R. K. New J. Chem. 2013, 37, 2654–2662. doi:10.1039/c3nj00317e |
| 49. | Bhat, H. R.; Singh, U. P.; Gahtori, P.; Ghosh, S. K.; Gogoi, K.; Prakash, A.; Singh, R. K. RSC Adv. 2013, 3, 2942–2952. doi:10.1039/c2ra21915h |
| 23. | Yagai, S.; Usui, M.; Seki, T.; Murayama, H.; Kikkawa, Y.; Uemura, S.; Karatsu, T.; Kitamura, A.; Asano, A.; Seki, S. J. Am. Chem. Soc. 2012, 134, 7983–7994. doi:10.1021/ja302574b |
| 25. | Kim, D.-Y.; Lee, S.-A.; Choi, Y.-J.; Hwang, S.-H.; Kuo, S.-W.; Nah, C.; Lee, M.-H.; Jeong, K.-U. Chem. – Eur. J. 2014, 20, 5689–5695. doi:10.1002/chem.201303924 |
| 27. | López de Murillas, D.; Piñol, R.; Ros, M. B.; Serrano, J. L.; Sierra, T.; de la Fuente, M. R. J. Mater. Chem. 2004, 14, 1117–1127. doi:10.1039/B310955K |
| 29. | Jeong, M. J.; Park, J. H.; Lee, C.; Chang, J. Y. Org. Lett. 2006, 8, 2221–2224. doi:10.1021/ol060479l |
| 33. | Moldovan, O.; Nagy, I.; Lameiras, P.; Antheaume, C.; Sacalis, C.; Darabantu, M. Tetrahedron: Asymmetry 2015, 26, 683–701. doi:10.1016/j.tetasy.2015.05.003 |
| 23. | Yagai, S.; Usui, M.; Seki, T.; Murayama, H.; Kikkawa, Y.; Uemura, S.; Karatsu, T.; Kitamura, A.; Asano, A.; Seki, S. J. Am. Chem. Soc. 2012, 134, 7983–7994. doi:10.1021/ja302574b |
| 50. | Enciso, A. E.; Ramirez-Crescencio, F.; Zeiser, M.; Redón, R.; Simanek, E. E. Polym. Chem. 2015, 6, 5219–5224. doi:10.1039/C5PY00899A |
| 50. | Enciso, A. E.; Ramirez-Crescencio, F.; Zeiser, M.; Redón, R.; Simanek, E. E. Polym. Chem. 2015, 6, 5219–5224. doi:10.1039/C5PY00899A |
| 51. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 52. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 53. | Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009. |
| 54. | Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009 |
| 55. | Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n |
| 56. | Foster, J. P.; Weinhold, F. J. Am. Chem. Soc. 1980, 102, 7211–7218. doi:10.1021/ja00544a007 |
| 51. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 52. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 53. | Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009. |
| 54. | Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009 |
| 45. | Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G |
| 46. | Fazekas, M.; Pintea, M.; Lameiras, P.; Lesur, A.; Berghian, C.; Silaghi-Dumitrescu, I.; Plé, N.; Darabantu, M. Eur. J. Org. Chem. 2008, 2473–2494. doi:10.1002/ejoc.200700847 |
| 18. | Vögtle, F.; Richardt, G.; Werner, N. Dendrimer Chemistry: Concepts, Syntheses, Properties, Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2009; 3, 14, 299–300. doi:10.1002/9783527626953 |
| 58. | de Gennes, P. G.; Hervet, H. J. Phys., Lett. 1983, 44, 351–360. doi:10.1051/jphyslet:01983004409035100 |
| 59. | Tomalia, D. A. Aldrichimica Acta 2004, 37, 39–57. |
| 18. | Vögtle, F.; Richardt, G.; Werner, N. Dendrimer Chemistry: Concepts, Syntheses, Properties, Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2009; 3, 14, 299–300. doi:10.1002/9783527626953 |
| 58. | de Gennes, P. G.; Hervet, H. J. Phys., Lett. 1983, 44, 351–360. doi:10.1051/jphyslet:01983004409035100 |
| 59. | Tomalia, D. A. Aldrichimica Acta 2004, 37, 39–57. |
| 60. | Friebolin, H. Basic One- and Two Dimensional NMR Spectroscopy; VCH Verlagsgesellschaft: Weinheim, New York, 1991; 147–165, 263–291. |
| 61. | Eliel, E. L.; Wilen, H. S. Stereochemistry of the Organic Compounds; John Wiley & Sons: New York, 1994; pp 642 ff. |
| 30. | Pintea, M.; Fazekas, M.; Lameiras, P.; Cadis, I.; Berghian, C.; Silaghi-Dumitrescu, I.; Popa, F.; Bele, C.; Plé, N.; Darabantu, M. Tetrahedron 2008, 64, 8851–8870. doi:10.1016/j.tet.2008.06.071 |
| 31. | Popa, F.; Lameiras, P.; Moldovan, O.; Tomoaia-Cotisel, M.; Hénon, E.; Martinez, A.; Sacalis, C.; Mocanu, A.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2012, 68, 8945–8967. doi:10.1016/j.tet.2012.07.096 |
| 32. | Moldovan, O.; Lameiras, P.; Nagy, I.; Opruta, T.; Popa, F.; Antheaume, C.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2013, 69, 2199–2213. doi:10.1016/j.tet.2012.12.059 |
| 33. | Moldovan, O.; Nagy, I.; Lameiras, P.; Antheaume, C.; Sacalis, C.; Darabantu, M. Tetrahedron: Asymmetry 2015, 26, 683–701. doi:10.1016/j.tetasy.2015.05.003 |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 45. | Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G |
| 46. | Fazekas, M.; Pintea, M.; Lameiras, P.; Lesur, A.; Berghian, C.; Silaghi-Dumitrescu, I.; Plé, N.; Darabantu, M. Eur. J. Org. Chem. 2008, 2473–2494. doi:10.1002/ejoc.200700847 |
| 35. | Deac, A. R.; Morar, C.; Turdean, G. L.; Darabantu, M.; Gál, E.; Bende, A.; Muresan, L. M. J. Solid State Electrochem. 2016, 20, 3071–3081. doi:10.1007/s10008-016-3298-0 |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 54. | Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009 |
| 56. | Foster, J. P.; Weinhold, F. J. Am. Chem. Soc. 1980, 102, 7211–7218. doi:10.1021/ja00544a007 |
| 57. | Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735–746. doi:10.1063/1.449486 |
| 62. |
Kessler, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 512–523. doi:10.1002/anie.198205121
Angew. Chem. 1982, 94, 509–520. doi:10.1002/ange.19820940705 |
| 47. | Moreno, X. K.; Simanek, E. E. Macromolecules 2008, 41, 4108–4114. doi:10.1021/ma702143f |
| 31. | Popa, F.; Lameiras, P.; Moldovan, O.; Tomoaia-Cotisel, M.; Hénon, E.; Martinez, A.; Sacalis, C.; Mocanu, A.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2012, 68, 8945–8967. doi:10.1016/j.tet.2012.07.096 |
| 4. |
Steffensen, M. B.; Simanek, E. E. Angew. Chem., Int. Ed. 2004, 43, 5178–5180. doi:10.1002/anie.200460031
Angew. Chem. 2004, 39, 5290–5292. doi:10.1002/ange.200460031 |
| 7. | Chen, H.-T.; Neerman, M. F.; Parrish, A. R.; Simanek, E. E. J. Am. Chem. Soc. 2004, 126, 10044–10048. doi:10.1021/ja048548j |
| 8. | Neerman, M. F.; Zhang, W.; Parrish, A. R.; Simanek, E. E. Int. J. Pharm. 2004, 281, 129–132. doi:10.1016/j.ijpharm.2004.04.023 |
| 9. | Lim, J.; Simanek, E. E. Mol. Pharmaceutics 2005, 2, 273–277. doi:10.1021/mp050030e |
| 10. | Merkel, M. O.; Mintzer, M. A.; Sitterberg, J.; Bakowsky, U.; Simanek, E. E.; Kissel, T. Bioconjugate Chem. 2009, 20, 1799–1806. doi:10.1021/bc900243r |
| 11. | Lim, J.; Simanek, E. E. Adv. Drug Delivery Rev. 2012, 64, 826–835. doi:10.1016/j.addr.2012.03.008 |
| 12. | Simanek, E. E.; Enciso, A. E.; Pavan, G. M. Expert Opin. Drug Discovery 2013, 8, 1057–1069. doi:10.1517/17460441.2013.813479 |
| 34. | Lates, V.; Gligor, D.; Darabantu, M.; Muresan, L. M. J. Appl. Electrochem. 2007, 37, 631–636. doi:10.1007/s10800-007-9293-5 |
| 35. | Deac, A. R.; Morar, C.; Turdean, G. L.; Darabantu, M.; Gál, E.; Bende, A.; Muresan, L. M. J. Solid State Electrochem. 2016, 20, 3071–3081. doi:10.1007/s10008-016-3298-0 |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 63. | Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A |
| 1. | Zhang, W.; Simanek, E. E. Org. Lett. 2000, 2, 843–845. doi:10.1021/ol005585g |
| 5. | Hollink, E.; Simanek, E. E. Org. Lett. 2006, 8, 2293–2295. doi:10.1021/ol060559p |
| 6. | Lim, J.; Mintzer, M. A.; Perez, L. M.; Simanek, E. E. Org. Lett. 2010, 12, 1148–1151. doi:10.1021/ol902669g |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 1. | Zhang, W.; Simanek, E. E. Org. Lett. 2000, 2, 843–845. doi:10.1021/ol005585g |
| 3. | Zhang, W.; Nowlan, D. T.; Thomson, L. M.; Lackowski, M. W.; Simanek, E. E. J. Am. Chem. Soc. 2001, 123, 8914–8922. doi:10.1021/ja0041369 |
| 4. |
Steffensen, M. B.; Simanek, E. E. Angew. Chem., Int. Ed. 2004, 43, 5178–5180. doi:10.1002/anie.200460031
Angew. Chem. 2004, 39, 5290–5292. doi:10.1002/ange.200460031 |
| 26. | Okuno, T.; Izuoka, A.; Ito, T.; Kubo, S.; Sugawara, T.; Sato, N.; Sugawara, Y. J. Chem. Soc., Perkin Trans. 2 1998, 889–896. doi:10.1039/a707737h |
| 27. | López de Murillas, D.; Piñol, R.; Ros, M. B.; Serrano, J. L.; Sierra, T.; de la Fuente, M. R. J. Mater. Chem. 2004, 14, 1117–1127. doi:10.1039/B310955K |
| 28. | Li, W.; Bu, W.; Li, H.; Wu, L.; Lib, M. Chem. Commun. 2005, 3785–3787. doi:10.1039/b503550c |
| 29. | Jeong, M. J.; Park, J. H.; Lee, C.; Chang, J. Y. Org. Lett. 2006, 8, 2221–2224. doi:10.1021/ol060479l |
| 63. | Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A |
| 2. | Takagi, K.; Hattori, T.; Kunisada, H.; Yuki, Y. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 4385–4395. doi:10.1002/1099-0518(20001215)38:24<4385::AID-POLA70>3.0.CO;2-Y |
| 30. | Pintea, M.; Fazekas, M.; Lameiras, P.; Cadis, I.; Berghian, C.; Silaghi-Dumitrescu, I.; Popa, F.; Bele, C.; Plé, N.; Darabantu, M. Tetrahedron 2008, 64, 8851–8870. doi:10.1016/j.tet.2008.06.071 |
| 31. | Popa, F.; Lameiras, P.; Moldovan, O.; Tomoaia-Cotisel, M.; Hénon, E.; Martinez, A.; Sacalis, C.; Mocanu, A.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2012, 68, 8945–8967. doi:10.1016/j.tet.2012.07.096 |
| 32. | Moldovan, O.; Lameiras, P.; Nagy, I.; Opruta, T.; Popa, F.; Antheaume, C.; Ramondenc, Y.; Darabantu, M. Tetrahedron 2013, 69, 2199–2213. doi:10.1016/j.tet.2012.12.059 |
| 33. | Moldovan, O.; Nagy, I.; Lameiras, P.; Antheaume, C.; Sacalis, C.; Darabantu, M. Tetrahedron: Asymmetry 2015, 26, 683–701. doi:10.1016/j.tetasy.2015.05.003 |
| 64. | Stewart, W. E.; Siddall, T. H. Chem. Rev. 1970, 70, 517–551. doi:10.1021/cr60267a001 |
| 22. | Beltrán, E.; Serrano, J. L.; Sierra, T.; Giménez, R. Org. Lett. 2010, 12, 1404–1407. doi:10.1021/ol902900y |
| 24. | Shi, Z.; Lu, H.; Chen, Z.; Cheng, R.; Chen, D. Polymer 2012, 53, 359–369. doi:10.1016/j.polymer.2011.11.047 |
| 42. | Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048 |
| 19. | Lai, L.-L.; Wang, L.-Y.; Lee, C.-H.; Lin, Y.-C.; Cheng, K.-L. Org. Lett. 2006, 8, 1541–1544. doi:10.1021/ol0531076 |
| 20. | Lai, L.-L.; Lee, C.-H.; Wang, L.-Y.; Cheng, K.-L.; Hsu, H.-F. J. Org. Chem. 2008, 73, 485–490. doi:10.1021/jo701990w |
| 21. | Lai, L.-L.; Wang, S.-W.; Cheng, K.-L.; Lee, J.-J.; Wang, T.-H.; Hsu, H.-F. Chem. – Eur. J. 2012, 18, 15361–15367. doi:10.1002/chem.201200933 |
| 25. | Kim, D.-Y.; Lee, S.-A.; Choi, Y.-J.; Hwang, S.-H.; Kuo, S.-W.; Nah, C.; Lee, M.-H.; Jeong, K.-U. Chem. – Eur. J. 2014, 20, 5689–5695. doi:10.1002/chem.201303924 |
| 63. | Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A |
| 18. | Vögtle, F.; Richardt, G.; Werner, N. Dendrimer Chemistry: Concepts, Syntheses, Properties, Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2009; 3, 14, 299–300. doi:10.1002/9783527626953 |
| 45. | Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G |
| 13. | Zhang, W.; Gonzalez, S. O.; Simanek, E. E. Macromolecules 2002, 35, 9015–9021. doi:10.1021/ma021101f |
| 14. | Acosta, E. J.; Deng, Y.; White, G. N.; Dixon, J. B.; McInnes, K. J.; Senseman, S. A.; Frantzen, A. S.; Simanek, E. E. Chem. Mater. 2003, 15, 2903–2909. doi:10.1021/cm0302328 |
| 15. | Acosta, E. J.; Gonzalez, S. O.; Simanek, E. E. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 168–177. doi:10.1002/pola.20493 |
| 16. | Yoo, Y.; Lunn, D. J.; Gonzalez, S.; Ristich, J. A.; Simanek, E. E.; Shantz, F. D. Chem. Mater. 2006, 18, 2935–2942. doi:10.1021/cm0520564 |
| 17. | Lalwani, S.; Venditto, V. J.; Chouai, A.; Rivera, E. G.; Shaunak, S.; Simanek, E. E. Macromolecules 2009, 42, 3152–3161. doi:10.1021/ma802250c |
| 23. | Yagai, S.; Usui, M.; Seki, T.; Murayama, H.; Kikkawa, Y.; Uemura, S.; Karatsu, T.; Kitamura, A.; Asano, A.; Seki, S. J. Am. Chem. Soc. 2012, 134, 7983–7994. doi:10.1021/ja302574b |
| 42. | Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048 |
| 37. | Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V. K. J. Org. Chem. 1985, 50, 2003–2004. doi:10.1021/jo00211a052 |
| 35. | Deac, A. R.; Morar, C.; Turdean, G. L.; Darabantu, M.; Gál, E.; Bende, A.; Muresan, L. M. J. Solid State Electrochem. 2016, 20, 3071–3081. doi:10.1007/s10008-016-3298-0 |
| 1. | Zhang, W.; Simanek, E. E. Org. Lett. 2000, 2, 843–845. doi:10.1021/ol005585g |
| 2. | Takagi, K.; Hattori, T.; Kunisada, H.; Yuki, Y. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 4385–4395. doi:10.1002/1099-0518(20001215)38:24<4385::AID-POLA70>3.0.CO;2-Y |
| 3. | Zhang, W.; Nowlan, D. T.; Thomson, L. M.; Lackowski, M. W.; Simanek, E. E. J. Am. Chem. Soc. 2001, 123, 8914–8922. doi:10.1021/ja0041369 |
| 4. |
Steffensen, M. B.; Simanek, E. E. Angew. Chem., Int. Ed. 2004, 43, 5178–5180. doi:10.1002/anie.200460031
Angew. Chem. 2004, 39, 5290–5292. doi:10.1002/ange.200460031 |
| 5. | Hollink, E.; Simanek, E. E. Org. Lett. 2006, 8, 2293–2295. doi:10.1021/ol060559p |
| 6. | Lim, J.; Mintzer, M. A.; Perez, L. M.; Simanek, E. E. Org. Lett. 2010, 12, 1148–1151. doi:10.1021/ol902669g |
| 7. | Chen, H.-T.; Neerman, M. F.; Parrish, A. R.; Simanek, E. E. J. Am. Chem. Soc. 2004, 126, 10044–10048. doi:10.1021/ja048548j |
| 8. | Neerman, M. F.; Zhang, W.; Parrish, A. R.; Simanek, E. E. Int. J. Pharm. 2004, 281, 129–132. doi:10.1016/j.ijpharm.2004.04.023 |
| 9. | Lim, J.; Simanek, E. E. Mol. Pharmaceutics 2005, 2, 273–277. doi:10.1021/mp050030e |
| 10. | Merkel, M. O.; Mintzer, M. A.; Sitterberg, J.; Bakowsky, U.; Simanek, E. E.; Kissel, T. Bioconjugate Chem. 2009, 20, 1799–1806. doi:10.1021/bc900243r |
| 11. | Lim, J.; Simanek, E. E. Adv. Drug Delivery Rev. 2012, 64, 826–835. doi:10.1016/j.addr.2012.03.008 |
| 12. | Simanek, E. E.; Enciso, A. E.; Pavan, G. M. Expert Opin. Drug Discovery 2013, 8, 1057–1069. doi:10.1517/17460441.2013.813479 |
| 13. | Zhang, W.; Gonzalez, S. O.; Simanek, E. E. Macromolecules 2002, 35, 9015–9021. doi:10.1021/ma021101f |
| 14. | Acosta, E. J.; Deng, Y.; White, G. N.; Dixon, J. B.; McInnes, K. J.; Senseman, S. A.; Frantzen, A. S.; Simanek, E. E. Chem. Mater. 2003, 15, 2903–2909. doi:10.1021/cm0302328 |
| 15. | Acosta, E. J.; Gonzalez, S. O.; Simanek, E. E. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 168–177. doi:10.1002/pola.20493 |
| 16. | Yoo, Y.; Lunn, D. J.; Gonzalez, S.; Ristich, J. A.; Simanek, E. E.; Shantz, F. D. Chem. Mater. 2006, 18, 2935–2942. doi:10.1021/cm0520564 |
| 17. | Lalwani, S.; Venditto, V. J.; Chouai, A.; Rivera, E. G.; Shaunak, S.; Simanek, E. E. Macromolecules 2009, 42, 3152–3161. doi:10.1021/ma802250c |
| 65. | Parker, F. S. Applications of Infrared Spectroscopy in Biochemistry, Biology and Medicine; Plenum Press: New York, 1971; pp 35 ff. doi:10.1007/978-1-4684-1872-9 |
| 66. | Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. Identification Spectrométrique de Composés Organiques; De Boeck & Larcier s.a., 2007; pp 95–96, 101. |
| 66. | Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. Identification Spectrométrique de Composés Organiques; De Boeck & Larcier s.a., 2007; pp 95–96, 101. |
| 67. | Heacock, R. A.; Marion, L. Can. J. Chem. 1956, 34, 1782–1795. doi:10.1139/v56-231 |
| 60. | Friebolin, H. Basic One- and Two Dimensional NMR Spectroscopy; VCH Verlagsgesellschaft: Weinheim, New York, 1991; 147–165, 263–291. |
| 63. | Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A |
| 65. | Parker, F. S. Applications of Infrared Spectroscopy in Biochemistry, Biology and Medicine; Plenum Press: New York, 1971; pp 35 ff. doi:10.1007/978-1-4684-1872-9 |
| 66. | Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. Identification Spectrométrique de Composés Organiques; De Boeck & Larcier s.a., 2007; pp 95–96, 101. |
| 42. | Morar, C.; Cost, L.; Lameiras, P.; Antheaume, C.; Darabantu, M. Synth. Commun. 2015, 45, 1688–1695. doi:10.1080/00397911.2015.1041048 |
| 43. | Katritzky, A. R.; Oniciu, D. C.; Ghiviriga, I.; Barkock, A. J. Chem. Soc., Perkin Trans. 2 1995, 785–792. doi:10.1039/P29950000785 |
| 44. | Katritzky, A. R.; Ghiviriga, I.; Steel, P. G.; Oniciu, D. C. J. Chem. Soc., Perkin Trans. 2 1996, 443–447. doi:10.1039/p29960000443 |
| 45. | Ghiviriga, I.; Oniciu, D. C. Chem. Commun. 2002, 2718–2719. doi:10.1039/B206811G |
| 40. | Negoro, M.; Kawata, K. Process for Producing 2,4,6-Tris(hydroxyphenylamino)-1,3,5-triazines and 2,4,6-Tris(substituted phenylamino)-1,3,5-triazines. U.S. Patent 2005/0209453 A1, Sept 22, 2005. |
| 63. | Fedeli, E.; Hernández-Aínsa, S.; Lancelot, A.; González-Pastor, R.; Calvo, P.; Sierra, T.; Serrano, J. L. Soft Matter 2015, 11, 6009–6017. doi:10.1039/C5SM01153A |
| 36. | Morar, C.; Turdean, G. L.; Bende, A.; Lameiras, P.; Antheaume, C.; Muresan, L. M.; Darabantu, M. C. R. Chim. 2017, 20, 402–414. doi:10.1016/j.crci.2016.07.002 |
| 71. | Bhatia, S. Nanoparticles types, classification, characterization, fabrication methods and drug delivery applications. Natural Polymer Drug Delivery Systems; Springer International Publishing: Switzerland , 2016; pp 33–93. doi:10.1007/978-3-319-41129-3_2 |
| 40. | Negoro, M.; Kawata, K. Process for Producing 2,4,6-Tris(hydroxyphenylamino)-1,3,5-triazines and 2,4,6-Tris(substituted phenylamino)-1,3,5-triazines. U.S. Patent 2005/0209453 A1, Sept 22, 2005. |
| 69. | van Dongen, M. A.; Orr, B. G.; Banaszak Holl, M. M. J. Phys. Chem. B 2014, 118, 7195–7202. doi:10.1021/jp504059p |
| 41. | de Hoog, P.; Gamez, P.; Driessen, W. L.; Reedijk, J. Tetrahedron Lett. 2002, 43, 6783–6786. doi:10.1016/S0040-4039(02)01498-3 |
| 38. | Hernandez, B. A.; Chang, V.; Villanueva, I.; Heagy, M. D. J. Org. Chem. 1999, 64, 6905–6906. doi:10.1021/jo990560k |
| 68. | Simanek, E. E.; Enciso, A. E. CHAPTER 10 Cationic Triazine Dendrimers: Synthesis, Characterization, and Biological Applications. Cationic Polymers in Regenerative Medicine; The Royal Society of Chemistry, 2015; pp 249–267. doi:10.1039/9781782620105-00249 |
| 39. | Ricken, S.; Osinski, P. W.; Eilbracht, P.; Haag, R. J. Mol. Catal. A: Chem. 2006, 257, 78–88. doi:10.1016/j.molcata.2006.04.037 |
| 35. | Deac, A. R.; Morar, C.; Turdean, G. L.; Darabantu, M.; Gál, E.; Bende, A.; Muresan, L. M. J. Solid State Electrochem. 2016, 20, 3071–3081. doi:10.1007/s10008-016-3298-0 |
© 2018 Morar et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)