

Abstract
Two routes to the title compounds are evaluated. First, a ca. 0.01 M CH2Cl2 solution of H3B·P((CH2)6CH=CH2)3 (1·BH3) is treated with 5 mol % of Grubbs' first generation catalyst (0 °C to reflux), followed by H2 (5 bar) and Wilkinson's catalyst (55 °C). Column chromatography affords H3B·P(n-C8H17)3 (1%), H3B·P((CH2)13CH2)(n-C8H17) (8%; see text for tie bars that indicate additional phosphorus–carbon linkages, which are coded in the abstract with italics), H3B·P((CH2)13CH2)((CH2)14)P((CH2)13CH2)·BH3 (6·2BH3, 10%), in,out-H3B·P((CH2)14)3P·BH3 (in,out-2·2BH3, 4%) and the stereoisomer (in,in/out,out)-2·2BH3 (2%). Four of these structures are verified by independent syntheses. Second, 1,14-tetradecanedioic acid is converted (reduction, bromination, Arbuzov reaction, LiAlH4) to H2P((CH2)14)PH2 (10; 76% overall yield). The reaction with H3B·SMe2 gives 10·2BH3, which is treated with n-BuLi (4.4 equiv) and Br(CH2)6CH=CH2 (4.0 equiv) to afford the tetraalkenyl precursor (H2C=CH(CH2)6)2(H3B)P((CH2)14)P(BH3)((CH2)6CH=CH2)2 (11·2BH3; 18%). Alternative approaches to 11·2BH3 (e.g., via 11) were unsuccessful. An analogous metathesis/hydrogenation/chromatography sequence with 11·2BH3 (0.0010 M in CH2Cl2) gives 6·2BH3 (5%), in,out-2·2BH3 (6%), and (in,in/out,out)-2·2BH3 (7%). Despite the doubled yield of 2·2BH3, the longer synthesis of 11·2BH3 vs 1·BH3 renders the two routes a toss-up; neither compares favorably with precious metal templated syntheses.
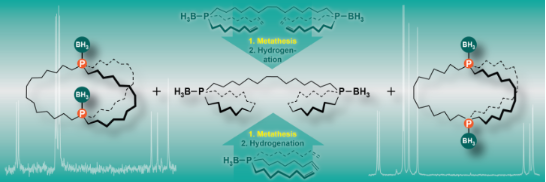
Graphical Abstract
Introduction
We have found that a variety of metal complexes with trans-phosphine ligands of the formula P((CH2)mCH=CH2)3 (1; m = 4–14) undergo threefold interligand ring closing alkene metatheses to give, after hydrogenations, metal complexes of in,in isomers of macrocyclic dibridgehead diphosphines [1-13]. Representative examples with square planar complexes are shown in Scheme 1. Analogous sequences with trigonal bipyramidal substrates proceed in somewhat higher overall yields, as analyzed elsewhere [1-4]. Setaka has developed a similar chemistry in which the phosphorus atoms are replaced by silicon and the metal fragment by p-phenylene (p-C6H4) or related aromatic moieties [14-19]. These types of compounds are viewed as promising candidates for molecular gyroscopes [14-21].
![[1860-5397-14-211-i1]](/bjoc/content/inline/1860-5397-14-211-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Syntheses of gyroscope like platinum and rhodium complexes and dibridgehead diphosphines derived therefrom.
Scheme 1: Syntheses of gyroscope like platinum and rhodium complexes and dibridgehead diphosphines derived th...
We subsequently developed an interest in the free dibridgehead diphosphine ligands P((CH2)n)3P (n = 14, 2; 18, 3), prompted in part by the unexpected discovery of the facile demetalations shown in Scheme 1 [5,6,10,22]. Such compounds were previously known only for much smaller ring sizes (n < 4) [23]. These reactions require excesses of certain nucleophiles, and the mechanisms remain under study. The yields are quite good, but the routes are stoichiometric in precious metals. Although the metals can be recovered as species such as K2Pt(CN)4 or RhCl(PMe3)3, we have nonetheless sought to develop more economical protocols.
The analogous Fe(CO)3 adducts are easily prepared [1-4], but in efforts to date it has not been possible to efficiently remove the dibridgehead diphosphine ligands from the low cost iron fragment. Oxidations that lead to the corresponding dibridgehead diphosphine dioxides (O=)P((CH2)n)3P(=O) have exhibited promise, but purification has been problematic [24]. Indeed, phosphine oxides are everyday precursors to phosphines, so we have considered various non-metal-templated routes to 2·2(=O), 3·2(=O), and related species. However, as described in the discussion section, the yields have not been competitive [25].
Another preliminary point concerns the ability of macrocyclic dibridgehead diphosphorus compounds to exhibit in/out isomerism [26]. As shown in Scheme 1, there are three limiting configurations for 2 and 3: in,in, out,out, and in,out (identical to out,in). The first two, as well as the degenerate in,out pair, can rapidly interconvert by a process termed homeomorphic isomerization [26,27], which is akin to turning the molecules inside out. Readers are referred to earlier publications in this series for additional details [22,25,28-30]. Interconversions between the in,in/out,out and in,out/out,in manifolds require phosphorus inversion and temperatures considerably in excess of 100 °C.
In this paper, we describe two non-metal-templated approaches to 2 that are based upon metatheses of phosphine boranes of alkene containing phosphines. The first involves the monophosphorus precursor H3B·P((CH2)6CH=CH2)3 (1·BH3) [31], and the second a diphosphorus precursor in which one of the methylene chains linking the two phosphorus atoms has already been installed. The advantages and limitations of each are analyzed in detail. Some of the results (Scheme 2) have appeared in the supporting information of a preliminary communication [28], and others in a dissertation [32].
Results
1. Monophosphorus precursors
As reported earlier [31], the alkene containing phosphine P((CH2)6CH=CH2)3 (1) can be prepared in 87% yield from the reaction of PCl3 and MgBr(CH2)6CH=CH2. Following the addition of H3B·SMe2, the phosphine borane 1·BH3 can be isolated in 65–85% yields [31], as shown in Scheme 2. It is critical to avoid an excess of H3B·SMe2, as this brings the C=C units into play. In fact, when substoichiometric amounts of H3B·SMe2 are added to THF solutions of purified 1·BH3, gels immediately form.
![[1860-5397-14-211-i2]](/bjoc/content/inline/1860-5397-14-211-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and alkene metathesis of the monophosphorus precursor 1·BH3.
Scheme 2: Synthesis and alkene metathesis of the monophosphorus precursor 1·BH3.
A ca. 0.01 M CH2Cl2 solution of 1·BH3 and a ca. 0.002 M CH2Cl2 solution of Grubbs' first generation catalyst (3 mol %) were combined at 0 °C. The mixture was warmed to room temperature, and a second charge of Grubbs' catalyst added (2 mol %). The sample was refluxed, and then filtered through silica gel. The filtrate was concentrated and treated with H2 (5 bar) and Wilkinson's catalyst (55 °C). The mixture was taken to dryness and the residue tediously chromatographed on a silica gel column. Numerous fractions were collected and analyzed by TLC. The mass recovery from the column was 33% of theory (for complete metathesis).
More than ten mobile products could be discerned, but only five could be isolated in pure form and ultimately identified. These are described in order of elution. Each was analyzed by NMR (1H, 31P{1H}, 13C{1H}; always CDCl3) and IR spectroscopy, mass spectrometry, and microanalysis, as summarized in the experimental section. The 13C{1H} NMR spectra proved to be most diagnostic of structure, and were analyzed in detail. The 31P{1H} NMR spectra were all very similar (broad apparent doublets due to phosphorus boron coupling).
First, traces of a colorless oil were obtained. The 1H NMR spectrum showed a characteristic triplet at 0.83 ppm consistent with a terminal methyl group. The 13C{1H} NMR spectrum exhibited eight signals, two of which were phosphorus coupled doublets. One of the singlets (14.0 ppm) was typical of a terminal methyl group. Based upon these data, and the integration of the 1H NMR spectrum, the oil was assigned as the hydrogenated phosphine borane H3B·P(n-C8H17)3 (4·BH3), a known compound [33]. The yield was only 1%.
Next, another colorless oil eluted. The 1H NMR and 13C{1H} NMR spectra again exhibited signals characteristic of a methyl group (0.86 ppm, t; 14.0 ppm, s). Integration of the 1H NMR spectrum established a 14:1 area ratio for the methylene (1.62–1.19 ppm) and methyl signals. The 13C{1H} NMR spectrum featured one set of seven signals and another set of eight with an intensity ratio of approximately 2:1. The less intense set resembled the signals arising from the n-octyl groups in 4·BH3. The more intense set was very similar to the signals arising from the cyclic ![[Graphic 1]](/bjoc/content/inline/1860-5397-14-211-i11.svg?max-width=637&scale=1.18182) substructures of 6·2BH3 (described below) and a phosphine borane
substructures of 6·2BH3 (described below) and a phosphine borane ![[Graphic 2]](/bjoc/content/inline/1860-5397-14-211-i12.svg?max-width=637&scale=1.18182) reported earlier [34]. The mass spectrum exhibited an intense ion at m/z 340 (5+, 93%), and no ions of higher mass. Hence, the oil was assigned as the monocyclic intramolecular metathesis product
reported earlier [34]. The mass spectrum exhibited an intense ion at m/z 340 (5+, 93%), and no ions of higher mass. Hence, the oil was assigned as the monocyclic intramolecular metathesis product ![[Graphic 3]](/bjoc/content/inline/1860-5397-14-211-i13.svg?max-width=637&scale=1.18182) (5·BH3; see Scheme 2). The yield was 8%.
(5·BH3; see Scheme 2). The yield was 8%.
The third product was also a colorless oil. The 13C{1H} NMR spectrum exhibited seven signals, three of which were phosphorus coupled doublets (second spectrum from top, Figure 1). Analogous coupling patterns are found with the free dibridgehead diphosphines 2 and 3 in Scheme 1. No NMR signals diagnostic of methyl groups were present, and further analysis is presented along with that for an isomer below.
![[1860-5397-14-211-1]](/bjoc/content/figures/1860-5397-14-211-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The 13C{1H} NMR spectra (CDCl3, 100 MHz) of in,out-2·2BH3, (in,in/out,out)-2·2BH3, 6·2BH3, and the crude reaction mixture after hydrogenation from Scheme 5 (top); doublets are marked with an asterisk.
Figure 1: The 13C{1H} NMR spectra (CDCl3, 100 MHz) of in,out-2·2BH3, (in,in/out,out)-2·2BH3, 6·2BH3, and the ...
A white powder was obtained next. The 13C{1H} NMR spectrum exhibited fourteen signals, half of which were approximately twice as intense as the others. Two signals of each set exhibited phosphorus coupling. The overall pattern was quite similar to those shown by metal complexes with cis or trans coordinating diphosphine ligands of the formula ![[Graphic 4]](/bjoc/content/inline/1860-5397-14-211-i14.svg?max-width=637&scale=1.18182) (6) [6,7,12,13,35]. This suggested the diphosphine diborane structure 6·2BH3 (see Scheme 2), which is derived from one metathesis involving alkenyl moieties on different phosphorus atoms, and two metatheses of alkenyl moieties on identical phosphorus atoms. The yield was 10%. The structure has been confirmed by an independent synthesis (detachment of the diphosphine from a platinum complex followed by borane addition) and a crystal structure [6].
(6) [6,7,12,13,35]. This suggested the diphosphine diborane structure 6·2BH3 (see Scheme 2), which is derived from one metathesis involving alkenyl moieties on different phosphorus atoms, and two metatheses of alkenyl moieties on identical phosphorus atoms. The yield was 10%. The structure has been confirmed by an independent synthesis (detachment of the diphosphine from a platinum complex followed by borane addition) and a crystal structure [6].
Finally, another white powder was obtained. As with the previous oil isolated above, the 13C{1H} NMR spectrum exhibited seven signals, three of which were phosphorus coupled doublets (third spectrum from top, Figure 1). Both spectra were consistent with dibridgehead diphosphine diboranes H3B·P((CH2)14)3P·BH3 (2·2BH3) derived from threefold intermolecular metatheses of 1·BH3. Based upon independent syntheses from the dibridgehead diphosphines 2 obtained in Scheme 1 [6], they were assigned as in,out-2·2BH3 (4%) and the stereoisomer (in,in/out,out)-2·2BH3 (2%), as shown in Scheme 2. The depiction of the latter as an out,out (vs in,in) isomer in Scheme 2 is arbitrary, but represents the form found in a confirming crystal structure [6].
Parallel reactions were conducted with Grubbs' second generation catalyst and the nitro-Grela catalyst [36]. However, the combined yields of 2 diminished.
2. Diphosphorus precursors
Since the yields of the cage like diphosphine diboranes 2·2BH3 in Scheme 2 were – as expected – very low, alternative strategies were considered. The poor mass balance was attributed, at least in part, to the formation of oligomeric products that were retained on the column. Improvements might be expected from precursors in which one of the methylene chains tethering the two phosphorus atoms was pre-formed. Thus, we set out to prepare a tetraalkenyl metathesis precursor as shown in Scheme 3.
![[1860-5397-14-211-i3]](/bjoc/content/inline/1860-5397-14-211-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the diphosphorus precursor 11·2BH3.
Scheme 3: Synthesis of the diphosphorus precursor 11·2BH3.
The first step, a previously reported reduction of commercial 1,14-tetradecanedioic acid to 1,14-tetradecanediol (7) [37], was followed by an Appel reaction to give 1,14-dibromotetradecane (8) [38-43]. An Arbuzov reaction then afforded the diphosphonate (EtO)2(O=)P((CH2)14)P(=O)(OEt)2 (9) [44]. Subsequent reduction with LiAlH4 gave the diprimary diphosphine H2P((CH2)14)PH2 (10) in 76% yield from 7 as a foul smelling white powder.
It has been shown that borane adducts of primary phosphines can be doubly deprotonated, and that the resulting phosphorus dianions can be bis(alkylated) [45-47]. Thus, the diphosphine 10 and H3B·SMe2 were reacted to give the diphosphine diborane H2(H3B)P((CH2)14)P(BH3)H2 (10·2BH3) as a white solid in 87% yield. A subsequent reaction with n-BuLi (4.4 equiv) and Br(CH2)6CH=CH2 (4.0 equiv) gave the tetraalkenyl target (H2C=CH(CH2)6)2(H3B)P((CH2)14)P(BH3)((CH2)6CH=CH2)2 (11·2BH3), but in only 18% yield.
Accordingly, two alternative routes to 11·2BH3 were considered. The initial step for the first is depicted in Scheme 4. Primary phosphines can be doubly deprotonated, analogously to borane adducts, and the phosphorus dianions subsequently bis(alkylated) [34,48]. Thus, 10 was treated with n-BuLi (4.1 equiv) and then Br(CH2)6CH=CH2 (4.0 equiv). Work-up gave the target compound (H2C=CH(CH2)6)2P((CH2)14)P((CH2)6CH=CH2)2 (11) in 72% yield. However, all attempts to convert 11 to 11·2BH3 gave only traces of the latter. Mainly insoluble material formed, which was presumed to be oligomeric and possibly derived from B–H additions to the alkenyl groups.
![[1860-5397-14-211-i4]](/bjoc/content/inline/1860-5397-14-211-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Truncated approaches to the diphosphorus precursor 11·2BH3 from 10.
Scheme 4: Truncated approaches to the diphosphorus precursor 11·2BH3 from 10.
In the second approach, 10 was first converted to the tetrachloride Cl2P((CH2)14)PCl2 (12) in 94% yield using triphosgene, a standard reagent for the chlorination of phosphorus–hydrogen bonds [49]. Since a direct reaction with an excess of the Grignard reagent BrMg(CH2)6CH=CH2 would give 11, a dead end, initial conversion to the bis(borane) adduct 12·2BH3 was envisioned. However, reactions of 12 and H3B·SMe2 (2.1 equiv) afforded only insoluble material.
Thus, despite the low yield of the final step in Scheme 3, reasonable quantities of the diphosphine diborane 11·2BH3 could be stockpiled. As shown in Scheme 5, 11·2BH3 was subjected to a metathesis/hydrogenation/column chromatography sequence similar to that for 1·BH3 in Scheme 2. However, a tenfold higher dilution was used in the metathesis step (0.0010 M as compared to 0.010 M).
![[1860-5397-14-211-i5]](/bjoc/content/inline/1860-5397-14-211-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Alkene metathesis of the diphosphorus precursor 11·2BH3.
Scheme 5: Alkene metathesis of the diphosphorus precursor 11·2BH3.
Figure 1 shows a 13C{1H} NMR spectrum of the crude product after hydrogenation stacked above spectra of the three products that could be isolated after the rather tedious column chromatography: the dibridgehead diphosphine diborane in,out-2·2BH3, its constitutional isomer 6·2BH3, and its stereoisomer (in,in/out,out)-2·2BH3. It can be inferred from the top spectrum that the three products were the major components and moreover present in approximately equal amounts. However, the isolated yields were affected by the challenging separation. In particular, in,out-2·2BH3 and 6·2BH3 eluted very closely, rendering some mixed fractions unavoidable and lowering the amounts of pure products.
Compared to the metathesis/hydrogenation sequence for 1·BH3 (Scheme 2) the yields of in,out-2·2BH3 and (in,in/out,out)-2·2BH3 (Scheme 5) are higher but still poor. Taking into account the overall yields (three steps from PCl3 and BrMg(CH2)6CH=CH2 in the first synthesis vs seven steps from 1,14-tetradecanedioic acid in the second), the latter route does not offer any advantage, even if one were to improve the conversion of 10·2BH3 to 11·2BH3.
Discussion
As contrasted in Scheme 6, Scheme 2 and Scheme 5 present two conceptually related routes to the isomeric title compound 2·2BH3. In the first, two trialkenylphosphine boranes (1·BH3 = I) must undergo metathesis. The first productive step is intermolecular, giving a diphosphorus compound with a P(CH2)6CH=CH(CH2)6P tether II that is positioned for subsequent intramolecular ring closing steps. Those involving alkenyl groups from different phosphorus atoms are productive (leading to 2·2BH3 via hydrogenation of IIIa), and those involving groups from the same phosphorus atoms are non-productive (leading to 6·2BH3 via hydrogenation of IVa). In the second, the starting material has a preformed P(CH2)14P tether (11·2BH3 = V), and the four alkenyl groups have reactivity options (→ IIIb or IVb) analogous to those of intermediate II with the P(CH2)6CH=CH(CH2)6P tether. Importantly, all of these steps are presumed to be largely under kinetic control, consistent with experience with the types of metatheses in Scheme 1 [1-13,34].
![[1860-5397-14-211-i6]](/bjoc/content/inline/1860-5397-14-211-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Schematic comparison of the key alkene metathesis steps in Scheme 2 and Scheme 5.
Scheme 6: Schematic comparison of the key alkene metathesis steps in Scheme 2 and Scheme 5.
Although the second route intuitively seems more favorable, after the initial intermolecular metathesis of 1·BH3 (I), both require an equivalent series of steps to reach (after hydrogenation) 2·2BH3. One reason 1·BH3 is an inferior substrate is that following the initial generation of a P(CH2)6CH=Ru species, two P(CH2)6CH=CH2 moieties remain available for non-productive intramolecular ring closing metathesis (giving VI). In contrast, with the analogous intermediate derived from 11·2BH3 (V), there is only one P(CH2)6CH=CH2 moiety that can give non-productive chemistry. It is also worth noting that high dilution provides less of an advantage in Scheme 2, as one wants to favor intermolecular over intramolecular metatheses in the first step. In Scheme 5, one wants to avoid intermolecular metatheses at all stages.
At present, we have no rationale for the in,out vs (in,in/out,out) isomer ratios for 2·2BH3. However, it is easy to map the sequence leading to each, as shown in Scheme 7. When there is only one tether between the two phosphorus atoms, the phosphorus–boron bonds can be arrayed in an anti fashion, as depicted in VII. When subsequent metatheses join alkenyl groups in the syn positions on each phosphorus atom (front to front and rear to rear), (in,in/out,out)-2·2BH3 must result (as drawn in Scheme 7, the out,out isomer would be the kinetic product). When the first metathesis does not join the syn positions, as in VIII (front to rear), one phosphorus–boron bond must subsequently be rotated by 180° to create a syn orientation for the second metathesis.
![[1860-5397-14-211-i7]](/bjoc/content/inline/1860-5397-14-211-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Steps that set the in,in/out,out vs in,out stereochemistry of 2·2BH3 in Scheme 2 and Scheme 5.
Scheme 7: Steps that set the in,in/out,out vs in,out stereochemistry of 2·2BH3 in Scheme 2 and Scheme 5.
Of course, if the first metathesis step does not require a syn relationship (per VIII), the same possibility can be entertained for the second (see IX). This would lead to an isomeric bicyclic compound with "crossed chains". We have sought to access such species by conducting metatheses of substrates of the types in Scheme 1 that give thirty-three membered macrocycles (n = 30) [7]. However, none have so far been detected. Other types of crossed chain in/out isomer systems have in fact been realized [25,30].
As communicated earlier [28] and will be described more fully in a later paper, both isomers of 2·2BH3 are easily deprotected to give the respective isomers of the dibridgehead diphosphine 2 in high yields. Since phosphine oxides are also easily converted to phosphines, one could consider parallel approaches to 2 via metatheses of the phosphine oxide (O=)P((CH2)6CH=CH2)3 (1(=O)) or diphosphine dioxide (H2C=CH(CH2)6)2(O=)P((CH2)14)P(=O)((CH2)6CH=CH2)2 (11·2(=O)). Given the poor results with 1·BH3 in Scheme 2, no attempt has been made to explore similar reactions with 1(=O).
However, as shown in Scheme 8, it has proved possible to synthesize the diphosphine dioxides 14, in which the two phosphorus atoms are tethered by a methylene chain, in two steps in 66–68% overall yields from diethyl phosphonate ((O=)PH(OEt)2), Grignard reagents BrMg(CH2)mCH=CH2, base (NaH), and appropriate α,ω-dibromides Br(CH2)nBr [25]. Following metathesis and hydrogenation, these afford dibridgehead diphosphine oxides 15 and 16 in 14–19% yields. This is slightly better than the combined yield of in,out- and (in,in/out,out)-2·2BH3 in Scheme 5, although the data are not strictly comparable as the ring sizes differ. It has not yet proved possible to efficiently separate the in/out isomers of 15 and 16. However, byproducts derived from metatheses of alkenyl groups on the same phosphorus atom – such as 17 (comparable to 6·2BH3) – appear to form in much smaller amounts.
![[1860-5397-14-211-i8]](/bjoc/content/inline/1860-5397-14-211-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Another non-metal-templated approach to dibridgehead diphosphorus compounds.
Scheme 8: Another non-metal-templated approach to dibridgehead diphosphorus compounds.
To our knowledge, only one macrocyclic dibridgehead diphosphine diborane has been previously reported, (in,in/out,out)-18·2BH3 in Scheme 9 [50,51]. This features triarylphosphorus bridgeheads and p-phenylene containing tethers that are long enough to allow rapid homeomorphic isomerization. The precursor 18·2(=O) was prepared by a threefold Williamson ether synthesis in surprisingly high yields (61% in,in/out,out and in,out combined) [50,51], likely aided by the geminal dialkyl effect associated with the quaternary centers [52].
![[1860-5397-14-211-i9]](/bjoc/content/inline/1860-5397-14-211-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Previously synthesized dibridgehead diphosphine diboranes.
Scheme 9: Previously synthesized dibridgehead diphosphine diboranes.
Finally, it should be noted that a number of alkene containing phosphine boranes have been employed in metathesis reactions [53,54]. In particular, the tetraalkenyl diphosphine diborane 19·2BH3 in Scheme 10 represents a downsized version of 11·2BH3. A species analogous to 6·2BH3, 20·2BH3, is obtained in much higher yield than any of the products in Scheme 5 [53]. Hence, selectivities can strongly depend upon the lengths of the methylene segments in the precursor.
![[1860-5397-14-211-i10]](/bjoc/content/inline/1860-5397-14-211-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Alkene metathesis of the tetraalkenyldiphosphine diborane 19·2BH3.
Scheme 10: Alkene metathesis of the tetraalkenyldiphosphine diborane 19·2BH3.
Conclusion
In conclusion, this work constitutes a further installment in the evolution of synthetic strategies for dibridgehead diphosphorus compounds that employ alkene metathesis. The new approaches (Scheme 2; Scheme 3 and Scheme 5) lack metal templates, which differentiates them from the routes presented in Scheme 1. However, neither is competitive with Scheme 1, despite eliminating the requirement for stoichiometric amounts of precious metals. Furthermore, preassembling a diphosphine diborane substrate per Scheme 3 and Scheme 5 is not competitive with the "shotgun" approach in Scheme 2, and both routes require comparably demanding preparative column chromatography. Hence, the most promising direction for future research would seem to be templated syntheses via non-precious metals [55]. This remains an area of ongoing investigation in our laboratory and further results will be reported in due course.
Experimental
General. Reactions (except hydrogenations) were conducted under inert atmospheres using standard Schlenk techniques. All chromatography was carried out under aerobic conditions. Additional data are supplied in Supporting Information File 1.
Metathesis/hydrogenation of H3B·P((CH2)6CH=CH2)3 (1·BH3; Scheme 2 [32]). A Schlenk flask was charged with 1·BH3 (1.177 g, 3.110 mmol) [31] and CH2Cl2 (320 mL; the resulting solution was 0.0097 M in 1·BH3) and cooled to 0 °C. A solution of Grubbs' first generation catalyst (0.077 g, 0.094 mmol, 3 mol %) in CH2Cl2 (50 mL) was added dropwise via syringe with stirring over 1 h. The cooling bath was removed. After 2 h, additional Grubbs' first generation catalyst was added as a solid (0.051 g, 0.062 mmol, 2 mol %). The flask was fitted with a condenser and the mixture was refluxed overnight, cooled to room temperature, and passed through a SiO2 pad (3 cm), which was rinsed with CH2Cl2. The eluate was concentrated to ca. 20 mL by rotary evaporation, and transferred to a Fischer–Porter bottle. Wilkinson's catalyst (0.086 g, 0.093 mmol, 3 mol %) was added, and the bottle was partially evacuated and charged with hydrogen (5 bar). The sample was kept at 55 ºC for 60 h. The solvent was removed and the residue was placed at the top of a chromatography column (SiO2, 3.5 × 36 cm), which was eluted with hexanes/CH2Cl2 (3:1 to 1:3 v/v) and then CH2Cl2. Fractions were assayed by TLC, combined where appropriate, and slowly evaporated to dryness in a fume hood. Some fractions (0.091 g total out of the recovered mass of 0.344 g) consisted of unidentified and/or impure products, or oligomers and polymers. Products that could be characterized are as follows (in order of elution).
H3B·P(n-C8H17)3 (4·BH3 [33]; 0.007 g, 0.018 mmol, 1%), colorless oil. Anal. calcd for C24H54BP (384.47): C, 74.98; H, 14.16; found: C, 74.93; H, 14.02; 1H NMR (400 MHz, CDCl3) δ 1.53–1.37 (m, 12H, CH2), 1.33–1.30 (m, 6H, CH2), 1.26–1.23 (m, 24H, CH2), 0.83 (t, 3JHH = 6.9 Hz, 9H, CH3), 0.47 and 0.19 (br apparent d, 3H, BH3); 13C{1H} NMR (101 MHz, CDCl3) δ 31.7 (s, CH2), 31.1 (d, JCP = 12.0 Hz, CH2), 29.0 (s, CH2), 28.9 (s, CH2), 22.9 (d, JCP = 34.3 Hz, CH2), 22.50 (s, CH2), 22.48 (s, CH2), 14.0 (s, CH3); 31P{1H} NMR (162 MHz, CDCl3) δ 15.9 and 15.5 (br apparent d); IR (oil film): 2926 (s), 2856 (m), 2366 (m), 1463 (m), 1413 (w), 1378 (w), 1135 (w), 1061 (m), 1034 (w), 807 (w), 764 (w), 722 (m) cm−1; MS (EI) [56]: 384 (M+, <1%), 370 ([M − BH3]+, 79%).
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-211-i15.svg?max-width=637&scale=1.18182) (5·BH3; 0.090 g, 0.25 mmol, 8%), colorless oil. Anal. calcd for C22H48BP (354.40): C, 74.56; H, 13.65; found: C, 74.27; H, 13.52; 1H NMR (500 MHz, CDCl3) δ 1.62–1.19 (m, 42H, CH2), 0.86 (t, 3H, 3JHH = 7.0 Hz, CH3), 0.48 and 0.26 (br apparent d, 3H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 31.7 (s, CH2), 31.2 (d, JCP = 12.6 Hz, CH2), 29.03 (s, CH2), 29.01 (s, CH2), 28.9 (d, JCP = 11.1 Hz, 2CH2), 26.7 (s, 2CH2), 26.53 (s, 2CH2), 26.48 (s, 2CH2), 26.1 (s, 2CH2), 23.8 (d, JCP = 35.4 Hz, CH2), 22.57 (d, JCP = 1.2 Hz, 2CH2), 22.55 (s, CH2), 22.3 (d, JCP = 33.6 Hz, CH2), 21.2 (d, JCP = 3.3 Hz, 2CH2), 14.0 (s, CH3); 31P {1H} NMR (202 MHz, CDCl3) δ 15.6 and 15.2 (br apparent d); IR (oil film): 2926 (s), 2856 (m), 2366 (m), 1459 (m), 1417 (w), 1135 (w), 1061 (m), 811 (m), 760 (m), 722 (m) cm−1; MS (EI) [56]: 340 ([M − BH3]+, 93%), 228 ([M − BH3 − C8H17 + 1]+, 100%).
(5·BH3; 0.090 g, 0.25 mmol, 8%), colorless oil. Anal. calcd for C22H48BP (354.40): C, 74.56; H, 13.65; found: C, 74.27; H, 13.52; 1H NMR (500 MHz, CDCl3) δ 1.62–1.19 (m, 42H, CH2), 0.86 (t, 3H, 3JHH = 7.0 Hz, CH3), 0.48 and 0.26 (br apparent d, 3H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 31.7 (s, CH2), 31.2 (d, JCP = 12.6 Hz, CH2), 29.03 (s, CH2), 29.01 (s, CH2), 28.9 (d, JCP = 11.1 Hz, 2CH2), 26.7 (s, 2CH2), 26.53 (s, 2CH2), 26.48 (s, 2CH2), 26.1 (s, 2CH2), 23.8 (d, JCP = 35.4 Hz, CH2), 22.57 (d, JCP = 1.2 Hz, 2CH2), 22.55 (s, CH2), 22.3 (d, JCP = 33.6 Hz, CH2), 21.2 (d, JCP = 3.3 Hz, 2CH2), 14.0 (s, CH3); 31P {1H} NMR (202 MHz, CDCl3) δ 15.6 and 15.2 (br apparent d); IR (oil film): 2926 (s), 2856 (m), 2366 (m), 1459 (m), 1417 (w), 1135 (w), 1061 (m), 811 (m), 760 (m), 722 (m) cm−1; MS (EI) [56]: 340 ([M − BH3]+, 93%), 228 ([M − BH3 − C8H17 + 1]+, 100%).
in,out-H3B·P((CH2)14)3P·BH3 (in,out-2·2BH3; 039 g, 0.057 mmol, 4%), colorless oil. Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found: C, 73.86; H, 13.49; 1H NMR (500 MHz, CDCl3) δ 1.56–1.51 (m, 12H, PCH2), 1.49–1.42 (m, 12H, CH2), 1.39–1.33 (m, 12H, CH2), 1.31–1.21 (m, 48H, CH2), 0.45 and 0.27 (br apparent d, 6H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 30.5 (d, JCP = 11.3 Hz, CH2), 28.35 (s, CH2), 28.28 (s, CH2), 28.2 (s, CH2), 28.1 (s, CH2), 23.0 (d, JCP = 34.3 Hz, CH2), 22.2 (d, JCP = 1.9 Hz, CH2); 31P{1H} NMR (202 MHz, CDCl3) δ 15.6 and 15.4 (br apparent d); IR (oil film): 2926 (s), 2853 (m), 2366 (w), 1459 (w), 1413 (w), 1135 (w), 1061 (m), 803 (w), 722 (w) cm−1; MS (MALDI+, THAP) [56]: 651.6 ([M – 2BH3 + 1]+, 100%).
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-211-i16.svg?max-width=637&scale=1.18182) (6·2BH3; 0.101 g, 0.149 mmol, 10%), white solid, mp 96 °C (capillary). Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found: C, 73.92; H, 13.47. The identity of this compound, which has been independently synthesized, has been confirmed crystallographically [6]. 1H NMR (500 MHz, CDCl3) δ 1.65–1.14 (br m, 84H, CH2), 0.49 and 0.26 (br apparent d, 6H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 31.3 (d, JCP = 12.6 Hz, CH2), 29.54 (s, CH2), 29.53 (s, CH2), 29.4 (s, CH2), 29.1 (s, CH2), 29.0 (d, JCP = 11.1 Hz, 2CH2), 26.8 (s, 2CH2), 26.6 (s, 2CH2), 26.5 (s, 2CH2), 26.1 (s, 2CH2), 23.8 (d, JCP = 35.3 Hz, CH2), 22.6 (d, JCP = 1.0 Hz, CH2), 22.3 (d, JCP = 33.5 Hz, 2CH2), 21.2 (d, JCP = 3.3 Hz, 2CH2); 31P {1H} NMR (202 MHz, CDCl3) δ 15.6 and 15.2 (br apparent d); IR (powder film): 2922 (s), 2853 (m), 2366 (m), 1459 (m), 1417 (w), 1135 (w), 1061 (m), 791 (w), 722 (m) cm−1; MS (EI) [56]: 678 (M+, 9%), 665 ([M − BH3]+, 100%), 652 ([M − 2BH3 + 1]+, 72%).
(6·2BH3; 0.101 g, 0.149 mmol, 10%), white solid, mp 96 °C (capillary). Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found: C, 73.92; H, 13.47. The identity of this compound, which has been independently synthesized, has been confirmed crystallographically [6]. 1H NMR (500 MHz, CDCl3) δ 1.65–1.14 (br m, 84H, CH2), 0.49 and 0.26 (br apparent d, 6H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 31.3 (d, JCP = 12.6 Hz, CH2), 29.54 (s, CH2), 29.53 (s, CH2), 29.4 (s, CH2), 29.1 (s, CH2), 29.0 (d, JCP = 11.1 Hz, 2CH2), 26.8 (s, 2CH2), 26.6 (s, 2CH2), 26.5 (s, 2CH2), 26.1 (s, 2CH2), 23.8 (d, JCP = 35.3 Hz, CH2), 22.6 (d, JCP = 1.0 Hz, CH2), 22.3 (d, JCP = 33.5 Hz, 2CH2), 21.2 (d, JCP = 3.3 Hz, 2CH2); 31P {1H} NMR (202 MHz, CDCl3) δ 15.6 and 15.2 (br apparent d); IR (powder film): 2922 (s), 2853 (m), 2366 (m), 1459 (m), 1417 (w), 1135 (w), 1061 (m), 791 (w), 722 (m) cm−1; MS (EI) [56]: 678 (M+, 9%), 665 ([M − BH3]+, 100%), 652 ([M − 2BH3 + 1]+, 72%).
(in,in/out,out)-H3B·P((CH2)14)3P·BH3 ((in,in/out,out)-2·2BH3; 0.016 g, 0.024 mmol, 2%), colorless oil that solidified to give a white powder, mp 112 °C. Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found: C, 74.71; H, 13.34; 1H NMR (500 MHz, CDCl3) δ 1.55–1.50 (m, 12H, CH2), 1.47–1.39 (m, 12H, CH2), 1.37–1.32 (m, 12H, CH2), 1.29–1.21 (m, 48H, CH2), 0.38 and 0.26 (br apparent d, 6H, BH3); 13C{1H} NMR (126 MHz, CDCl3) δ 30.6 (d, JCP = 12.1 Hz, CH2), 29.23 (s, CH2), 29.17 (s, CH2), 28.9 (s, CH2), 28.4 (s, CH2), 22.5 (d, JCP = 34.1 Hz, CH2), 22.1 (d, JCP = 2.7 Hz, CH2); 31P{1H} NMR (202 MHz, CDCl3) δ 14.9 and 14.7 (br apparent d); IR (powder film): 2922 (s), 2853 (s), 2366 (m), 1467 (m), 1413 (w), 1131 (w), 1061 (m), 807 (w), 760 (w), 718 (m) cm−1; MS (MALDI+, THAP) [56]: 702.0 ([M + Na]+, 98%), 666.0 ([M − BH3 + 1]+, 100%).
Metathesis/hydrogenation of (H2C=CH(CH2)6)2(H3B)P((CH2)14)P(BH3)((CH2)6CH=CH2)2 (11·2BH3; Scheme 5 [32]). Diphosphine diborane 11·2BH3 (1.222 g, 1.672 mmol), CH2Cl2 (1700 mL; the resulting solution was 0.0010 M in 11·2BH3), Grubbs' first generation catalyst (0.069 g, 0.083 mmol, 5 mol %), Wilkinson's catalyst (0.046 g, 0.050 mmol, ca. 3 mol %), and H2 were combined in a procedure analogous to that used for 1·BH3. An identical work-up gave in,out-2·2BH3 (0.072 g, 0.106 mmol, 6%, minor impurities evident by 13C{1H} NMR), 6·2BH3 (0.056 g, 0.083 mmol, 5%, minor impurities evident by 13C{1H} NMR), and (in,in/out,out)-2·2BH3 (0.075 g, 0.111 mmol, 7%), along with several fractions consisting of unidentified and/or impure products, or oligomers and polymers. Spectroscopic data for in,out-2·2BH3, (in,in/out,out)-2·2BH3, and 6·2BH3 matched those reported above.
Supporting Information
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 240.6 KB | Download |
References
-
Shima, T.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2004, 43, 5537–5540. doi:10.1002/anie.200460534
Angew. Chem. 2004, 116, 5653–5656. doi:10.1002/ange.200460534
Return to citation in text: [1] [2] [3] [4] -
Lang, G. M.; Shima, T.; Wang, L.; Cluff, K. J.; Skopek, K.; Hampel, F.; Blümel, J.; Gladysz, J. A. J. Am. Chem. Soc. 2016, 138, 7649–7663. doi:10.1021/jacs.6b03178
Return to citation in text: [1] [2] [3] [4] -
Lang, G. M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2016, 35, 2873–2889. doi:10.1021/acs.organomet.6b00447
Return to citation in text: [1] [2] [3] [4] -
Lang, G. M.; Skaper, D.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2016, 45, 16190–16204. doi:10.1039/C6DT03258C
Return to citation in text: [1] [2] [3] [4] -
Nawara, A. J.; Shima, T.; Hampel, F.; Gladysz, J. A. J. Am. Chem. Soc. 2006, 128, 4962–4963. doi:10.1021/ja061044w
Return to citation in text: [1] [2] [3] -
Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Kharel, S.; Joshi, H.; Bhuvanesh, N.; Gladysz, J. A. Organometallics 2018, 173, in press. doi:10.1021/acs.organomet.8b00345
Return to citation in text: [1] [2] [3] [4] -
Wang, L.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2006, 45, 4372–4375. doi:10.1002/anie.200601191
Angew. Chem. 2006, 118, 4479–4482. doi:10.1002/ange.200601191
Return to citation in text: [1] [2] -
Wang, L.; Shima, T.; Hampel, F.; Gladysz, J. A. Chem. Commun. 2006, 4075–4077. doi:10.1039/B606728J
Return to citation in text: [1] [2] -
Estrada, A. L.; Jia, T.; Bhuvanesh, N.; Blümel, J.; Gladysz, J. A. Eur. J. Inorg. Chem. 2015, 5318–5321. doi:10.1002/ejic.201500953
Return to citation in text: [1] [2] [3] -
Hess, G. D.; Hampel, F.; Gladysz, J. A. Organometallics 2007, 26, 5129–5131. doi:10.1021/om700625u
Return to citation in text: [1] [2] -
Fiedler, T.; Bhuvanesh, N.; Hampel, F.; Reibenspies, J. H.; Gladysz, J. A. Dalton Trans. 2016, 45, 7131–7147. doi:10.1039/C6DT00692B
Return to citation in text: [1] [2] [3] -
Hess, G. D.; Fiedler, T.; Hampel, F.; Gladysz, J. A. Inorg. Chem. 2017, 56, 7454–7469. doi:10.1021/acs.inorgchem.7b00909
Return to citation in text: [1] [2] [3] -
Setaka, W.; Yamaguchi, K. J. Am. Chem. Soc. 2013, 135, 14560–14563. doi:10.1021/ja408405f
and earlier work cited therein.
Return to citation in text: [1] [2] -
Setaka, W.; Inoue, K.; Higa, S.; Yoshigai, S.; Kono, H.; Yamaguchi, K. J. Org. Chem. 2014, 79, 8288–8295. doi:10.1021/jo501539h
Return to citation in text: [1] [2] -
Setaka, W.; Higa, S.; Yamaguchi, K. Org. Biomol. Chem. 2014, 12, 3354–3357. doi:10.1039/C4OB00470A
Return to citation in text: [1] [2] -
Shionari, H.; Inagaki, Y.; Yamaguchi, K.; Setaka, W. Org. Biomol. Chem. 2015, 13, 10511–10516. doi:10.1039/C5OB01644D
Return to citation in text: [1] [2] -
Nishiyama, Y.; Inagaki, Y.; Yamguchi, K.; Setaka, W. J. Org. Chem. 2015, 80, 9959–9966. doi:10.1021/acs.joc.5b01489
Return to citation in text: [1] [2] -
Masuda, T.; Arase, J.; Inagaki, Y.; Kawahata, M.; Yamaguchi, K.; Ohhara, T.; Nakao, A.; Momma, H.; Kwon, E.; Setaka, W. Cryst. Growth Des. 2016, 16, 4392–4401. doi:10.1021/acs.cgd.6b00508
Return to citation in text: [1] [2] -
Kottas, G. S.; Clarke, L. I.; Horinek, D.; Michl, J. Chem. Rev. 2005, 105, 1281–1376. doi:10.1021/cr0300993
Return to citation in text: [1] -
Vogelsberg, C. S.; Garcia-Garibay, M. A. Chem. Soc. Rev. 2012, 41, 1892–1910. doi:10.1039/C1CS15197E
Return to citation in text: [1] -
Kharel, S.; Joshi, H.; Bierschenk, S.; Stollenz, M.; Taher, D.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2017, 139, 2172–2175. doi:10.1021/jacs.6b12788
Return to citation in text: [1] [2] -
Alder, R. W.; Butts, C. P.; Orpen, A. G.; Read, D.; Oliva, J. M. J. Chem. Soc., Perkin Trans. 2 2001, 282–287. doi:10.1039/b008903f
and references cited therein.
Return to citation in text: [1] -
Hilliard, C. R. Diphosphine Dioxide Cages and Hydrogen Peroxide Adducts of Phosphine Oxides: Syntheses and Applications in Surface Science. Ph.D. Thesis, Texas A&M University, College Station, USA, 2013.
Return to citation in text: [1] -
Kharel, S.; Jia, T.; Bhuvanesh, N.; Reibenspies, J. H.; Blümel, J.; Gladysz, J. A. Chem. – Asian J. 2018, 13, in press. doi:10.1002/asia.201800739
Return to citation in text: [1] [2] [3] [4] -
Alder, R. W.; East, S. P. Chem. Rev. 1996, 96, 2097–2112. doi:10.1021/cr940246k
Return to citation in text: [1] [2] -
Park, C. H.; Simmons, H. E. J. Am. Chem. Soc. 1968, 90, 2429–2431. doi:10.1021/ja01011a046
Return to citation in text: [1] -
Stollenz, M.; Barbasiewicz, M.; Nawara-Hultzsch, A. J.; Fiedler, T.; Laddusaw, R. M.; Bhuvanesh, N.; Gladysz, J. A. Angew. Chem., Int. Ed. 2011, 50, 6647–6651. doi:10.1002/anie.201100893
Angew. Chem. 2011, 123, 6777–6781. doi:10.1002/ange.201100893
Return to citation in text: [1] [2] [3] -
Stollenz, M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2011, 30, 6510–6513. doi:10.1021/om200924g
Return to citation in text: [1] -
Stollenz, M.; Taher, D.; Bhuvanesh, N.; Reibenspies, J. H.; Baranová, Z.; Gladysz, J. A. Chem. Commun. 2015, 51, 16053–16056. doi:10.1039/C5CC05620A
Return to citation in text: [1] [2] -
Nawara-Hultzsch, A. J.; Skopek, K.; Shima, T.; Barbasiewicz, M.; Hess, G. D.; Skaper, D.; Gladysz, J. A. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 414–424. doi:10.1515/znb-2010-0327
The yield range given for 1·BH3 reflects the results of repeated preparations since this original report.
Return to citation in text: [1] [2] [3] [4] -
Fiedler, T. Syntheses of Gyroscope-like Osmium Complexes and Cage-like Diphosphines. Doctoral Thesis, Friedrich-Alexander-University, Erlangen, Germany, 2011.
Return to citation in text: [1] [2] [3] -
Berthod, M.; Favre-Réguillon, A.; Mohamad, J.; Mignani, G.; Docherty, G.; Lemaire, M. Synlett 2007, 1545–1548. doi:10.1055/s-2007-982536
Return to citation in text: [1] [2] -
Shima, T.; Bauer, E. B.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2004, 1012–1028. doi:10.1039/b400156g
Return to citation in text: [1] [2] [3] -
Joshi, H.; Kharel, S.; Ehnbom, A.; Skopek, K.; Hess, G. D.; Fiedler, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2018, 140, 8463–8478. doi:10.1021/jacs.8b02846
Return to citation in text: [1] -
Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. doi:10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0
Angew. Chem. 2002, 114, 4210–4212. doi:10.1002/1521-3757(20021104)114:21<4210::AID-ANGE4210>3.0.CO;2-J
Return to citation in text: [1] -
Nguyen, T. B.; Castanet, A.-S.; Nguyen, T.-H.; Nguyen, K. P. P.; Bardeau, J.-F.; Gibaud, A.; Mortier, J. Tetrahedron 2006, 62, 647–651. doi:10.1016/j.tet.2005.10.003
Return to citation in text: [1] -
For previous syntheses and characterization of 8, see references [39-41].
Return to citation in text: [1] -
Woolford, R. G. Can. J. Chem. 1962, 40, 1846–1850. doi:10.1139/v62-280
Return to citation in text: [1] [2] -
Taffa, D.; Kathiresan, M.; Walder, L. Langmuir 2009, 25, 5371–5379. doi:10.1021/la8038126
See Supporting Information.
Return to citation in text: [1] [2] -
Matsubara, H.; Tsukida, M.; Ishihara, D.; Kuniyoshi, K.; Ryu, I. Synlett 2010, 2014–2018. doi:10.1055/s-0030-1258482
Return to citation in text: [1] [2] -
Soomro, S. A.; Benmouna, R.; Berger, R.; Meier, H. Eur. J. Org. Chem. 2005, 3586–3593. doi:10.1002/ejoc.200500185
See for the methodology used to prepare 8.
Return to citation in text: [1] -
de Andrade, V. S. C.; de Mattos, M. C. S. Curr. Org. Synth. 2015, 12, 309–327. doi:10.2174/1570179412666150305231358
Return to citation in text: [1] -
For a related Arbuzov reaction of 8, see reference [40].
Return to citation in text: [1] -
Stranne, R.; Vasse, J.-L.; Moberg, C. Org. Lett. 2001, 3, 2525–2528. doi:10.1021/ol016193s
Return to citation in text: [1] -
Staubitz, A.; Robertson, A. P. M.; Sloan, M. E.; Manners, I. Chem. Rev. 2010, 110, 4023–4078. doi:10.1021/cr100105a
(see section 4.2.1.1).
Return to citation in text: [1] -
Gaumont, A.-C.; Bourumeau, K.; Denis, J.-M.; Guenot, P. J. Organomet. Chem. 1994, 484, 9–12. doi:10.1016/0022-328X(94)87178-7
Return to citation in text: [1] -
Pinto, P.; Götz, A. W.; Marconi, G.; Hess, B. A.; Marinetti, A.; Heinemann, F. W.; Zenneck, U. Organometallics 2006, 25, 2607–2616. doi:10.1021/om050461z
Return to citation in text: [1] -
Dahlenburg, L.; Kaunert, A. Eur. J. Inorg. Chem. 1998, 885–887. doi:10.1002/(SICI)1099-0682(199807)1998:7<885::AID-EJIC885>3.0.CO;2-6
Return to citation in text: [1] -
Däbritz, F.; Theumer, G.; Gruner, M.; Bauer, I. Tetrahedron 2009, 65, 2995–3002. doi:10.1016/j.tet.2009.01.102
Return to citation in text: [1] [2] -
Bauer, I.; Habicher, W. D. Collect. Czech. Chem. Commun. 2004, 69, 1195–1230. doi:10.1135/cccc20041195
Return to citation in text: [1] [2] -
Jung, M. E.; Piizzi, G. Chem. Rev. 2005, 105, 1735–1766. doi:10.1021/cr940337h
Return to citation in text: [1] -
Schuman, M.; Trevitt, M.; Redd, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2000, 39, 2491–2493. doi:10.1002/1521-3773(20000717)39:14<2491::AID-ANIE2491>3.0.CO;2-H
Angew. Chem. 2000, 112, 2604–2607. doi:10.1002/1521-3757(20000717)112:14<2604::AID-ANGE2604>3.0.CO;2-#
Return to citation in text: [1] [2] -
Wu, X.; O'Brien, P.; Ellwood, S.; Secci, F.; Kelly, B. Org. Lett. 2013, 15, 192–195. doi:10.1021/ol303253h
Return to citation in text: [1] -
The NiCl2 analog of the PtCl2 adduct of in,in-2 in Scheme 1 is known, but is so far only available from the free dibridgehead diphosphine 2 [22].
Return to citation in text: [1] -
The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix.
Return to citation in text: [1] [2] [3] [4] [5]
| 45. | Stranne, R.; Vasse, J.-L.; Moberg, C. Org. Lett. 2001, 3, 2525–2528. doi:10.1021/ol016193s |
| 46. |
Staubitz, A.; Robertson, A. P. M.; Sloan, M. E.; Manners, I. Chem. Rev. 2010, 110, 4023–4078. doi:10.1021/cr100105a
(see section 4.2.1.1). |
| 47. | Gaumont, A.-C.; Bourumeau, K.; Denis, J.-M.; Guenot, P. J. Organomet. Chem. 1994, 484, 9–12. doi:10.1016/0022-328X(94)87178-7 |
| 34. | Shima, T.; Bauer, E. B.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2004, 1012–1028. doi:10.1039/b400156g |
| 48. | Pinto, P.; Götz, A. W.; Marconi, G.; Hess, B. A.; Marinetti, A.; Heinemann, F. W.; Zenneck, U. Organometallics 2006, 25, 2607–2616. doi:10.1021/om050461z |
| 49. | Dahlenburg, L.; Kaunert, A. Eur. J. Inorg. Chem. 1998, 885–887. doi:10.1002/(SICI)1099-0682(199807)1998:7<885::AID-EJIC885>3.0.CO;2-6 |
| 50. | Däbritz, F.; Theumer, G.; Gruner, M.; Bauer, I. Tetrahedron 2009, 65, 2995–3002. doi:10.1016/j.tet.2009.01.102 |
| 51. | Bauer, I.; Habicher, W. D. Collect. Czech. Chem. Commun. 2004, 69, 1195–1230. doi:10.1135/cccc20041195 |
| 52. | Jung, M. E.; Piizzi, G. Chem. Rev. 2005, 105, 1735–1766. doi:10.1021/cr940337h |
| 25. | Kharel, S.; Jia, T.; Bhuvanesh, N.; Reibenspies, J. H.; Blümel, J.; Gladysz, J. A. Chem. – Asian J. 2018, 13, in press. doi:10.1002/asia.201800739 |
| 50. | Däbritz, F.; Theumer, G.; Gruner, M.; Bauer, I. Tetrahedron 2009, 65, 2995–3002. doi:10.1016/j.tet.2009.01.102 |
| 51. | Bauer, I.; Habicher, W. D. Collect. Czech. Chem. Commun. 2004, 69, 1195–1230. doi:10.1135/cccc20041195 |
| 25. | Kharel, S.; Jia, T.; Bhuvanesh, N.; Reibenspies, J. H.; Blümel, J.; Gladysz, J. A. Chem. – Asian J. 2018, 13, in press. doi:10.1002/asia.201800739 |
| 30. | Stollenz, M.; Taher, D.; Bhuvanesh, N.; Reibenspies, J. H.; Baranová, Z.; Gladysz, J. A. Chem. Commun. 2015, 51, 16053–16056. doi:10.1039/C5CC05620A |
| 28. |
Stollenz, M.; Barbasiewicz, M.; Nawara-Hultzsch, A. J.; Fiedler, T.; Laddusaw, R. M.; Bhuvanesh, N.; Gladysz, J. A. Angew. Chem., Int. Ed. 2011, 50, 6647–6651. doi:10.1002/anie.201100893
Angew. Chem. 2011, 123, 6777–6781. doi:10.1002/ange.201100893 |
| 1. |
Shima, T.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2004, 43, 5537–5540. doi:10.1002/anie.200460534
Angew. Chem. 2004, 116, 5653–5656. doi:10.1002/ange.200460534 |
| 2. | Lang, G. M.; Shima, T.; Wang, L.; Cluff, K. J.; Skopek, K.; Hampel, F.; Blümel, J.; Gladysz, J. A. J. Am. Chem. Soc. 2016, 138, 7649–7663. doi:10.1021/jacs.6b03178 |
| 3. | Lang, G. M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2016, 35, 2873–2889. doi:10.1021/acs.organomet.6b00447 |
| 4. | Lang, G. M.; Skaper, D.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2016, 45, 16190–16204. doi:10.1039/C6DT03258C |
| 5. | Nawara, A. J.; Shima, T.; Hampel, F.; Gladysz, J. A. J. Am. Chem. Soc. 2006, 128, 4962–4963. doi:10.1021/ja061044w |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 7. | Kharel, S.; Joshi, H.; Bhuvanesh, N.; Gladysz, J. A. Organometallics 2018, 173, in press. doi:10.1021/acs.organomet.8b00345 |
| 8. |
Wang, L.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2006, 45, 4372–4375. doi:10.1002/anie.200601191
Angew. Chem. 2006, 118, 4479–4482. doi:10.1002/ange.200601191 |
| 9. | Wang, L.; Shima, T.; Hampel, F.; Gladysz, J. A. Chem. Commun. 2006, 4075–4077. doi:10.1039/B606728J |
| 10. | Estrada, A. L.; Jia, T.; Bhuvanesh, N.; Blümel, J.; Gladysz, J. A. Eur. J. Inorg. Chem. 2015, 5318–5321. doi:10.1002/ejic.201500953 |
| 11. | Hess, G. D.; Hampel, F.; Gladysz, J. A. Organometallics 2007, 26, 5129–5131. doi:10.1021/om700625u |
| 12. | Fiedler, T.; Bhuvanesh, N.; Hampel, F.; Reibenspies, J. H.; Gladysz, J. A. Dalton Trans. 2016, 45, 7131–7147. doi:10.1039/C6DT00692B |
| 13. | Hess, G. D.; Fiedler, T.; Hampel, F.; Gladysz, J. A. Inorg. Chem. 2017, 56, 7454–7469. doi:10.1021/acs.inorgchem.7b00909 |
| 34. | Shima, T.; Bauer, E. B.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2004, 1012–1028. doi:10.1039/b400156g |
| 7. | Kharel, S.; Joshi, H.; Bhuvanesh, N.; Gladysz, J. A. Organometallics 2018, 173, in press. doi:10.1021/acs.organomet.8b00345 |
| 53. |
Schuman, M.; Trevitt, M.; Redd, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2000, 39, 2491–2493. doi:10.1002/1521-3773(20000717)39:14<2491::AID-ANIE2491>3.0.CO;2-H
Angew. Chem. 2000, 112, 2604–2607. doi:10.1002/1521-3757(20000717)112:14<2604::AID-ANGE2604>3.0.CO;2-# |
| 54. | Wu, X.; O'Brien, P.; Ellwood, S.; Secci, F.; Kelly, B. Org. Lett. 2013, 15, 192–195. doi:10.1021/ol303253h |
| 53. |
Schuman, M.; Trevitt, M.; Redd, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2000, 39, 2491–2493. doi:10.1002/1521-3773(20000717)39:14<2491::AID-ANIE2491>3.0.CO;2-H
Angew. Chem. 2000, 112, 2604–2607. doi:10.1002/1521-3757(20000717)112:14<2604::AID-ANGE2604>3.0.CO;2-# |
| 55. | The NiCl2 analog of the PtCl2 adduct of in,in-2 in Scheme 1 is known, but is so far only available from the free dibridgehead diphosphine 2 [22]. |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 56. | The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix. |
| 56. | The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix. |
| 56. | The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix. |
| 33. | Berthod, M.; Favre-Réguillon, A.; Mohamad, J.; Mignani, G.; Docherty, G.; Lemaire, M. Synlett 2007, 1545–1548. doi:10.1055/s-2007-982536 |
| 56. | The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix. |
| 32. | Fiedler, T. Syntheses of Gyroscope-like Osmium Complexes and Cage-like Diphosphines. Doctoral Thesis, Friedrich-Alexander-University, Erlangen, Germany, 2011. |
| 31. |
Nawara-Hultzsch, A. J.; Skopek, K.; Shima, T.; Barbasiewicz, M.; Hess, G. D.; Skaper, D.; Gladysz, J. A. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 414–424. doi:10.1515/znb-2010-0327
The yield range given for 1·BH3 reflects the results of repeated preparations since this original report. |
| 32. | Fiedler, T. Syntheses of Gyroscope-like Osmium Complexes and Cage-like Diphosphines. Doctoral Thesis, Friedrich-Alexander-University, Erlangen, Germany, 2011. |
| 39. | Woolford, R. G. Can. J. Chem. 1962, 40, 1846–1850. doi:10.1139/v62-280 |
| 40. |
Taffa, D.; Kathiresan, M.; Walder, L. Langmuir 2009, 25, 5371–5379. doi:10.1021/la8038126
See Supporting Information. |
| 41. | Matsubara, H.; Tsukida, M.; Ishihara, D.; Kuniyoshi, K.; Ryu, I. Synlett 2010, 2014–2018. doi:10.1055/s-0030-1258482 |
| 56. | The most intense peak of the isotope envelope is given; m/z (relative intensity, %). THAP = 2.4.6-trihydroxyacetophenone matrix. |
| 1. |
Shima, T.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2004, 43, 5537–5540. doi:10.1002/anie.200460534
Angew. Chem. 2004, 116, 5653–5656. doi:10.1002/ange.200460534 |
| 2. | Lang, G. M.; Shima, T.; Wang, L.; Cluff, K. J.; Skopek, K.; Hampel, F.; Blümel, J.; Gladysz, J. A. J. Am. Chem. Soc. 2016, 138, 7649–7663. doi:10.1021/jacs.6b03178 |
| 3. | Lang, G. M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2016, 35, 2873–2889. doi:10.1021/acs.organomet.6b00447 |
| 4. | Lang, G. M.; Skaper, D.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2016, 45, 16190–16204. doi:10.1039/C6DT03258C |
| 5. | Nawara, A. J.; Shima, T.; Hampel, F.; Gladysz, J. A. J. Am. Chem. Soc. 2006, 128, 4962–4963. doi:10.1021/ja061044w |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 7. | Kharel, S.; Joshi, H.; Bhuvanesh, N.; Gladysz, J. A. Organometallics 2018, 173, in press. doi:10.1021/acs.organomet.8b00345 |
| 8. |
Wang, L.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2006, 45, 4372–4375. doi:10.1002/anie.200601191
Angew. Chem. 2006, 118, 4479–4482. doi:10.1002/ange.200601191 |
| 9. | Wang, L.; Shima, T.; Hampel, F.; Gladysz, J. A. Chem. Commun. 2006, 4075–4077. doi:10.1039/B606728J |
| 10. | Estrada, A. L.; Jia, T.; Bhuvanesh, N.; Blümel, J.; Gladysz, J. A. Eur. J. Inorg. Chem. 2015, 5318–5321. doi:10.1002/ejic.201500953 |
| 11. | Hess, G. D.; Hampel, F.; Gladysz, J. A. Organometallics 2007, 26, 5129–5131. doi:10.1021/om700625u |
| 12. | Fiedler, T.; Bhuvanesh, N.; Hampel, F.; Reibenspies, J. H.; Gladysz, J. A. Dalton Trans. 2016, 45, 7131–7147. doi:10.1039/C6DT00692B |
| 13. | Hess, G. D.; Fiedler, T.; Hampel, F.; Gladysz, J. A. Inorg. Chem. 2017, 56, 7454–7469. doi:10.1021/acs.inorgchem.7b00909 |
| 5. | Nawara, A. J.; Shima, T.; Hampel, F.; Gladysz, J. A. J. Am. Chem. Soc. 2006, 128, 4962–4963. doi:10.1021/ja061044w |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 10. | Estrada, A. L.; Jia, T.; Bhuvanesh, N.; Blümel, J.; Gladysz, J. A. Eur. J. Inorg. Chem. 2015, 5318–5321. doi:10.1002/ejic.201500953 |
| 22. | Kharel, S.; Joshi, H.; Bierschenk, S.; Stollenz, M.; Taher, D.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2017, 139, 2172–2175. doi:10.1021/jacs.6b12788 |
| 32. | Fiedler, T. Syntheses of Gyroscope-like Osmium Complexes and Cage-like Diphosphines. Doctoral Thesis, Friedrich-Alexander-University, Erlangen, Germany, 2011. |
| 14. |
Setaka, W.; Yamaguchi, K. J. Am. Chem. Soc. 2013, 135, 14560–14563. doi:10.1021/ja408405f
and earlier work cited therein. |
| 15. | Setaka, W.; Inoue, K.; Higa, S.; Yoshigai, S.; Kono, H.; Yamaguchi, K. J. Org. Chem. 2014, 79, 8288–8295. doi:10.1021/jo501539h |
| 16. | Setaka, W.; Higa, S.; Yamaguchi, K. Org. Biomol. Chem. 2014, 12, 3354–3357. doi:10.1039/C4OB00470A |
| 17. | Shionari, H.; Inagaki, Y.; Yamaguchi, K.; Setaka, W. Org. Biomol. Chem. 2015, 13, 10511–10516. doi:10.1039/C5OB01644D |
| 18. | Nishiyama, Y.; Inagaki, Y.; Yamguchi, K.; Setaka, W. J. Org. Chem. 2015, 80, 9959–9966. doi:10.1021/acs.joc.5b01489 |
| 19. | Masuda, T.; Arase, J.; Inagaki, Y.; Kawahata, M.; Yamaguchi, K.; Ohhara, T.; Nakao, A.; Momma, H.; Kwon, E.; Setaka, W. Cryst. Growth Des. 2016, 16, 4392–4401. doi:10.1021/acs.cgd.6b00508 |
| 20. | Kottas, G. S.; Clarke, L. I.; Horinek, D.; Michl, J. Chem. Rev. 2005, 105, 1281–1376. doi:10.1021/cr0300993 |
| 21. | Vogelsberg, C. S.; Garcia-Garibay, M. A. Chem. Soc. Rev. 2012, 41, 1892–1910. doi:10.1039/C1CS15197E |
| 31. |
Nawara-Hultzsch, A. J.; Skopek, K.; Shima, T.; Barbasiewicz, M.; Hess, G. D.; Skaper, D.; Gladysz, J. A. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 414–424. doi:10.1515/znb-2010-0327
The yield range given for 1·BH3 reflects the results of repeated preparations since this original report. |
| 14. |
Setaka, W.; Yamaguchi, K. J. Am. Chem. Soc. 2013, 135, 14560–14563. doi:10.1021/ja408405f
and earlier work cited therein. |
| 15. | Setaka, W.; Inoue, K.; Higa, S.; Yoshigai, S.; Kono, H.; Yamaguchi, K. J. Org. Chem. 2014, 79, 8288–8295. doi:10.1021/jo501539h |
| 16. | Setaka, W.; Higa, S.; Yamaguchi, K. Org. Biomol. Chem. 2014, 12, 3354–3357. doi:10.1039/C4OB00470A |
| 17. | Shionari, H.; Inagaki, Y.; Yamaguchi, K.; Setaka, W. Org. Biomol. Chem. 2015, 13, 10511–10516. doi:10.1039/C5OB01644D |
| 18. | Nishiyama, Y.; Inagaki, Y.; Yamguchi, K.; Setaka, W. J. Org. Chem. 2015, 80, 9959–9966. doi:10.1021/acs.joc.5b01489 |
| 19. | Masuda, T.; Arase, J.; Inagaki, Y.; Kawahata, M.; Yamaguchi, K.; Ohhara, T.; Nakao, A.; Momma, H.; Kwon, E.; Setaka, W. Cryst. Growth Des. 2016, 16, 4392–4401. doi:10.1021/acs.cgd.6b00508 |
| 31. |
Nawara-Hultzsch, A. J.; Skopek, K.; Shima, T.; Barbasiewicz, M.; Hess, G. D.; Skaper, D.; Gladysz, J. A. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 414–424. doi:10.1515/znb-2010-0327
The yield range given for 1·BH3 reflects the results of repeated preparations since this original report. |
| 1. |
Shima, T.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2004, 43, 5537–5540. doi:10.1002/anie.200460534
Angew. Chem. 2004, 116, 5653–5656. doi:10.1002/ange.200460534 |
| 2. | Lang, G. M.; Shima, T.; Wang, L.; Cluff, K. J.; Skopek, K.; Hampel, F.; Blümel, J.; Gladysz, J. A. J. Am. Chem. Soc. 2016, 138, 7649–7663. doi:10.1021/jacs.6b03178 |
| 3. | Lang, G. M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2016, 35, 2873–2889. doi:10.1021/acs.organomet.6b00447 |
| 4. | Lang, G. M.; Skaper, D.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2016, 45, 16190–16204. doi:10.1039/C6DT03258C |
| 28. |
Stollenz, M.; Barbasiewicz, M.; Nawara-Hultzsch, A. J.; Fiedler, T.; Laddusaw, R. M.; Bhuvanesh, N.; Gladysz, J. A. Angew. Chem., Int. Ed. 2011, 50, 6647–6651. doi:10.1002/anie.201100893
Angew. Chem. 2011, 123, 6777–6781. doi:10.1002/ange.201100893 |
| 25. | Kharel, S.; Jia, T.; Bhuvanesh, N.; Reibenspies, J. H.; Blümel, J.; Gladysz, J. A. Chem. – Asian J. 2018, 13, in press. doi:10.1002/asia.201800739 |
| 26. | Alder, R. W.; East, S. P. Chem. Rev. 1996, 96, 2097–2112. doi:10.1021/cr940246k |
| 27. | Park, C. H.; Simmons, H. E. J. Am. Chem. Soc. 1968, 90, 2429–2431. doi:10.1021/ja01011a046 |
| 24. | Hilliard, C. R. Diphosphine Dioxide Cages and Hydrogen Peroxide Adducts of Phosphine Oxides: Syntheses and Applications in Surface Science. Ph.D. Thesis, Texas A&M University, College Station, USA, 2013. |
| 22. | Kharel, S.; Joshi, H.; Bierschenk, S.; Stollenz, M.; Taher, D.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2017, 139, 2172–2175. doi:10.1021/jacs.6b12788 |
| 25. | Kharel, S.; Jia, T.; Bhuvanesh, N.; Reibenspies, J. H.; Blümel, J.; Gladysz, J. A. Chem. – Asian J. 2018, 13, in press. doi:10.1002/asia.201800739 |
| 28. |
Stollenz, M.; Barbasiewicz, M.; Nawara-Hultzsch, A. J.; Fiedler, T.; Laddusaw, R. M.; Bhuvanesh, N.; Gladysz, J. A. Angew. Chem., Int. Ed. 2011, 50, 6647–6651. doi:10.1002/anie.201100893
Angew. Chem. 2011, 123, 6777–6781. doi:10.1002/ange.201100893 |
| 29. | Stollenz, M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2011, 30, 6510–6513. doi:10.1021/om200924g |
| 30. | Stollenz, M.; Taher, D.; Bhuvanesh, N.; Reibenspies, J. H.; Baranová, Z.; Gladysz, J. A. Chem. Commun. 2015, 51, 16053–16056. doi:10.1039/C5CC05620A |
| 1. |
Shima, T.; Hampel, F.; Gladysz, J. A. Angew. Chem., Int. Ed. 2004, 43, 5537–5540. doi:10.1002/anie.200460534
Angew. Chem. 2004, 116, 5653–5656. doi:10.1002/ange.200460534 |
| 2. | Lang, G. M.; Shima, T.; Wang, L.; Cluff, K. J.; Skopek, K.; Hampel, F.; Blümel, J.; Gladysz, J. A. J. Am. Chem. Soc. 2016, 138, 7649–7663. doi:10.1021/jacs.6b03178 |
| 3. | Lang, G. M.; Bhuvanesh, N.; Reibenspies, J. H.; Gladysz, J. A. Organometallics 2016, 35, 2873–2889. doi:10.1021/acs.organomet.6b00447 |
| 4. | Lang, G. M.; Skaper, D.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2016, 45, 16190–16204. doi:10.1039/C6DT03258C |
| 40. |
Taffa, D.; Kathiresan, M.; Walder, L. Langmuir 2009, 25, 5371–5379. doi:10.1021/la8038126
See Supporting Information. |
| 23. |
Alder, R. W.; Butts, C. P.; Orpen, A. G.; Read, D.; Oliva, J. M. J. Chem. Soc., Perkin Trans. 2 2001, 282–287. doi:10.1039/b008903f
and references cited therein. |
| 26. | Alder, R. W.; East, S. P. Chem. Rev. 1996, 96, 2097–2112. doi:10.1021/cr940246k |
| 22. | Kharel, S.; Joshi, H.; Bierschenk, S.; Stollenz, M.; Taher, D.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2017, 139, 2172–2175. doi:10.1021/jacs.6b12788 |
| 34. | Shima, T.; Bauer, E. B.; Hampel, F.; Gladysz, J. A. Dalton Trans. 2004, 1012–1028. doi:10.1039/b400156g |
| 31. |
Nawara-Hultzsch, A. J.; Skopek, K.; Shima, T.; Barbasiewicz, M.; Hess, G. D.; Skaper, D.; Gladysz, J. A. Z. Naturforsch., B: J. Chem. Sci. 2010, 65, 414–424. doi:10.1515/znb-2010-0327
The yield range given for 1·BH3 reflects the results of repeated preparations since this original report. |
| 33. | Berthod, M.; Favre-Réguillon, A.; Mohamad, J.; Mignani, G.; Docherty, G.; Lemaire, M. Synlett 2007, 1545–1548. doi:10.1055/s-2007-982536 |
| 38. | For previous syntheses and characterization of 8, see references [39-41]. |
| 39. | Woolford, R. G. Can. J. Chem. 1962, 40, 1846–1850. doi:10.1139/v62-280 |
| 40. |
Taffa, D.; Kathiresan, M.; Walder, L. Langmuir 2009, 25, 5371–5379. doi:10.1021/la8038126
See Supporting Information. |
| 41. | Matsubara, H.; Tsukida, M.; Ishihara, D.; Kuniyoshi, K.; Ryu, I. Synlett 2010, 2014–2018. doi:10.1055/s-0030-1258482 |
| 42. |
Soomro, S. A.; Benmouna, R.; Berger, R.; Meier, H. Eur. J. Org. Chem. 2005, 3586–3593. doi:10.1002/ejoc.200500185
See for the methodology used to prepare 8. |
| 43. | de Andrade, V. S. C.; de Mattos, M. C. S. Curr. Org. Synth. 2015, 12, 309–327. doi:10.2174/1570179412666150305231358 |
| 36. |
Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. doi:10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0
Angew. Chem. 2002, 114, 4210–4212. doi:10.1002/1521-3757(20021104)114:21<4210::AID-ANGE4210>3.0.CO;2-J |
| 37. | Nguyen, T. B.; Castanet, A.-S.; Nguyen, T.-H.; Nguyen, K. P. P.; Bardeau, J.-F.; Gibaud, A.; Mortier, J. Tetrahedron 2006, 62, 647–651. doi:10.1016/j.tet.2005.10.003 |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
| 7. | Kharel, S.; Joshi, H.; Bhuvanesh, N.; Gladysz, J. A. Organometallics 2018, 173, in press. doi:10.1021/acs.organomet.8b00345 |
| 12. | Fiedler, T.; Bhuvanesh, N.; Hampel, F.; Reibenspies, J. H.; Gladysz, J. A. Dalton Trans. 2016, 45, 7131–7147. doi:10.1039/C6DT00692B |
| 13. | Hess, G. D.; Fiedler, T.; Hampel, F.; Gladysz, J. A. Inorg. Chem. 2017, 56, 7454–7469. doi:10.1021/acs.inorgchem.7b00909 |
| 35. | Joshi, H.; Kharel, S.; Ehnbom, A.; Skopek, K.; Hess, G. D.; Fiedler, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. J. Am. Chem. Soc. 2018, 140, 8463–8478. doi:10.1021/jacs.8b02846 |
| 6. | Nawara-Hultzsch, A. J.; Stollenz, M.; Barbasiewicz, M.; Szafert, S.; Lis, T.; Hampel, F.; Bhuvanesh, N.; Gladysz, J. A. Chem. – Eur. J. 2014, 20, 4617–4637. doi:10.1002/chem.201304419 |
© 2018 Fiedler et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
