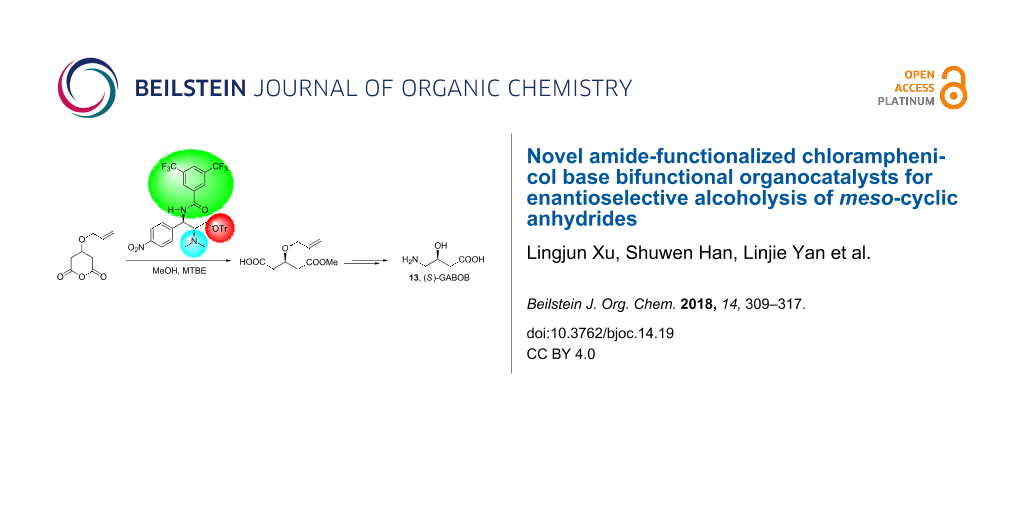
Abstract
A family of novel chloramphenicol base-amide organocatalysts possessing a NH functionality at C-1 position as monodentate hydrogen bond donor were developed and evaluated for enantioselective organocatalytic alcoholysis of meso-cyclic anhydrides. These structural diversified organocatalysts were found to induce high enantioselectivity in alcoholysis of anhydrides and was successfully applied to the asymmetric synthesis of (S)-GABOB.

Graphical Abstract
Introduction
Over the past decade, remarkable advances in the utilization of natural products as chiral structural motifs for the design of bifunctional organocatalysts have been achieved. A high stereocontrol in alcoholytic catalytic asymmetric desymmetrizations of meso-cyclic anhydrides is of special interest [1-7]. Major families originating from natural products include cinchona alkaloids [8-17] and proteinogenic α-amino acids such as proline [18-20] and valine [21] etc. [22-29], while fully synthetic catalysts are rare, only involving Brønsted acid/base catalysts [30], cyclohexanediamine catalysts [31,32], and diphenylethylenediamine catalysts [33,34]. Significant progress in our laboratory has been made in the development of chiral bifunctional urea 1 [35], thiourea 2 and 3 [36,37], sulfonamide 4 [38-40] and squaramide 5 [38-40] catalysts derived from chloramphenicol base (Figure 1), which showed excellent reactivity and enantioselectivity for this asymmetric transformation. A typical example of the great utility of chloramphenicol base scaffold in industrial production of enantiopure compounds is the synthesis of a precursor of biotin (vitamin H) by the chloramphenicol base-promoted asymmetric methanolysis of meso-cyclic anhydrides [41-43].
![[1860-5397-14-19-1]](/bjoc/content/figures/1860-5397-14-19-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chloramphenicol-base-derived bifunctional organocatalysts.
Figure 1: Chloramphenicol-base-derived bifunctional organocatalysts.
Despite extensive studies on chloramphenicol-base-derived bifunctional derivatives catalyzed asymmetric alcoholysis of anhydride, the exploration of new effective and easily accessible fully synthetic organocatalysts through further modification of this privilege motif are always needed. Our present design is inspired by cinchona-derived sulfonamides, first developed by Song and co-workers [44-46]. Because of its easy availability and unique stereochemical aspect, we were therefore intrigued by the possibility of modifying the chloramphenicol base structural backbone by virtue of substituting the hydroxy group at C-1 position by a simple amide moiety as monodentate hydrogen bond donor to activate the electrophile (anhydride), whilst retaining the tertiary amine functionality to activate the nucleophile (alcohol, Figure 2). As part of our ongoing research program on chloramphenicol base organocatalysis, herein, we report a new class of chloramphenicol-derived bifunctional amide organocatalysts and their application for enantioselective alcoholysis of meso-cyclic anhydrides.
![[1860-5397-14-19-2]](/bjoc/content/figures/1860-5397-14-19-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Design of new chloramphenicol base amide organocatalysts.
Figure 2: Design of new chloramphenicol base amide organocatalysts.
In the precedented urea- or thiourea-based organocatalytic methanolysis of anhydrides, one big problem is the homo-aggregation of the catalysts via hydrogen bonding, which decreased the reactivity and enantioselectivity and required diluted concentrations with low temperatures [47]. With the new chloramphenicol base amide organocatalysts we envisioned that the presence of a bulky oxygen group could avoid the intermolecular aggregation of the catalyst, while keeping the desired bifunctional Brønsted acid/base reactivity for enantioselective desymmetrization of anhydrides [48,49].
Results and Discussion
A series of new chloramphenicol based-amide bifunctional catalysts 7a–q (Scheme 1) were synthesized from the appropriate optically pure (1R,2R)-diamine 6, prepared via several steps from chloramphenicol base [50]. In general, the synthesis is straightforward and well-compatible to electronically-diversified amides. Particularly, electron-deficient amides performed better. With 3,5-(CF3)2-disubstituted amide, the corresponding pruduct 7i was achieved with 90% yield. Different protecting groups for the nitrogen at the C-2 position and oxygen at C-3 are also investigated to yield the desired chiral catalysts using a simple procedure.
![[1860-5397-14-19-i1]](/bjoc/content/inline/1860-5397-14-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of bifunctional amide catalysts 7a–q.
Scheme 1: Synthesis of bifunctional amide catalysts 7a–q.
With various bifunctional catalysts in hand, assessment of their catalytic behavior in enantioselective alcoholysis of meso-cyclic anhydride 8a was carried out in various conditions with 10 equivalents of methanol. Resultingly, this novel Brønsted acid/base catalysts 7a–q proved to be sufficiently active and gave the monoester product with high yield and superior enantioselectivity, suggesting the unique reactivity of this amide-based chloramphenicol scaffold.
Electronic effects were first investigated. Generally, electron deficient catalysts gave better reactivity and enantioselectivity than electron-rich variants (Table 1, entries 1–9). Among them, 7i performed best with 95% yield and 92% ee in 17 h (Table 1, entry 9). The surprising reactivity with this electron-poor 7i suggested that the pKa is crucial for this catalysis. Further modifications of the chloramphenicol skeleton with various protecting groups on the nitrogen at C-2 position and oxygen at C-3 did not improve the reaction but with lowered enantioselectivity (Table 1, entries 10–16). Notably, with methyl-protected catalyst 7q, this reaction proceeded with longer reaction time but with decreased enantioselectivity, suggesting the effects of a bulky ether group (Table 1, entry 17). Solvent effects were then evaluated and aprotic solvent performed better than protic solvents (Table 1, entries 18 and 19), which is in accordance with literature precedence [45]. Furthermore, changing concentration or low down the temperature of the reaction didn’t alter the yield or enantioselectivity, suggesting the concentration/temperature independence of this catalytic system (Table 1, entries 20–23). These results implicated that there is no significant aggregation of catalysts in this system.
Table 1: Conditions screening for asymmetric methanolysis of meso-cyclic anhydrides.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-19-i3.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Cat. | Solvent | Conc. (M) | T (°C) | Time (h) | Yielda (%) | eeb,c (%) |
| 1 | 7a | MTBE | 0.05 | 20 | 34 | 91 | 84 |
| 2 | 7b | MTBE | 0.05 | 20 | 36 | 94 | 80 |
| 3 | 7c | MTBE | 0.05 | 20 | 40 | 95 | 77 |
| 4 | 7d | MTBE | 0.05 | 20 | 46 | 94 | 80 |
| 5 | 7e | MTBE | 0.05 | 20 | 48 | 99 | 83 |
| 6 | 7f | MTBE | 0.05 | 20 | 58 | 100 | 78 |
| 7 | 7g | MTBE | 0.05 | 20 | 96 | 99 | 72 |
| 8 | 7h | MTBE | 0.05 | 20 | 48 | 100 | 76 |
| 9 | 7i | MTBE | 0.05 | 20 | 17 | 95 | 92 |
| 10 | 7j | MTBE | 0.05 | 20 | 20 | 85 | 80 |
| 11 | 7k | MTBE | 0.05 | 20 | 58 | 99 | 71 |
| 12 | 7l | MTBE | 0.05 | 20 | 216 | 100 | 58 |
| 13 | 7m | MTBE | 0.05 | 20 | 28 | 95 | 84 |
| 14 | 7n | MTBE | 0.05 | 20 | 24 | 91 | 76 |
| 15 | 7o | MTBE | 0.05 | 20 | 192 | 97 | 82 |
| 16 | 7p | MTBE | 0.05 | 20 | 240 | 98 | 68 |
| 17 | 7q | MTBE | 0.05 | 20 | 58 | 93 | 54 |
| 18 | 7i | Toluene | 0.05 | 20 | 14 | 96 | 79 |
| 19 | 7i | MeOH | 0.05 | 20 | 18 | 91 | 10 |
| 20 | 7i | MTBE | 0.0125 | 20 | 86 | 99 | 94 |
| 21 | 7i | MTBE | 0.1 | 20 | 12 | 95 | 87 |
| 22 | 7i | MTBE | 0.05 | 0 | 120 | 99 | 95 |
| 23 | 7i | MTBE | 0.05 | −20 | 384 | 92 | 96 |
aIsolated yield. bDetermined by HPLC after conversion to amide derivatives with (S)-1-phenylethylamine. cThe absolute configuration was determined by comparing with literature report.
This bifunctional Brønsted acid/base catalyst 7i proved to be highly active toward asymmetric methanolysis of various meso-cyclic anhydrides. As shown in Table 2, various cyclic anhydrides were conveniently converted into the corresponding monoester products in high yields and enantioselectivities. Notably, bicyclic anhydrides (Table 2, entries 1 and 2) and tricyclic anhydrides (Table 2, entries 3 and 4) were more reactive than mono-cyclic anhydrides (Table 2, entries 5–12). In the case of mono-substituted cyclic substrates, including alkyl- (Table 2, entries 5–7), ether- (entry 8), aryl-substitution (entries 9–11), moderate enantioselectivities were detected. Moreover, the chiral mono-substituted products are versatile key building blocks for the synthesis of a variety of the industrially important pharmaceuticals, such as γ-aminobutyric acid (GABA) and γ-amino-β-hydroxybutyric acid (GABOB) analogues (baclofen HCl and pregabalin) [51-54], HMG-CoA reductase inhibitors (“statins”) [55-57], etc.
Table 2: Asymmetric methanolysis of meso-cyclic anhydrides.a,b.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-19-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Anhydride | Product | Time (h) | Yieldc (%) | eed (%) |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-19-i5.svg?max-width=637&scale=1.0)
8a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-19-i6.svg?max-width=637&scale=1.0)
9a |
17 | 98 | 95 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-19-i7.svg?max-width=637&scale=1.0)
8b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-19-i8.svg?max-width=637&scale=1.0)
9b |
60 | 97 | 90 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-19-i9.svg?max-width=637&scale=1.0)
8c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-19-i10.svg?max-width=637&scale=1.0)
9c |
66 | 94 | 73 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-19-i11.svg?max-width=637&scale=1.0)
8d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-19-i12.svg?max-width=637&scale=1.0)
9d |
66 | 95 | 84 |
| 5 e |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-19-i13.svg?max-width=637&scale=1.0)
8e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-19-i14.svg?max-width=637&scale=1.0)
9e |
84 | 87 | 81 |
| 6 e |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-19-i15.svg?max-width=637&scale=1.0)
8f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-19-i16.svg?max-width=637&scale=1.0)
9f |
84 | 91 | 80 |
| 7 e |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-19-i17.svg?max-width=637&scale=1.0)
8g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-19-i18.svg?max-width=637&scale=1.0)
9g |
84 | 90 | 94 |
| 8e |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-19-i19.svg?max-width=637&scale=1.0)
8h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-19-i20.svg?max-width=637&scale=1.0)
9h |
84 | 97 | 81 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-19-i21.svg?max-width=637&scale=1.0)
8i |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-19-i22.svg?max-width=637&scale=1.0)
9i |
60 | 89 | 77 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-19-i23.svg?max-width=637&scale=1.0)
8j |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-19-i24.svg?max-width=637&scale=1.0)
9j |
72 | 87 | 78 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-19-i25.svg?max-width=637&scale=1.0)
8k |
![[Graphic 24]](/bjoc/content/inline/1860-5397-14-19-i26.svg?max-width=637&scale=1.0)
9k |
84 | 92 | 79 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-14-19-i27.svg?max-width=637&scale=1.0)
8l |
![[Graphic 26]](/bjoc/content/inline/1860-5397-14-19-i28.svg?max-width=637&scale=1.0)
9l |
96 | 91 | 80 |
aUnless otherwise noted, reactions were carried out with anhydride (0.5 mmol), MeOH (5.0 mmol), and 7i (10 mol %) in MTBE (10 mL) at rt. bAbsolute configuration was determined by comparing with literature report. cIsolated yield. dDetermined by HPLC after after derivatization. eIn MTBE (40 mL) at 0 °C.
The generality and scope of this methodology was further demonstrated in the alcoholysis of 8a with different alcohols (Table 3). Excellent yields with high enantioselectivities were obtained in all cases. The sterically more bulky 2-propanol worked well in this reaction. Allylic alcohol, benzyl alcohol and cinnamyl alcohol were also well compatible in this bifunctional organocatalysis conditions to furnish the desired products.
Table 3: Asymmetric alcoholysis of anhydride 8a with various alcohols.a
![[Graphic 27]](/bjoc/content/inline/1860-5397-14-19-i29.svg?max-width=637&scale=1.0)
|
|||||
| Entry | ROH | Monoester | Time (h) | Yieldb (%) | eec,d (%) |
| 1 | methanol | 9a | 17 | 98 | 95 |
| 2 | ethanol | 9m | 96 | 97 | 93 |
| 3 | 2-propanol | 9n | 98 | 93 | 89 |
| 4 | allylic alcohol | 9o | 84 | 90 | 85 |
| 5 | benzyl alcohol | 9p | 66 | 98 | 86 |
| 6 | cinamyl alcohol | 9q | 66 | 99 | 90 |
aUnless otherwise noted, above reactions were carried out with anhydride (0.5 mmol), MeOH (5.0 mmol), and catalyst 7i (10 mol %) in MTBE (40 mL) at rt. bIsolated yield. cDetermined by HPLC after derivatization. dAbsolute configuration was determined by comparing with the literature report.
Application of this methodology to the synthesis of (S)-GABOB (13), a metabolic derivative of the neurotransmitter γ-aminobutanoic acid, was performed to illustrate the synthetic utility [58,59] (Scheme 2). From commercially available diethyl 3-hydroxyglutarate (10), anhydride 8h was prepared in three steps in 54% overall yield. The crucial chloramphenicol base amide 7i catalyzed enantioselective alcoholysis gave monoester 9h in multigram scale. The resulting monoester with the desired stereocenter at C-3 was converted to triprotected derivative of GABOB 11 via Curtius rearrangement in 75% yield over two steps. After Pd/C-catalyzed hydrogenative deprotection of hydroxy group in methanol under acidic conditions, the corresponding alcohol 12 was transferred to the product 13 via acid-promoted deprotection. A single recrystallization from ethanol allowed 13 to be isolated with 96% ee and 73% yield.
![[1860-5397-14-19-i2]](/bjoc/content/inline/1860-5397-14-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Asymmetric synthesis of (S)-GABOB (13).
Scheme 2: Asymmetric synthesis of (S)-GABOB (13).
Conclusion
In conclusion, we developed a unique bifunctional chiral organocatalyst via introducing a simple amide functionality at C-1 of the easily available chloramphenicol scaffold, which shows good catalytic reactivity and enantioselectivity in the alcoholytic desymmetrization of meso-cyclic anhydrides. This method proved to be generally applicable on various anhydrides or alcohols to generate the desired monoesters in good yield and enantioselectivity. The application of this method to the synthesis of (S)-GABOB demonstrated the great synthetic potential for pharmaceutically useful chemicals. Further modification of this chloramphenicol scaffold as well as total synthesis of other natural chemicals are right now ongoing in our lab.
Experimental
General procedure for the synthesis of 9
Similarly as described in [37], with stirring under an atmosphere of nitrogen, alcohol (5 mmol, 10 equiv) was added dropwise to a solution of an anhydride 8 (0.5 mmol, 1 equiv) and 7i (0.05 mmol, 10 mol %) in MTBE (20 mL) at room temperature. The reaction was monitored by using thin-layer chromatography. Once the reaction was completed, the solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2 (10 mL). The solution was successively washed with saturated Na2CO3 (2 × 5 mL), acidified with excess 2 N HCl, followed by extraction with EtOAc (3 × 10 mL). The combined organic phase were dried over Na2SO4 and concentrated to afford the corresponding monoester, without further purification by flash chromatography.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, 1H NMR files. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Borissov, A.; Davies, T. Q.; Ellis, S. R.; Fleming, T. A.; Richardson, M. S. W.; Dixon, D. J. Chem. Soc. Rev. 2016, 45, 5474. doi:10.1039/C5CS00015G
Return to citation in text: [1] -
Zheng, C.; Chen, F.-E. Chin. Chem. Lett. 2014, 25, 1. doi:10.1016/j.cclet.2013.11.025
Return to citation in text: [1] -
Rodriguez-Docampo, Z.; Connon, S. J. ChemCatChem 2012, 4, 151. doi:10.1002/cctc.201100266
Return to citation in text: [1] -
Díaz de Villegas, M. D.; Gálvez, J. A.; Etayo, P.; Badorrey, R.; López-Ram-de-Víu, P. Chem. Soc. Rev. 2011, 40, 5564. doi:10.1039/c1cs15120g
Return to citation in text: [1] -
Atodiresei, I.; Schiffers, I.; Bolm, C. Chem. Rev. 2007, 107, 5683. doi:10.1021/cr068369f
Return to citation in text: [1] -
Chen, Y.; McDaid, P.; Deng, L. Chem. Rev. 2003, 103, 2965. doi:10.1021/cr020037x
Return to citation in text: [1] -
Spivey, A. C.; Andrews, B. I. Angew. Chem., Int. Ed. 2001, 40, 3131. doi:10.1002/1521-3773(20010903)40:17<3131::AID-ANIE3131>3.0.CO;2-Z
Return to citation in text: [1] -
Du, F.; Zhou, J.; Peng, Y. Org. Lett. 2017, 19, 1310. doi:10.1021/acs.orglett.7b00128
Return to citation in text: [1] -
Liu, X.; Wang, Y.; Yang, D.; Zhang, J.; Liu, D.; Su, W. Angew. Chem., Int. Ed. 2016, 55, 8100. doi:10.1002/anie.201602880
Return to citation in text: [1] -
Miyaji, R.; Asano, K..; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151
Return to citation in text: [1] -
Lam, Y.-h.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 9556. doi:10.1021/ja504714m
Return to citation in text: [1] -
Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841. doi:10.1002/anie.200702187
Return to citation in text: [1] -
Song, J.; Wang, Y.; Deng, L. J. Am. Chem. Soc. 2006, 128, 6048. doi:10.1021/ja060716f
Return to citation in text: [1] -
McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367. doi:10.1002/anie.200501721
Return to citation in text: [1] -
Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481. doi:10.1039/b508833j
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967. doi:10.1021/ol050431s
Return to citation in text: [1] -
Li, H.; Wang, Y.; Tang, L.; Deng, L. J. Am. Chem. Soc. 2004, 126, 9906. doi:10.1021/ja047281l
Return to citation in text: [1] -
Dong, S.; Liu, X.; Zhu, Y.; He, P.; Lin, L.; Feng, X. J. Am. Chem. Soc. 2013, 135, 10026. doi:10.1021/ja404379n
Return to citation in text: [1] -
Dong, S.; Liu, X.; Chen, X.; Mei, F.; Zhang, Y.; Gao, B.; Lin, L.; Feng, X. J. Am. Chem. Soc. 2010, 132, 10650. doi:10.1021/ja1046928
Return to citation in text: [1] -
List, B. Tetrahedron 2002, 58, 5573. doi:10.1016/S0040-4020(02)00516-1
Return to citation in text: [1] -
Berkessel, A.; Cleemann, F.; Mukherjee, S. Angew. Chem., Int. Ed. 2005, 44, 7466. doi:10.1002/anie.200502003
Return to citation in text: [1] -
Matsunaga, H.; Tajima, D.; Kawauchi, T.; Yasuyama, T.; Ando, S.; Ishizuka, T. Org. Biomol. Chem. 2017, 15, 2892. doi:10.1039/C6OB02743A
Return to citation in text: [1] -
Novacek, J.; Izzo, J. A.; Vetticatt, M. J.; Waser, M. Chem. – Eur. J. 2016, 22, 17339. doi:10.1002/chem.201604153
Return to citation in text: [1] -
Wang, S.-G.; Liu, X.-J.; Zhao, Q.-C.; Wang, S.-B.; You, S.-L. Angew. Chem., Int. Ed. 2015, 54, 14929. doi:10.1002/anie.201507998
Return to citation in text: [1] -
Gajulapalli V, P. R.; Vinayagam, P.; Kesavan, V. Org. Biomol. Chem. 2014, 12, 4186. doi:10.1039/C4OB00271G
Return to citation in text: [1] -
Fang, Y.-Q.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 5660. doi:10.1021/ja801344w
Return to citation in text: [1] -
Berkessel, A.; Cleemann, F.; Mukherjee, S.; Müller, T. N.; Lex, J. Angew. Chem., Int. Ed. 2005, 44, 807. doi:10.1002/anie.200461442
Return to citation in text: [1] -
Berkessel, A.; Mukherjee, S.; Cleemann, F.; Müller, T. N.; Lex, J. Chem. Commun. 2005, 1898. doi:10.1039/B418666D
Return to citation in text: [1] -
Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Org. Lett. 2004, 6, 625. doi:10.1021/ol0364531
Return to citation in text: [1] -
Wakchaure, V. N.; List, B. Angew. Chem., Int. Ed. 2010, 49, 4136. doi:10.1002/anie.201000637
Return to citation in text: [1] -
Smith, E.; Schiffers, I.; Bolm, C. Tetrahedron 2010, 66, 6349. doi:10.1016/j.tet.2010.04.121
Return to citation in text: [1] -
Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672. doi:10.1021/ja036972z
Return to citation in text: [1] -
Honjo, T.; Tsumura, T.; Sano, S.; Nagao, Y.; Yamaguchi, K.; Sei, Y. Synlett 2009, 3279. doi:10.1055/s-0029-1218374
Return to citation in text: [1] -
Honjo, T.; Sano, S.; Shiro, M.; Nagao, Y. Angew. Chem., Int. Ed. 2005, 44, 5838. doi:10.1002/anie.200501408
Return to citation in text: [1] -
Wang, H.; Yan, L.; Wu, Y.; Lu, Y.; Chen, F. Org. Lett. 2015, 17, 5452. doi:10.1021/acs.orglett.5b02813
Return to citation in text: [1] -
Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2009, 351, 547. doi:10.1002/adsc.200800761
Return to citation in text: [1] -
Yan, L.-J.; Wang, H.-F.; Chen, W.-X.; Tao, Y.; Jin, K.-J.; Chen, F.-E. ChemCatChem 2016, 8, 2249. doi:10.1002/cctc.201600228
Return to citation in text: [1] [2] -
Yang, H.-J.; Xiong, F.-J.; Chen, X.-F.; Chen, F.-E. Eur. J. Org. Chem. 2013, 4495. doi:10.1002/ejoc.201300464
Return to citation in text: [1] [2] -
Chen, X.; Xiong, F.; Chen, W.; He, Q.; Chen, F. J. Org. Chem. 2014, 79, 2723. doi:10.1021/jo402829b
Return to citation in text: [1] [2] -
Yang, H.-J.; Xiong, F.-J.; Li, J.; Chen, F.-E. Chin. Chem. Lett. 2013, 24, 553. doi:10.1016/j.cclet.2013.04.009
Return to citation in text: [1] [2] -
Chen, F.-E.; Dai, H.-F.; Kuang, Y.-Y.; Jia, H.-Q. Tetrahedron: Asymmetry 2003, 14, 3667. doi:10.1016/j.tetasy.2003.08.034
Return to citation in text: [1] -
Chen, F. Synthesis method of [3aS,6aR]-1,3-dibenzyl-tetrahydro-4H-furo[3,4-d]-imidazolyl-2,4[1H]-diketone. Chin. Pat. CN1473832A, Feb 11, 2004.
Return to citation in text: [1] -
Shioiri, T.; Izawa, K.; Konoike, T., Eds. Pharmaceutical Process Chemistry; Wiley-VCH: Weinheim, 2010; p 296. doi:10.1002/9783527633678
Return to citation in text: [1] -
Park, S. E.; Nam, E. H.; Jang, H. B.; Oh, J. S.; Some, S.; Lee, Y. S.; Song, C. E. Adv. Synth. Catal. 2010, 352, 2211. doi:10.1002/adsc.201000289
Return to citation in text: [1] -
Rho, H. S.; Oh, S. H.; Lee, J. W.; Lee, J. Y.; Chin, J.; Song, C. E. Chem. Commun. 2008, 1208. doi:10.1039/b719811f
Return to citation in text: [1] [2] -
Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem., Int. Ed. 2008, 47, 7872. doi:10.1002/anie.200801636
Return to citation in text: [1] -
One major breakthrough was derived from Song’s group in 2008 using a thermally robust sulfonamide-based cinchona alkaloids, see reference [46].
Return to citation in text: [1] -
Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun. 2009, 7224. doi:10.1039/B917882A
Return to citation in text: [1] -
Li, H.; Liu, X.; Wu, F.; Tang, L.; Deng, L. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20625. doi:10.1073/pnas.1004439107
Return to citation in text: [1] -
For a detailed experimental procedure for the synthesis of (1S,2S)-diamine 6, see reference [36].
Return to citation in text: [1] -
Ordóñez, M.; Cativiela, C.; Romero-Estudillo, I. Tetrahedron: Asymmetry 2016, 27, 999. doi:10.1016/j.tetasy.2016.08.011
Return to citation in text: [1] -
Trabocchi, A.; Menchi, G.; Guarna, A. In Amino Acids, Peptides and Proteins in Organic Chemsitry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; Vol. 1, pp 527 ff.
Return to citation in text: [1] -
Hanrahan, J. R.; Johnston, G. A. R. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; Vol. 1, pp 573 ff.
Return to citation in text: [1] -
Ordóňez, M.; Cativiela, C. Tetrahedron: Asymmetry 2007, 18, 3. doi:10.1016/j.tetasy.2006.12.001
Return to citation in text: [1] -
Wu, Y.; Xiong, F.-J.; Chen, F.-E. Tetrahedron 2015, 71, 8487. doi:10.1016/j.tet.2015.07.059
Return to citation in text: [1] -
Časar, Z. Curr. Org. Chem. 2010, 14, 816. doi:10.2174/138527210791111858
Return to citation in text: [1] -
Miyachi, N.; Suzuki, M.; Ohara, Y.; Hiyama, T. J. J. Synth. Org. Chem., Jpn. 1995, 53, 186. doi:10.5059/yukigoseikyokaishi.53.186
Return to citation in text: [1] -
Valderas, C.; Casarrubios, L.; de la Torre, M. C.; Sierra, M. A. Tetrahedron Lett. 2017, 58, 326. doi:10.1016/j.tetlet.2016.12.025
Return to citation in text: [1] -
Ivšić, T.; Dokli, I.; Rimac, A.; Hameršak, Z. Eur. J. Org. Chem. 2014, 631. doi:10.1002/ejoc.201301374
Return to citation in text: [1]
| 51. | Ordóñez, M.; Cativiela, C.; Romero-Estudillo, I. Tetrahedron: Asymmetry 2016, 27, 999. doi:10.1016/j.tetasy.2016.08.011 |
| 52. | Trabocchi, A.; Menchi, G.; Guarna, A. In Amino Acids, Peptides and Proteins in Organic Chemsitry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; Vol. 1, pp 527 ff. |
| 53. | Hanrahan, J. R.; Johnston, G. A. R. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; Vol. 1, pp 573 ff. |
| 54. | Ordóňez, M.; Cativiela, C. Tetrahedron: Asymmetry 2007, 18, 3. doi:10.1016/j.tetasy.2006.12.001 |
| 50. | For a detailed experimental procedure for the synthesis of (1S,2S)-diamine 6, see reference [36]. |
| 45. | Rho, H. S.; Oh, S. H.; Lee, J. W.; Lee, J. Y.; Chin, J.; Song, C. E. Chem. Commun. 2008, 1208. doi:10.1039/b719811f |
| 1. | Borissov, A.; Davies, T. Q.; Ellis, S. R.; Fleming, T. A.; Richardson, M. S. W.; Dixon, D. J. Chem. Soc. Rev. 2016, 45, 5474. doi:10.1039/C5CS00015G |
| 2. | Zheng, C.; Chen, F.-E. Chin. Chem. Lett. 2014, 25, 1. doi:10.1016/j.cclet.2013.11.025 |
| 3. | Rodriguez-Docampo, Z.; Connon, S. J. ChemCatChem 2012, 4, 151. doi:10.1002/cctc.201100266 |
| 4. | Díaz de Villegas, M. D.; Gálvez, J. A.; Etayo, P.; Badorrey, R.; López-Ram-de-Víu, P. Chem. Soc. Rev. 2011, 40, 5564. doi:10.1039/c1cs15120g |
| 5. | Atodiresei, I.; Schiffers, I.; Bolm, C. Chem. Rev. 2007, 107, 5683. doi:10.1021/cr068369f |
| 6. | Chen, Y.; McDaid, P.; Deng, L. Chem. Rev. 2003, 103, 2965. doi:10.1021/cr020037x |
| 7. | Spivey, A. C.; Andrews, B. I. Angew. Chem., Int. Ed. 2001, 40, 3131. doi:10.1002/1521-3773(20010903)40:17<3131::AID-ANIE3131>3.0.CO;2-Z |
| 22. | Matsunaga, H.; Tajima, D.; Kawauchi, T.; Yasuyama, T.; Ando, S.; Ishizuka, T. Org. Biomol. Chem. 2017, 15, 2892. doi:10.1039/C6OB02743A |
| 23. | Novacek, J.; Izzo, J. A.; Vetticatt, M. J.; Waser, M. Chem. – Eur. J. 2016, 22, 17339. doi:10.1002/chem.201604153 |
| 24. | Wang, S.-G.; Liu, X.-J.; Zhao, Q.-C.; Wang, S.-B.; You, S.-L. Angew. Chem., Int. Ed. 2015, 54, 14929. doi:10.1002/anie.201507998 |
| 25. | Gajulapalli V, P. R.; Vinayagam, P.; Kesavan, V. Org. Biomol. Chem. 2014, 12, 4186. doi:10.1039/C4OB00271G |
| 26. | Fang, Y.-Q.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 5660. doi:10.1021/ja801344w |
| 27. | Berkessel, A.; Cleemann, F.; Mukherjee, S.; Müller, T. N.; Lex, J. Angew. Chem., Int. Ed. 2005, 44, 807. doi:10.1002/anie.200461442 |
| 28. | Berkessel, A.; Mukherjee, S.; Cleemann, F.; Müller, T. N.; Lex, J. Chem. Commun. 2005, 1898. doi:10.1039/B418666D |
| 29. | Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Org. Lett. 2004, 6, 625. doi:10.1021/ol0364531 |
| 47. | One major breakthrough was derived from Song’s group in 2008 using a thermally robust sulfonamide-based cinchona alkaloids, see reference [46]. |
| 21. | Berkessel, A.; Cleemann, F.; Mukherjee, S. Angew. Chem., Int. Ed. 2005, 44, 7466. doi:10.1002/anie.200502003 |
| 48. | Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun. 2009, 7224. doi:10.1039/B917882A |
| 49. | Li, H.; Liu, X.; Wu, F.; Tang, L.; Deng, L. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20625. doi:10.1073/pnas.1004439107 |
| 18. | Dong, S.; Liu, X.; Zhu, Y.; He, P.; Lin, L.; Feng, X. J. Am. Chem. Soc. 2013, 135, 10026. doi:10.1021/ja404379n |
| 19. | Dong, S.; Liu, X.; Chen, X.; Mei, F.; Zhang, Y.; Gao, B.; Lin, L.; Feng, X. J. Am. Chem. Soc. 2010, 132, 10650. doi:10.1021/ja1046928 |
| 20. | List, B. Tetrahedron 2002, 58, 5573. doi:10.1016/S0040-4020(02)00516-1 |
| 41. | Chen, F.-E.; Dai, H.-F.; Kuang, Y.-Y.; Jia, H.-Q. Tetrahedron: Asymmetry 2003, 14, 3667. doi:10.1016/j.tetasy.2003.08.034 |
| 42. | Chen, F. Synthesis method of [3aS,6aR]-1,3-dibenzyl-tetrahydro-4H-furo[3,4-d]-imidazolyl-2,4[1H]-diketone. Chin. Pat. CN1473832A, Feb 11, 2004. |
| 43. | Shioiri, T.; Izawa, K.; Konoike, T., Eds. Pharmaceutical Process Chemistry; Wiley-VCH: Weinheim, 2010; p 296. doi:10.1002/9783527633678 |
| 36. | Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2009, 351, 547. doi:10.1002/adsc.200800761 |
| 8. | Du, F.; Zhou, J.; Peng, Y. Org. Lett. 2017, 19, 1310. doi:10.1021/acs.orglett.7b00128 |
| 9. | Liu, X.; Wang, Y.; Yang, D.; Zhang, J.; Liu, D.; Su, W. Angew. Chem., Int. Ed. 2016, 55, 8100. doi:10.1002/anie.201602880 |
| 10. | Miyaji, R.; Asano, K..; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi:10.1021/jacs.5b04151 |
| 11. | Lam, Y.-h.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 9556. doi:10.1021/ja504714m |
| 12. | Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841. doi:10.1002/anie.200702187 |
| 13. | Song, J.; Wang, Y.; Deng, L. J. Am. Chem. Soc. 2006, 128, 6048. doi:10.1021/ja060716f |
| 14. | McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367. doi:10.1002/anie.200501721 |
| 15. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481. doi:10.1039/b508833j |
| 16. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967. doi:10.1021/ol050431s |
| 17. | Li, H.; Wang, Y.; Tang, L.; Deng, L. J. Am. Chem. Soc. 2004, 126, 9906. doi:10.1021/ja047281l |
| 44. | Park, S. E.; Nam, E. H.; Jang, H. B.; Oh, J. S.; Some, S.; Lee, Y. S.; Song, C. E. Adv. Synth. Catal. 2010, 352, 2211. doi:10.1002/adsc.201000289 |
| 45. | Rho, H. S.; Oh, S. H.; Lee, J. W.; Lee, J. Y.; Chin, J.; Song, C. E. Chem. Commun. 2008, 1208. doi:10.1039/b719811f |
| 46. | Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem., Int. Ed. 2008, 47, 7872. doi:10.1002/anie.200801636 |
| 35. | Wang, H.; Yan, L.; Wu, Y.; Lu, Y.; Chen, F. Org. Lett. 2015, 17, 5452. doi:10.1021/acs.orglett.5b02813 |
| 38. | Yang, H.-J.; Xiong, F.-J.; Chen, X.-F.; Chen, F.-E. Eur. J. Org. Chem. 2013, 4495. doi:10.1002/ejoc.201300464 |
| 39. | Chen, X.; Xiong, F.; Chen, W.; He, Q.; Chen, F. J. Org. Chem. 2014, 79, 2723. doi:10.1021/jo402829b |
| 40. | Yang, H.-J.; Xiong, F.-J.; Li, J.; Chen, F.-E. Chin. Chem. Lett. 2013, 24, 553. doi:10.1016/j.cclet.2013.04.009 |
| 37. | Yan, L.-J.; Wang, H.-F.; Chen, W.-X.; Tao, Y.; Jin, K.-J.; Chen, F.-E. ChemCatChem 2016, 8, 2249. doi:10.1002/cctc.201600228 |
| 33. | Honjo, T.; Tsumura, T.; Sano, S.; Nagao, Y.; Yamaguchi, K.; Sei, Y. Synlett 2009, 3279. doi:10.1055/s-0029-1218374 |
| 34. | Honjo, T.; Sano, S.; Shiro, M.; Nagao, Y. Angew. Chem., Int. Ed. 2005, 44, 5838. doi:10.1002/anie.200501408 |
| 38. | Yang, H.-J.; Xiong, F.-J.; Chen, X.-F.; Chen, F.-E. Eur. J. Org. Chem. 2013, 4495. doi:10.1002/ejoc.201300464 |
| 39. | Chen, X.; Xiong, F.; Chen, W.; He, Q.; Chen, F. J. Org. Chem. 2014, 79, 2723. doi:10.1021/jo402829b |
| 40. | Yang, H.-J.; Xiong, F.-J.; Li, J.; Chen, F.-E. Chin. Chem. Lett. 2013, 24, 553. doi:10.1016/j.cclet.2013.04.009 |
| 46. | Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem., Int. Ed. 2008, 47, 7872. doi:10.1002/anie.200801636 |
| 31. | Smith, E.; Schiffers, I.; Bolm, C. Tetrahedron 2010, 66, 6349. doi:10.1016/j.tet.2010.04.121 |
| 32. | Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672. doi:10.1021/ja036972z |
| 55. | Wu, Y.; Xiong, F.-J.; Chen, F.-E. Tetrahedron 2015, 71, 8487. doi:10.1016/j.tet.2015.07.059 |
| 56. | Časar, Z. Curr. Org. Chem. 2010, 14, 816. doi:10.2174/138527210791111858 |
| 57. | Miyachi, N.; Suzuki, M.; Ohara, Y.; Hiyama, T. J. J. Synth. Org. Chem., Jpn. 1995, 53, 186. doi:10.5059/yukigoseikyokaishi.53.186 |
| 30. | Wakchaure, V. N.; List, B. Angew. Chem., Int. Ed. 2010, 49, 4136. doi:10.1002/anie.201000637 |
| 36. | Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2009, 351, 547. doi:10.1002/adsc.200800761 |
| 37. | Yan, L.-J.; Wang, H.-F.; Chen, W.-X.; Tao, Y.; Jin, K.-J.; Chen, F.-E. ChemCatChem 2016, 8, 2249. doi:10.1002/cctc.201600228 |
| 58. | Valderas, C.; Casarrubios, L.; de la Torre, M. C.; Sierra, M. A. Tetrahedron Lett. 2017, 58, 326. doi:10.1016/j.tetlet.2016.12.025 |
| 59. | Ivšić, T.; Dokli, I.; Rimac, A.; Hameršak, Z. Eur. J. Org. Chem. 2014, 631. doi:10.1002/ejoc.201301374 |
© 2018 Xu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)