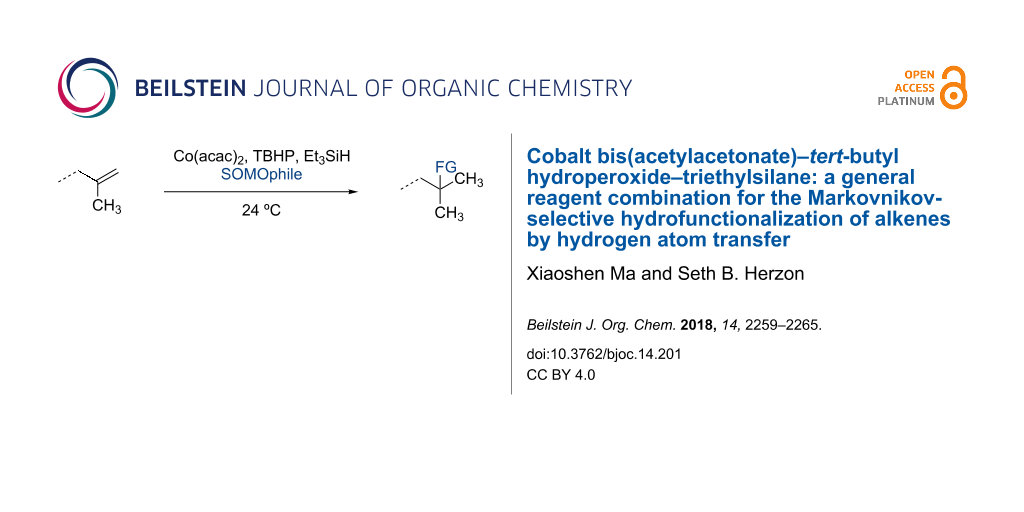
Abstract
We show that cobalt bis(acetylacetonate) [Co(acac)2], tert-butyl hydroperoxide (TBHP), and triethylsilane (Et3SiH) constitute an inexpensive, general, and practical reagent combination to initiate a broad range of Markovnikov-selective alkene hydrofunctionalization reactions. These transformations are believed to proceed by cobalt-mediated hydrogen atom transfer (HAT) to the alkene substrate, followed by interception of the resulting alkyl radical intermediate with a SOMOphile. In addition, we report the first reductive couplings of unactivated alkenes and aryldiazonium salts by an HAT pathway. The simplicity and generality of the Co(acac)2–TBHP–Et3SiH reagent combination suggests it as a useful starting point to develop HAT reactions in complex settings.

Graphical Abstract
Introduction
Many powerful methods to effect alkene hydrogenation [1-4] and Markovnikov-selective hydroheterofunctionalization (H–X addition, X = O [5-9], I [3], Br [3], Se [3], S [8-10], Cl [8,11], F [12,13], and N [8,14-17]) by metal-mediated hydrogen atom transfer (HAT) [18-21] are now known. Additionally, methods to achieve carbon–carbon bond formation to alkenes by HAT have been developed (e.g., reductive coupling [22-28], formal hydromethylation [29], cycloisomerization [8,30,31], hydrooximation [32], hydroheteroarylation [28,33-35], hydroarylation [36-38], and cross-coupling [37]). Many of these transformations have found applications in synthesis [6,39-47]. Although mechanistically-complex [28] the outcome of these reactions can be rationalized as initiating by HAT to the alkene, to form the kinetically- and thermodynamically-favored alkyl radical intermediate, which may be in equilibrium with the corresponding metal alkyl complex. This radical then undergoes addition to a second reagent (SOMOphile) to form the functionalized product (Scheme 1).
![[1860-5397-14-201-i1]](/bjoc/content/inline/1860-5397-14-201-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General mechanism of alkene hydrofunctionalization via HAT.
Scheme 1: General mechanism of alkene hydrofunctionalization via HAT.
A wide range of manganese, cobalt, or iron-based complexes containing diverse supporting ligands have found use in these reactions. To the best of our knowledge, the iron oxalate–sodium borohydride system, introduced by Boger and co-workers [8], is the only reagent combination shown to accommodate a broad range of SOMOphiles. However, the cobalt–salen complexes that are commonly employed [10,11,13,15,16,30-32,36,37,48] contain many different ligand architectures [21], and often need to be prepared by multistep sequences. Here we report a uniform set of reaction conditions to achieve a broad range of HAT hydrofunctionalization reactions using the simple reagents cobalt acetoacetonate [Co(acac)2], tert-butyl hydroperoxide (TBHP), and triethylsilane (Et3SiH). The practicality and generality of this system should motivate its application in synthesis.
Results and Discussion
In 2014, we reported the reduction of alkenyl halides (e.g., 1, Scheme 2) utilizing Co(acac)2, TBHP, and two reductants, triethylsilane and 1,4-dihydrobenzene (DHB) [2]. Mechanistic studies showed that Et3SiH participates in the formation of a cobalt hydride intermediate that delivers a hydrogen atom to the less-substituted position of the alkene. The resulting alkyl radical is believed to abstract a second hydrogen atom from DHB to generate the reduced product [2]. This mechanism separates the alkyl radical formation and functionalization steps by employing two different reagents. Accordingly, we investigated the application of this system in other HAT reactions. In these studies, methallyl p-methoxybenzoate (3a) was used as substrate (Table 1).
![[1860-5397-14-201-i2]](/bjoc/content/inline/1860-5397-14-201-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reduction of the alkenyl chloride 1 by HAT.
Scheme 2: Reduction of the alkenyl chloride 1 by HAT.
Table 1: Markovnikov Hydrofunctionalization of 3a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-201-i4.svg?max-width=637&scale=1.0)
|
|||||||
| entry | x | y | SOMOphile | z | solvent | product | yieldb |
|---|---|---|---|---|---|---|---|
| 1 | 5.00 | 5.00 | – | – | n-propanol |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-201-i5.svg?max-width=637&scale=1.0)
4a |
86% |
| 2 | 2.50 | 10.0 | NFSI | 2.50 | CH2Cl2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-201-i6.svg?max-width=637&scale=1.0)
4b |
36% |
| 3 | 2.50 | 10.0 | TsCl | 2.50 | n-propanol |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-201-i7.svg?max-width=637&scale=1.0)
4c |
92% |
| 4 | 3.75 | 10.0 | TsBr | 2.50 | n-propanol |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-201-i8.svg?max-width=637&scale=1.0)
4d |
95% |
| 5 | 3.75 | 10.0 | CH2I2 | 15.0 | CH2Cl2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-201-i9.svg?max-width=637&scale=1.0)
4e |
89% |
| 6 | 10.0 | 10.0 | O2 | – | n-propanol |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-201-i10.svg?max-width=637&scale=1.0)
4f |
69% |
| 7 | 2.50 | 10.0 | PhSO2SPh | 2.50 | n-propanol |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-201-i11.svg?max-width=637&scale=1.0)
4g |
96% |
| 8 | 2.50 | 10.0 | TsSePh | 2.50 | n-propanol |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-201-i12.svg?max-width=637&scale=1.0)
4h |
89% |
| 9 | 1.00 | 10.0 | p-ABSA | 5.00 | CH3CN |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-201-i13.svg?max-width=637&scale=1.0)
4i |
79% |
| 10 | 0 | 6.25 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-201-i14.svg?max-width=637&scale=1.0)
5a |
1.50 | CH2Cl2 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-201-i15.svg?max-width=637&scale=1.0)
7a |
92% |
| 11 | 3.75 | 10.0 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-201-i16.svg?max-width=637&scale=1.0)
6a |
2.50 | n-propanol |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-201-i17.svg?max-width=637&scale=1.0)
4j |
60% |
| 12 | 3.75 | 10.0 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-201-i18.svg?max-width=637&scale=1.0)
6b |
2.50 | n-propanol |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-201-i19.svg?max-width=637&scale=1.0)
4k |
48% |
| 13 | 0 | 5.00 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-201-i20.svg?max-width=637&scale=1.0)
6c |
5.00 | CH2Cl2 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-201-i21.svg?max-width=637&scale=1.0)
4l |
66%
4.7:1 rrb |
| 14 | 0 | 5.00 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-201-i22.svg?max-width=637&scale=1.0)
6d |
1.00 | CH3CN |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-201-i23.svg?max-width=637&scale=1.0)
4m |
50% |
aFor detailed reaction conditions, see Supporting Information File 1. bIsolated yields after purification by flash-column chromatography. brr = ratio of regioisomers.
Under our standard reduction conditions, alkene 3a was transformed to isobutyl p-methoxybenzoate (4a) in 86% yield (entry 1, Table 1). Inspired by the methods of Boger [12] and Hiroya [13], a number of fluorination reagents were examined to achieve hydrofluorination. Although no product was observed using SelectFluor®, diethylaminosulfur trifluoride (DAST), or tosyl fluoride (see Supporting Information File 1, Table S1, entries 2–4), N-fluorobenzenesulfonimide (NFSI) provided the desired hydrofluorination product 4b in 36% yield (Table 1, entry 2). Carreira and co-workers reported the first hydrochlorination reaction via a cobalt-catalyzed HAT process [11]. By utilizing p-toluenesulfonyl chloride (TsCl) and p-toluenesulfonyl bromide (TsBr) under our conditions, the desired hydrochlorination and hydrobromination products 4c and 4d were obtained in 92% and 95% yields, respectively [3] (Table 1, entries 3 and 4). To our knowledge, the formation of 4d represents the first Markovnikov-selective alkene hydrobromination by an HAT pathway. Attempts to extend this reaction to hydroiodination using related reagents, p-toluenesulfonyl iodide, N-iodosuccinimide, or molecular iodine failed to provide the expected product (see Supporting Information File 1, Table S1, entries 9–11) [3]. Surprisingly, diiodomethane, possessed the desired reactivity and the hydroiodination product 4e was isolated in 89% yield (Table 1, entry 5) [3]. Ethyl iodoacetate, iodoacetonitrile, and 1,2-diiodoethane were also effective, but the yields of 4e and conversion of 3a were lower (see Supporting Information File 1, Table S1, entries 13–15). Mukaiyama and co-workers’ pioneering Markonikov-selective alkene hydration reaction [5,49-54] proceeds using dioxygen as the oxygen atom source. Exposure of the alkene 3a to similar conditions provided the tertiary alcohol 4f in 69% yield (Table 1, entry 6). Inspired by Girijavallabhan and co-workers’ report [10], we were able to trap the tertiary alkyl radical with S-phenyl benzene thiosulfonate (PhSO2SPh, Table 1, entry 7) and Se-phenyl 4-methylbenzenesulfonoselenoate (TsSePh, Table 1, entry 8) [3] to afford the corresponding products 4g and 4h in 96% and 89% yields, respectively. Carreira and co-workers reported the hydroazidation of alkenes using cobalt–salen complexes as hydrogen atom transfer agents and para-toluenesulfonyl azide as an azide source [16,48,55]. After careful optimization of the azidation reagent (p-acetamidobenzenesulfonyl azide (p-ABSA)) and additive equivalents (see Supporting Information File 1, Table S1, entries 19–28), the tertiary alkyl azide 4i was obtained in 79% yield (Table 1, entry 9).
To broaden the scope of C–N coupling process via HAT, we investigated other nitrogen-containing SOMOphiles in the HAT reaction. Employing 4-methoxyphenyldiazonium tetrafluoroborate (5a) in our HAT conditions, the alkyl aryl azo product 7a was obtained in 92% yield (Table 1, entry 10). Due to the importance of azo compounds in synthetic organic chemistry, industrial dyes, and medicinal chemistry [56,57], we investigated the scope of this transformation (Scheme 3). By varying the alkene substitution pattern, we determined that the coupling of tertiary radicals is more efficient than secondary radicals. For example, methallyl p-methoxybenzoate (3a) and prenyl p-methoxybenzoate (3c, not shown) afforded the azo compounds 7a and 7c in 92% and 91% yields, respectively. By comparison the yield of the azo product using allyl p-methoxybenzoate (3b, now shown) as substrate was somewhat lower (77%). Diazonium salts bearing substituents with different steric and electronic properties were examined. These experiments revealed that this coupling is compatible with a broad range of functional groups and aryl substitution patterns (7d–j, 52–97%). The naphthylazo derivative 7k was obtained in 69% yield using 1-naphthyldiazonium tetrafluoroborate. However, aryldiazonium salts bearing nitro substituents were not compatible with this HAT coupling. For example, the use of p-nitrobenzendiazonium tetrafluoroborate failed to provide the expected coupling product 7l. This may be due to alternative reaction pathways involving reduction of the nitro substituent [17].
![[1860-5397-14-201-i3]](/bjoc/content/inline/1860-5397-14-201-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of alkyl-aryl azo compound synthesis via HAT. Conditions: alkene (0.250 mmol), diazonium salt (1.50 equiv), Co(acac)2 (1.00 equiv), TBHP (1.00 equiv), Et3SiH (6.25 equiv), CH2Cl2 (0.2 M), argon, 15–120 min. All yields are isolated yields after flash-column chromatography. aReaction conducted on 1.00 mmol scale.
Scheme 3: Substrate scope of alkyl-aryl azo compound synthesis via HAT. Conditions: alkene (0.250 mmol), diaz...
Additional carbon–carbon bond formation strategies were also examined using the Co(acac)2 HAT system. Sulfonyl oximes 6a and 6b [32] afforded the carbon–carbon coupled products 4j and 4k in 60% and 48% yields, respectively (Table 1, entries 11 and 12, respectively). Recently, our laboratory reported a formal intermolecular hydroheteroarylation using N-methoxy heteroarenium salts by Co(acac)2-mediated HAT [33,34]. In the original reports, 36 discrete unactivated alkenes were coupled with 38 different heteroarenium salts under mild conditions [33,34]. A representative example comprises the coupling of methallyl p-methoxybenzoate (3a) with N-methoxypyridinium methyl sulfate (6c) to form the hydropyridylation product 4l in 66% yield and as a 4.7:1 ratio of regioisomers (Table 1, entry 13). We also demonstrated that the alkyl radical generated from the HAT process can be trapped by (η6-benzene)manganese tricarbonyl hexafluorophosphate (6d) to provide the reductive coupling product 4m in 50% yield (Table 1, entry 14).
Conclusion
In summary, we have demonstrated that under a consistent set of conditions, the Co(acac)2–TBHP–Et3SiH system effects a diverse array of Markovnikov-selective hydrofunctionalization reactions of unactivated alkenes (H–X addition, X = H, F, Cl, Br, I, O, S, Se, N, and C). We have also reported the first reductive coupling reactions of alkenes and aryldiazonium salts under HAT conditions. These transformations proceed in high regioselectivity and efficiency. Further efforts will focus on expanding the alkene scope and exploring the site-selectivity in polyene substrates.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and characterization data for all new compounds. | ||
| Format: PDF | Size: 4.1 MB | Download |
References
-
Iwasaki, K.; Wan, K. K.; Oppedisano, A.; Crossley, S. W. M.; Shenvi, R. A. J. Am. Chem. Soc. 2014, 136, 1300–1303. doi:10.1021/ja412342g
Return to citation in text: [1] -
King, S. M.; Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2014, 136, 6884–6887. doi:10.1021/ja502885c
Return to citation in text: [1] [2] [3] -
Ma, X.; Herzon, S. B. Chem. Sci. 2015, 6, 6250–6255. doi:10.1039/C5SC02476E
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Obradors, C.; Martinez, R. M.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 4962–4971. doi:10.1021/jacs.6b02032
Return to citation in text: [1] -
Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 573–576. doi:10.1246/cl.1989.573
Return to citation in text: [1] [2] -
Ishikawa, H.; Colby, D. A.; Boger, D. L. J. Am. Chem. Soc. 2008, 130, 420–421. doi:10.1021/ja078192m
Return to citation in text: [1] [2] -
Ishikawa, H.; Colby, D. A.; Seto, S.; Va, P.; Tam, A.; Kakei, H.; Rayl, T. J.; Hwang, I.; Boger, D. L. J. Am. Chem. Soc. 2009, 131, 4904–4916. doi:10.1021/ja809842b
Return to citation in text: [1] -
Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ma, X.; Herzon, S. B. J. Org. Chem. 2016, 81, 8673–8695. doi:10.1021/acs.joc.6b01709
Return to citation in text: [1] [2] -
Girijavallabhan, V.; Alvarez, C.; Njoroge, F. G. J. Org. Chem. 2011, 76, 6442–6446. doi:10.1021/jo201016z
Return to citation in text: [1] [2] [3] -
Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 5758–5760. doi:10.1002/anie.200801760
Return to citation in text: [1] [2] [3] -
Barker, T. J.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 13588–13591. doi:10.1021/ja3063716
Return to citation in text: [1] [2] -
Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h
Return to citation in text: [1] [2] [3] -
Kato, K.; Mukaiyama, T. Chem. Lett. 1992, 21, 1137–1140. doi:10.1246/cl.1992.1137
Return to citation in text: [1] -
Waser, J.; Carreira, E. M. J. Am. Chem. Soc. 2004, 126, 5676–5677. doi:10.1021/ja048698u
Return to citation in text: [1] [2] -
Waser, J.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2005, 127, 8294–8295. doi:10.1021/ja052164r
Return to citation in text: [1] [2] [3] -
Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J. C.; Lee, B. J.; Spergel, S. H.; Mertzman, M. E.; Pitts, W. J.; La Cruz, T. E.; Schmidt, M. A.; Darvatkar, N.; Natarajan, S. R.; Baran, P. S. Science 2015, 348, 886–891. doi:10.1126/science.aab0245
Return to citation in text: [1] [2] -
Eisenberg, D. C.; Norton, J. R. Isr. J. Chem. 1991, 31, 55–66. doi:10.1002/ijch.199100006
Return to citation in text: [1] -
Gansäuer, A.; Shi, L.; Otte, M.; Huth, I.; Rosales, A.; Sancho-Sanz, I.; Padial, N. M.; Oltra, J. E. Hydrogen Atom Donors: Recent Developments. In Radicals in Synthesis III; Heinrich, M.; Gansäuer, A., Eds.; Topics in Current Chemistry; Springer: Berlin Heidelberg, 2012. doi:10.1007/128_2011_124
Return to citation in text: [1] -
Hoffmann, R. W. Chem. Soc. Rev. 2016, 45, 577–583. doi:10.1039/C5CS00423C
Return to citation in text: [1] -
Crossley, S. W. M.; Obradors, C.; Martinez, R. M.; Shenvi, R. A. Chem. Rev. 2016, 116, 8912–9000. doi:10.1021/acs.chemrev.6b00334
Return to citation in text: [1] [2] -
Choi, J.; Tang, L.; Norton, J. R. J. Am. Chem. Soc. 2007, 129, 234–240. doi:10.1021/ja066325i
Return to citation in text: [1] -
Choi, J.; Pulling, M. E.; Smith, D. M.; Norton, J. R. J. Am. Chem. Soc. 2008, 130, 4250–4252. doi:10.1021/ja710455c
Return to citation in text: [1] -
Li, G.; Han, A.; Pulling, M. E.; Estes, D. P.; Norton, J. R. J. Am. Chem. Soc. 2012, 134, 14662–14665. doi:10.1021/ja306037w
Return to citation in text: [1] -
Lo, J. C.; Yabe, Y.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 1304–1307. doi:10.1021/ja4117632
Return to citation in text: [1] -
Lo, J. C.; Gui, J.; Yabe, Y.; Pan, C.-M.; Baran, P. S. Nature 2014, 516, 343–348. doi:10.1038/nature14006
Return to citation in text: [1] -
Kuo, J. L.; Hartung, J.; Han, A.; Norton, J. R. J. Am. Chem. Soc. 2015, 137, 1036–1039. doi:10.1021/ja511883b
Return to citation in text: [1] -
Lo, J. C.; Kim, D.; Pan, C.-M.; Edwards, J. T.; Yabe, Y.; Gui, J.; Qin, T.; Gutiérrez, S.; Giacoboni, J.; Smith, M. W.; Holland, P. L.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 2484–2503. doi:10.1021/jacs.6b13155
Return to citation in text: [1] [2] [3] -
Dao, H. T.; Li, C.; Michaudel, Q.; Maxwell, B. D.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 8046–8049. doi:10.1021/jacs.5b05144
Return to citation in text: [1] -
Crossley, S. W. M.; Barabé, F.; Shenvi, R. A. J. Am. Chem. Soc. 2014, 136, 16788–16791. doi:10.1021/ja5105602
Return to citation in text: [1] [2] -
Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2007, 46, 4519–4522. doi:10.1002/anie.200700575
Return to citation in text: [1] [2] -
Gaspar, B.; Carreira, E. M. J. Am. Chem. Soc. 2009, 131, 13214–13215. doi:10.1021/ja904856k
Return to citation in text: [1] [2] [3] -
Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2016, 138, 8718–8721. doi:10.1021/jacs.6b05271
Return to citation in text: [1] [2] [3] -
Ma, X.; Dang, H.; Rose, J. A.; Rablen, P.; Herzon, S. B. J. Am. Chem. Soc. 2017, 139, 5998–6007. doi:10.1021/jacs.7b02388
Return to citation in text: [1] [2] [3] -
Bordi, S.; Starr, J. T. Org. Lett. 2017, 19, 2290–2293. doi:10.1021/acs.orglett.7b00833
Return to citation in text: [1] -
Crossley, S. W. M.; Martinez, R. M.; Guevara-Zuluaga, S.; Shenvi, R. A. Org. Lett. 2016, 18, 2620–2623. doi:10.1021/acs.orglett.6b01047
Return to citation in text: [1] [2] -
Green, S. A.; Matos, J. L. M.; Yagi, A.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 12779–12782. doi:10.1021/jacs.6b08507
Return to citation in text: [1] [2] [3] -
Green, S. A.; Vásquez-Céspedes, S.; Shenvi, R. A. J. Am. Chem. Soc. 2018. doi:10.1021/jacs.8b05868
Return to citation in text: [1] -
Shenvi, R. A.; Guerrero, C. A.; Shi, J.; Li, C.-C.; Baran, P. S. J. Am. Chem. Soc. 2008, 130, 7241–7243. doi:10.1021/ja8023466
Return to citation in text: [1] -
Schindler, C. S.; Stephenson, C. R. J.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 8852–8855. doi:10.1002/anie.200803655
Return to citation in text: [1] -
Jeker, O. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3474–3477. doi:10.1002/anie.201109175
Return to citation in text: [1] -
Barker, T. J.; Duncan, K. K.; Otrubova, K.; Boger, D. L. ACS Med. Chem. Lett. 2013, 4, 985–988. doi:10.1021/ml400281w
Return to citation in text: [1] -
Leggans, E. K.; Duncan, K. K.; Barker, T. J.; Schleicher, K. D.; Boger, D. L. J. Med. Chem. 2013, 56, 628–639. doi:10.1021/jm3015684
Return to citation in text: [1] -
Shigehisa, H.; Suwa, Y.; Furiya, N.; Nakaya, Y.; Fukushima, M.; Ichihashi, Y.; Hiroya, K. Angew. Chem., Int. Ed. 2013, 52, 3646–3649. doi:10.1002/anie.201210099
Return to citation in text: [1] -
George, D. T.; Kuenstner, E. J.; Pronin, S. V. J. Am. Chem. Soc. 2015, 137, 15410–15413. doi:10.1021/jacs.5b11129
Return to citation in text: [1] -
Ruider, S. A.; Sandmeier, T.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 2378–2382. doi:10.1002/anie.201410419
Return to citation in text: [1] -
Allemann, O.; Brutsch, M.; Lukesh, J. C.; Brody, D. M.; Boger, D. L. J. Am. Chem. Soc. 2016, 138, 8376–8379. doi:10.1021/jacs.6b04330
Return to citation in text: [1] -
Waser, J.; Gaspar, B.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2006, 128, 11693–11712. doi:10.1021/ja062355+
Return to citation in text: [1] [2] -
Mukaiyama, T.; Isayama, S.; Inoki, S.; Kato, K.; Yamada, T.; Takai, T. Chem. Lett. 1989, 18, 449–452. doi:10.1246/cl.1989.449
Return to citation in text: [1] -
Inoki, S.; Kato, K.; Takai, T.; Isayama, S.; Yamada, T.; Mukaiyama, T. Chem. Lett. 1989, 18, 515–518. doi:10.1246/cl.1989.515
Return to citation in text: [1] -
Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 569–572. doi:10.1246/cl.1989.569
Return to citation in text: [1] -
Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 1071–1074. doi:10.1246/cl.1989.1071
Return to citation in text: [1] -
Kato, K.; Yamada, T.; Takai, T.; Inoki, S.; Isayama, S. Bull. Chem. Soc. Jpn. 1990, 63, 179–186. doi:10.1246/bcsj.63.179
Return to citation in text: [1] -
Isayama, S. Bull. Chem. Soc. Jpn. 1990, 63, 1305–1310. doi:10.1246/bcsj.63.1305
Return to citation in text: [1] -
Gaspar, B.; Waser, J.; Carreira, E. M. Synthesis 2007, 3839–3845. doi:10.1055/s-2007-1000817
Return to citation in text: [1] -
Hunger, K.; Mischke, P.; Rieper, W.; Zhang, S. Azo Dyes. Ullmann's Encyclopedia of Industrial Chemistr; Wiley-VCH Verlag GmbH & Co. KGaA, 2000. doi:10.1002/14356007.o03_o07.pub2
Return to citation in text: [1] -
Acton, Q. A. Azo Compounds—Advances in Research and Application: 2013 Edition: ScholarlyBrief; ScholarlyEditions: Atlanta, GA, 2013.
Return to citation in text: [1]
| 1. | Iwasaki, K.; Wan, K. K.; Oppedisano, A.; Crossley, S. W. M.; Shenvi, R. A. J. Am. Chem. Soc. 2014, 136, 1300–1303. doi:10.1021/ja412342g |
| 2. | King, S. M.; Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2014, 136, 6884–6887. doi:10.1021/ja502885c |
| 3. | Ma, X.; Herzon, S. B. Chem. Sci. 2015, 6, 6250–6255. doi:10.1039/C5SC02476E |
| 4. | Obradors, C.; Martinez, R. M.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 4962–4971. doi:10.1021/jacs.6b02032 |
| 28. | Lo, J. C.; Kim, D.; Pan, C.-M.; Edwards, J. T.; Yabe, Y.; Gui, J.; Qin, T.; Gutiérrez, S.; Giacoboni, J.; Smith, M. W.; Holland, P. L.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 2484–2503. doi:10.1021/jacs.6b13155 |
| 33. | Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2016, 138, 8718–8721. doi:10.1021/jacs.6b05271 |
| 34. | Ma, X.; Dang, H.; Rose, J. A.; Rablen, P.; Herzon, S. B. J. Am. Chem. Soc. 2017, 139, 5998–6007. doi:10.1021/jacs.7b02388 |
| 35. | Bordi, S.; Starr, J. T. Org. Lett. 2017, 19, 2290–2293. doi:10.1021/acs.orglett.7b00833 |
| 32. | Gaspar, B.; Carreira, E. M. J. Am. Chem. Soc. 2009, 131, 13214–13215. doi:10.1021/ja904856k |
| 36. | Crossley, S. W. M.; Martinez, R. M.; Guevara-Zuluaga, S.; Shenvi, R. A. Org. Lett. 2016, 18, 2620–2623. doi:10.1021/acs.orglett.6b01047 |
| 37. | Green, S. A.; Matos, J. L. M.; Yagi, A.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 12779–12782. doi:10.1021/jacs.6b08507 |
| 38. | Green, S. A.; Vásquez-Céspedes, S.; Shenvi, R. A. J. Am. Chem. Soc. 2018. doi:10.1021/jacs.8b05868 |
| 33. | Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2016, 138, 8718–8721. doi:10.1021/jacs.6b05271 |
| 34. | Ma, X.; Dang, H.; Rose, J. A.; Rablen, P.; Herzon, S. B. J. Am. Chem. Soc. 2017, 139, 5998–6007. doi:10.1021/jacs.7b02388 |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 30. | Crossley, S. W. M.; Barabé, F.; Shenvi, R. A. J. Am. Chem. Soc. 2014, 136, 16788–16791. doi:10.1021/ja5105602 |
| 31. | Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2007, 46, 4519–4522. doi:10.1002/anie.200700575 |
| 56. | Hunger, K.; Mischke, P.; Rieper, W.; Zhang, S. Azo Dyes. Ullmann's Encyclopedia of Industrial Chemistr; Wiley-VCH Verlag GmbH & Co. KGaA, 2000. doi:10.1002/14356007.o03_o07.pub2 |
| 57. | Acton, Q. A. Azo Compounds—Advances in Research and Application: 2013 Edition: ScholarlyBrief; ScholarlyEditions: Atlanta, GA, 2013. |
| 5. | Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 573–576. doi:10.1246/cl.1989.573 |
| 6. | Ishikawa, H.; Colby, D. A.; Boger, D. L. J. Am. Chem. Soc. 2008, 130, 420–421. doi:10.1021/ja078192m |
| 7. | Ishikawa, H.; Colby, D. A.; Seto, S.; Va, P.; Tam, A.; Kakei, H.; Rayl, T. J.; Hwang, I.; Boger, D. L. J. Am. Chem. Soc. 2009, 131, 4904–4916. doi:10.1021/ja809842b |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 9. | Ma, X.; Herzon, S. B. J. Org. Chem. 2016, 81, 8673–8695. doi:10.1021/acs.joc.6b01709 |
| 32. | Gaspar, B.; Carreira, E. M. J. Am. Chem. Soc. 2009, 131, 13214–13215. doi:10.1021/ja904856k |
| 17. | Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J. C.; Lee, B. J.; Spergel, S. H.; Mertzman, M. E.; Pitts, W. J.; La Cruz, T. E.; Schmidt, M. A.; Darvatkar, N.; Natarajan, S. R.; Baran, P. S. Science 2015, 348, 886–891. doi:10.1126/science.aab0245 |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 14. | Kato, K.; Mukaiyama, T. Chem. Lett. 1992, 21, 1137–1140. doi:10.1246/cl.1992.1137 |
| 15. | Waser, J.; Carreira, E. M. J. Am. Chem. Soc. 2004, 126, 5676–5677. doi:10.1021/ja048698u |
| 16. | Waser, J.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2005, 127, 8294–8295. doi:10.1021/ja052164r |
| 17. | Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J. C.; Lee, B. J.; Spergel, S. H.; Mertzman, M. E.; Pitts, W. J.; La Cruz, T. E.; Schmidt, M. A.; Darvatkar, N.; Natarajan, S. R.; Baran, P. S. Science 2015, 348, 886–891. doi:10.1126/science.aab0245 |
| 22. | Choi, J.; Tang, L.; Norton, J. R. J. Am. Chem. Soc. 2007, 129, 234–240. doi:10.1021/ja066325i |
| 23. | Choi, J.; Pulling, M. E.; Smith, D. M.; Norton, J. R. J. Am. Chem. Soc. 2008, 130, 4250–4252. doi:10.1021/ja710455c |
| 24. | Li, G.; Han, A.; Pulling, M. E.; Estes, D. P.; Norton, J. R. J. Am. Chem. Soc. 2012, 134, 14662–14665. doi:10.1021/ja306037w |
| 25. | Lo, J. C.; Yabe, Y.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 1304–1307. doi:10.1021/ja4117632 |
| 26. | Lo, J. C.; Gui, J.; Yabe, Y.; Pan, C.-M.; Baran, P. S. Nature 2014, 516, 343–348. doi:10.1038/nature14006 |
| 27. | Kuo, J. L.; Hartung, J.; Han, A.; Norton, J. R. J. Am. Chem. Soc. 2015, 137, 1036–1039. doi:10.1021/ja511883b |
| 28. | Lo, J. C.; Kim, D.; Pan, C.-M.; Edwards, J. T.; Yabe, Y.; Gui, J.; Qin, T.; Gutiérrez, S.; Giacoboni, J.; Smith, M. W.; Holland, P. L.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 2484–2503. doi:10.1021/jacs.6b13155 |
| 12. | Barker, T. J.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 13588–13591. doi:10.1021/ja3063716 |
| 13. | Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h |
| 29. | Dao, H. T.; Li, C.; Michaudel, Q.; Maxwell, B. D.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 8046–8049. doi:10.1021/jacs.5b05144 |
| 16. | Waser, J.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2005, 127, 8294–8295. doi:10.1021/ja052164r |
| 48. | Waser, J.; Gaspar, B.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2006, 128, 11693–11712. doi:10.1021/ja062355+ |
| 55. | Gaspar, B.; Waser, J.; Carreira, E. M. Synthesis 2007, 3839–3845. doi:10.1055/s-2007-1000817 |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 11. | Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 5758–5760. doi:10.1002/anie.200801760 |
| 5. | Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 573–576. doi:10.1246/cl.1989.573 |
| 49. | Mukaiyama, T.; Isayama, S.; Inoki, S.; Kato, K.; Yamada, T.; Takai, T. Chem. Lett. 1989, 18, 449–452. doi:10.1246/cl.1989.449 |
| 50. | Inoki, S.; Kato, K.; Takai, T.; Isayama, S.; Yamada, T.; Mukaiyama, T. Chem. Lett. 1989, 18, 515–518. doi:10.1246/cl.1989.515 |
| 51. | Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 569–572. doi:10.1246/cl.1989.569 |
| 52. | Isayama, S.; Mukaiyama, T. Chem. Lett. 1989, 18, 1071–1074. doi:10.1246/cl.1989.1071 |
| 53. | Kato, K.; Yamada, T.; Takai, T.; Inoki, S.; Isayama, S. Bull. Chem. Soc. Jpn. 1990, 63, 179–186. doi:10.1246/bcsj.63.179 |
| 54. | Isayama, S. Bull. Chem. Soc. Jpn. 1990, 63, 1305–1310. doi:10.1246/bcsj.63.1305 |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 9. | Ma, X.; Herzon, S. B. J. Org. Chem. 2016, 81, 8673–8695. doi:10.1021/acs.joc.6b01709 |
| 10. | Girijavallabhan, V.; Alvarez, C.; Njoroge, F. G. J. Org. Chem. 2011, 76, 6442–6446. doi:10.1021/jo201016z |
| 18. | Eisenberg, D. C.; Norton, J. R. Isr. J. Chem. 1991, 31, 55–66. doi:10.1002/ijch.199100006 |
| 19. | Gansäuer, A.; Shi, L.; Otte, M.; Huth, I.; Rosales, A.; Sancho-Sanz, I.; Padial, N. M.; Oltra, J. E. Hydrogen Atom Donors: Recent Developments. In Radicals in Synthesis III; Heinrich, M.; Gansäuer, A., Eds.; Topics in Current Chemistry; Springer: Berlin Heidelberg, 2012. doi:10.1007/128_2011_124 |
| 20. | Hoffmann, R. W. Chem. Soc. Rev. 2016, 45, 577–583. doi:10.1039/C5CS00423C |
| 21. | Crossley, S. W. M.; Obradors, C.; Martinez, R. M.; Shenvi, R. A. Chem. Rev. 2016, 116, 8912–9000. doi:10.1021/acs.chemrev.6b00334 |
| 10. | Girijavallabhan, V.; Alvarez, C.; Njoroge, F. G. J. Org. Chem. 2011, 76, 6442–6446. doi:10.1021/jo201016z |
| 28. | Lo, J. C.; Kim, D.; Pan, C.-M.; Edwards, J. T.; Yabe, Y.; Gui, J.; Qin, T.; Gutiérrez, S.; Giacoboni, J.; Smith, M. W.; Holland, P. L.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 2484–2503. doi:10.1021/jacs.6b13155 |
| 37. | Green, S. A.; Matos, J. L. M.; Yagi, A.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 12779–12782. doi:10.1021/jacs.6b08507 |
| 33. | Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2016, 138, 8718–8721. doi:10.1021/jacs.6b05271 |
| 34. | Ma, X.; Dang, H.; Rose, J. A.; Rablen, P.; Herzon, S. B. J. Am. Chem. Soc. 2017, 139, 5998–6007. doi:10.1021/jacs.7b02388 |
| 6. | Ishikawa, H.; Colby, D. A.; Boger, D. L. J. Am. Chem. Soc. 2008, 130, 420–421. doi:10.1021/ja078192m |
| 39. | Shenvi, R. A.; Guerrero, C. A.; Shi, J.; Li, C.-C.; Baran, P. S. J. Am. Chem. Soc. 2008, 130, 7241–7243. doi:10.1021/ja8023466 |
| 40. | Schindler, C. S.; Stephenson, C. R. J.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 8852–8855. doi:10.1002/anie.200803655 |
| 41. | Jeker, O. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3474–3477. doi:10.1002/anie.201109175 |
| 42. | Barker, T. J.; Duncan, K. K.; Otrubova, K.; Boger, D. L. ACS Med. Chem. Lett. 2013, 4, 985–988. doi:10.1021/ml400281w |
| 43. | Leggans, E. K.; Duncan, K. K.; Barker, T. J.; Schleicher, K. D.; Boger, D. L. J. Med. Chem. 2013, 56, 628–639. doi:10.1021/jm3015684 |
| 44. | Shigehisa, H.; Suwa, Y.; Furiya, N.; Nakaya, Y.; Fukushima, M.; Ichihashi, Y.; Hiroya, K. Angew. Chem., Int. Ed. 2013, 52, 3646–3649. doi:10.1002/anie.201210099 |
| 45. | George, D. T.; Kuenstner, E. J.; Pronin, S. V. J. Am. Chem. Soc. 2015, 137, 15410–15413. doi:10.1021/jacs.5b11129 |
| 46. | Ruider, S. A.; Sandmeier, T.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 2378–2382. doi:10.1002/anie.201410419 |
| 47. | Allemann, O.; Brutsch, M.; Lukesh, J. C.; Brody, D. M.; Boger, D. L. J. Am. Chem. Soc. 2016, 138, 8376–8379. doi:10.1021/jacs.6b04330 |
| 13. | Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h |
| 11. | Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 5758–5760. doi:10.1002/anie.200801760 |
| 2. | King, S. M.; Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2014, 136, 6884–6887. doi:10.1021/ja502885c |
| 12. | Barker, T. J.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 13588–13591. doi:10.1021/ja3063716 |
| 21. | Crossley, S. W. M.; Obradors, C.; Martinez, R. M.; Shenvi, R. A. Chem. Rev. 2016, 116, 8912–9000. doi:10.1021/acs.chemrev.6b00334 |
| 2. | King, S. M.; Ma, X.; Herzon, S. B. J. Am. Chem. Soc. 2014, 136, 6884–6887. doi:10.1021/ja502885c |
| 8. | Leggans, E. K.; Barker, T. J.; Duncan, K. K.; Boger, D. L. Org. Lett. 2012, 14, 1428–1431. doi:10.1021/ol300173v |
| 10. | Girijavallabhan, V.; Alvarez, C.; Njoroge, F. G. J. Org. Chem. 2011, 76, 6442–6446. doi:10.1021/jo201016z |
| 11. | Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 5758–5760. doi:10.1002/anie.200801760 |
| 13. | Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h |
| 15. | Waser, J.; Carreira, E. M. J. Am. Chem. Soc. 2004, 126, 5676–5677. doi:10.1021/ja048698u |
| 16. | Waser, J.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2005, 127, 8294–8295. doi:10.1021/ja052164r |
| 30. | Crossley, S. W. M.; Barabé, F.; Shenvi, R. A. J. Am. Chem. Soc. 2014, 136, 16788–16791. doi:10.1021/ja5105602 |
| 31. | Gaspar, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2007, 46, 4519–4522. doi:10.1002/anie.200700575 |
| 32. | Gaspar, B.; Carreira, E. M. J. Am. Chem. Soc. 2009, 131, 13214–13215. doi:10.1021/ja904856k |
| 36. | Crossley, S. W. M.; Martinez, R. M.; Guevara-Zuluaga, S.; Shenvi, R. A. Org. Lett. 2016, 18, 2620–2623. doi:10.1021/acs.orglett.6b01047 |
| 37. | Green, S. A.; Matos, J. L. M.; Yagi, A.; Shenvi, R. A. J. Am. Chem. Soc. 2016, 138, 12779–12782. doi:10.1021/jacs.6b08507 |
| 48. | Waser, J.; Gaspar, B.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2006, 128, 11693–11712. doi:10.1021/ja062355+ |
© 2018 Ma and Herzon; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)