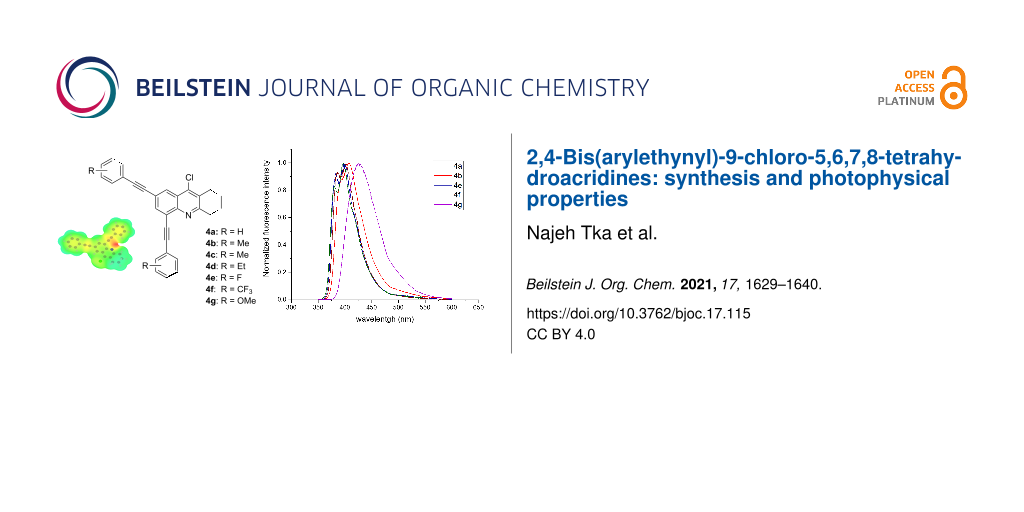
Abstract
Acridine derivatives have attracted considerable interest in numerous areas owing to their attractive physical and chemical properties. Herein, starting from readily available anthranilic acid, an efficient synthesis of 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine derivatives was accomplished via a one-pot double Sonogashira cross-coupling method. The UV-visible absorption and emission properties of the synthesized molecules have been examined. Additionally, theoretical studies based on density functional theory (DFT/B3LYP/6-31G(d)) were carried out.

Graphical Abstract
Introduction
The development and design of small π-conjugated molecules have attracted increasing attention for their inspiring applications in the fields of solar cells [1-3], organic devices [4-8], and as chemosensors [9,10]. The acridine core (Figure 1), formed by two benzenes fused to a pyridine ring, is among the most extensively studied heterocyclic aromatic compounds. It has first appeared as a side product during the synthesis of anthracene [11] and became an abundant scaffold in medicinal chemistry [12-14]. Acridine derivatives have exhibited a range of biological activities [15-20] and have been particularly explored in chemotherapeutic protocols against several types of tumors [21-31]. In recent years, much attention has been devoted to acridines in materials science due to their attractive photophysical and electrochemical properties [32-35]. They have been investigated in organic electronic devices [36-39] and were reported to be promising candidates for potential use as organic light emitting diodes [40]. Thus, various synthetic methodologies for the preparation of acridine-based molecules have been developed [41-47].
![[1860-5397-17-115-1]](/bjoc/content/figures/1860-5397-17-115-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Tetrahydroacridine, containing a partially hydrogenated ring, is another privileged scaffold which showed interesting biological activities [48-54]. As a typical example, 9-amino-1,2,3,4-tetrahydroacridine or tacrine was the first drug approved for the treatment of Alzheimer's disease [55-57]. Surprisingly, photophysical properties of tetrahydroacridines have, to the best of our knowledge, not been studied so far. Recently, our research group reported the synthesis of a large variety of acridine derivatives which showed promising fluorescence properties and high quantum yields [58-60]. In continuation of our previous studies and as a part of our interest in discovering new organic materials applications [61-63], we herein report the synthesis of new 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine derivatives. The investigation of their photophysical properties and theoretical DFT studies were achieved aiming to understand the influence of substituents at introduced arylethynyl groups.
Results and Discussion
Synthesis
At the outset of this study, we prepared 2,4-dibromo-9-chloro-5,6,7,8-tetrahydroacridine (2) following a two-step approach. We first prepared 3,5-dibromoanthranilic acid (1) by refluxing anthranilic acid with 2.2 equivalents of bromine in acetic acid as previously reported [64]. Subsequently, the POCl3-mediated cyclodehydration of 1 and cyclohexanone afforded 2 through an adapted reported procedure (Scheme 1) [65].
![[1860-5397-17-115-i1]](/bjoc/content/inline/1860-5397-17-115-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,4-dibromo-9-chloro-5,6,7,8-tetrahydroacridine (2).
Scheme 1: Synthesis of 2,4-dibromo-9-chloro-5,6,7,8-tetrahydroacridine (2).
Tetrahydroacridine 2 represents a novel synthetic building block for Pd catalysis. With this precursor in hand, we intended to expand the π-conjugation by introducing two arylethynyl groups by Sonogashira reactions [66-69]. For the optimization, we studied the reaction of 2 with phenylacetylene (3a) and we obtained the desired product 4a in up to 72% as best yield using 0.6 mol % of tetrakis(triphenylphosphine)palladium(0) and 1.2 mol % of copper iodide (Scheme 2, Table 1).
![[1860-5397-17-115-i2]](/bjoc/content/inline/1860-5397-17-115-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridines 4a–g.
Scheme 2: Synthesis of 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridines 4a–g.
Table 1: Effects of solvent, base, and temperature on the Sonogashira coupling between 2 and 3a.a
| Entry | Temperature | Base | Solvent | Yieldb (%) |
| 1 | 80 | Et3N | dioxane | 72 |
|
2
3 |
80
80 |
Et3N
Et3N |
toluene
DMF |
70
68 |
|
4
5 6 7 |
80
80 90 100 |
iPr2EtN
iPr2EtN iPr2EtN iPr2EtN |
dioxane
– – – |
80
90 87 88 |
aReagents and conditions: Pd(PPh3)4 (0.6 mol %), CuI (1.2 mol %), solvent (3 mL), base (0.5 mL), 2 (0.5 mmol), 3a (1.1 mmol), 80 °C, 3 h. bIsolated yield.
The reaction proceeded chemoselectively at the two carbon–bromine bonds giving 2,4-bis(phenylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4a). This result was not entirely predictable, as the chlorine atom is located at the more reactive electron-poor pyridine moiety of the heterocyclic core structure. In fact, the chlorine position proved to be quite unreactive and all attempts to isolate 2,4,9-tris(phenylethynyl)-5,6,7,8-tetrahydroacridine failed even after using an excess of phenylacetylene and prolonging the reaction time. In order to study the regioselectivity of the reaction, a series of experiments were carried out with decreasing amounts of phenylacetylene. Although we used one equivalent of phenylacetylene, we were not able to isolate the mono coupling product.
Concerning the catalyst performance, Pd(PPh3)4 was found to be a suitable catalyst. In contrast, PdCl2(PPh3)2 was slightly less effective and gave lower yields. The replacement of dioxane by toluene or DMF did not lead to any significant improvement of the yields (Table 1, entries 2 and 3). For further improvement of the coupling, we evaluated the effect of the organic base. We found that the use of DIPEA instead of Et3N afforded better yields (Table 1, entry 4). Besides, the use of DIPEA as base and solvent gave a significant improvement of the yield (Table 1, entry 5). Our final effort consisted in evaluating the effect of the temperature. We found that increasing the temperature to 90 or 100 °C did not lead to any improvement (Table 1, entries 6 and 7).
The best result for the Sonogashira coupling reaction between intermediate 2 and phenylacetylene (3a) was obtained using 0.6 mol % of Pd(PPh3)4, 1.2 mol % of CuI in DIPEA at 80 °C for three hours. With the optimized conditions in hand, we examined the scope of the coupling reaction of 2 with different phenylacetylenes 3b–g. As shown in Table 2, tetrahydroacridine derivatives 4a–g were obtained in moderate to good yields. The yields were better for acetylenes containing electron-donating substituents. For example, arylacetylene 3g, bearing a methoxy group, gave the best chemical yield of 93%. However, in case of the electron-attracting trifluoromethyl group (3e), we obtained a somewhat lower, but still good yield of 75%.
Table 2: Yields of 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine derivatives 4a–g.
| Entry | Arylacetylene | Product | Yielda (%) | Time (h) |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-115-i3.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-115-i4.svg?max-width=637&scale=1.0)
4a |
90 | 3 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-115-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-115-i6.svg?max-width=637&scale=1.0)
4b |
85 | 3 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-115-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-115-i8.svg?max-width=637&scale=1.0)
4c |
82 | 3 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-115-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-115-i10.svg?max-width=637&scale=1.0)
4d |
83 | 3 |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-115-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-115-i12.svg?max-width=637&scale=1.0)
4e |
80 | 4 |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-115-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-115-i14.svg?max-width=637&scale=1.0)
4f |
75 | 4 |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-115-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-115-i16.svg?max-width=637&scale=1.0)
4g |
93 | 4 |
aIsolated yields.
Photophysical properties
As a prominent blue fluorescence was observed for the prepared tetrahydroacridine derivatives, their photophysical properties were investigated. Absorption and emission spectra were measured at room temperature in diluted dichloromethane solution and are depicted in Figure 2 and Figure 3. All spectroscopic data, including the maximum of absorption and emission, fluorescence quantum yield, stokes shift, onset of the absorption wavelengths and optical band gap are summarized in Table 3. Aiming to understand the impact of substituents at arylethynyl groups, spectra of diversely substituted tetrahydroacridines were compared with unsubstituted derivative 4a taken as reference.
![[1860-5397-17-115-2]](/bjoc/content/figures/1860-5397-17-115-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis absorption spectra of 4a,b and 4e–g in diluted dichloromethane solutions at room temperature (c = 1 × 10−5 M).
Figure 2: UV–vis absorption spectra of 4a,b and 4e–g in diluted dichloromethane solutions at room temperature...
![[1860-5397-17-115-3]](/bjoc/content/figures/1860-5397-17-115-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Emission spectra of 4a,b and 4e–g in diluted dichloromethane solutions at room temperature (c = 1 × 10−5 M).
Figure 3: Emission spectra of 4a,b and 4e–g in diluted dichloromethane solutions at room temperature (c = 1 ×...
Table 3: Photophysical properties of 4a,b and 4e–g in dichloromethane solutions.
| Emission | Absorption | |||||||
| λem (nm) | FWHMa | Φfluob | ν̃ stokesc | λabs (nm ) | log ε | λonset (nm) | Egopt (eV)d | |
| 4a |
386
400 |
48 | 0.11 | 2400 |
346
365 |
4.83
4.71 |
389 | 3.18 |
| 4b |
394
406 |
55 | 0.10 | 4100 |
311
348 |
5.10
4.85 |
406 | 3.05 |
| 4e |
382
398 |
50 | 0.14 | 2600 |
343
361 |
5.08
4.98 |
385 | 3.22 |
| 4f |
380
396 |
50 | 0.11 | 2400 |
339
362 |
5.11
4.98 |
387 | 3.20 |
| 4g | 426 | 66 | 0.20 | 4300 |
321
360 |
4.96
4.73 |
415 | 2.98 |
aSpectrum full width at half maximum. bFluorescence standard: quinine bisulfate in 1 N H2SO4 (Φfluo= 0.54) [70]. cStokes shift in wavenumber (cm−1) = (1/λabsmax − 1/λemmax ) 107. dEstimated from the onset point of the absorption spectra: Egopt = 1240/λonset [71].
The UV–vis absorption was measured in a spectral rang of 300 nm to 600 nm. The optical absorption spectra of all compounds spread over the UV range and showed wide absorption bands. These bands are assigned to a π→π* electronic transitions of the quinoline core and its two arylethynyl groups. As shown in Figure 2, the unsubstituted derivative 4a exhibited wide bands with two maxima at 346 and 365 nm. A methyl group at the ortho position have a minor impact and derivative 4b showed similar optical transitions with a slight red shift. While, derivatives 4e and 4f bearing an electron-deficient fluoro or trifluoromethyl group show a hyperchromic shift of their bands located between 340 and 380 nm. In case of the electron-donating methoxy substituent (4g), a bathochromic shift was observed. Besides, a new band appeared at 321 nm which may be attributed to intermolecular charge transfer between the oxygen lone pair electrons and the quinoline core. The unsubstituted derivative 4a presents an onset of absorption (λonset) at 389 nm and its optical band gap was deduced to be around 3.18 eV. Tetrahydroacridines 4e and 4f showed approximately the same optical band gaps. However, the optical band gaps of 4b and 4g are lower (3.05 eV and 2.98 eV, respectively).
Emission spectra of synthesized tetrahydroacridine derivatives were measured in dichloromethane solutions under UV-laser excitation of 325 nm. The emission spectrum of compound 4a presents a profile with two transitions located at 386 and 400 nm. Methyl-substituted derivative 4b gave a slight red shift of 10 nm as compared to 4a. In contrast, fluorine and trifluoromethyl-substituted derivatives 4e and 4f show nearly the same emission. However, derivative 4g containing an electron-donating methoxy substituent exhibits a larger red shift of around 40 nm. Based on the absorption and emission spectra, the prepared tetrahydroacridine derivatives possess stokes shifts (wavenumber) ranging from 2400 to 4300 cm−1. Their fluorescence quantum yields range from 0.1 to 0.2 as measured according to a relative method using quinine sulfate [70]. Tetrahydroacridine derivative 4g containing an electron-donating methoxy substituent gave the highest fluorescence intensity as shown in Figure 3 and a quantum yield of 20%.
DFT studies
The arylethynyl substituents showed an impact on the absorption and emission. In order to elucidate these experimental observations, quantum chemical calculations based on density functional theory (DFT) methodology were performed. The estimated visualization of highest occupied and lowest unoccupied molecular orbitals, as well as the molecular electrostatic potential (MEP) of prepared products are given in Table 4.
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-115-i17.png?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-17-115-i18.png?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-17-115-i19.png?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-17-115-i20.png?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-17-115-i21.png?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-17-115-i22.png?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-17-115-i23.png?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-17-115-i24.png?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-17-115-i25.png?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-17-115-i26.png?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-17-115-i27.png?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-17-115-i28.png?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-17-115-i29.png?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-17-115-i30.png?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-17-115-i31.png?max-width=637&scale=1.0)
For phenylethynyl-substituted product 4a, the blue colored surface, located mainly at the cyclohexane ring, visualizes the electron deficiency. While the red region, localized essentially at the nitrogen atom and its closer ethynyl group, show the electron abundance. Due to their low donating effect, the methyl group in product 4b induce an addition of yellow regions into the external phenyl rings. However, the electron-deficient fluorine atom in derivative 4e results in a decrease of the electron density of the tetrahydroacridine core and the external phenyl rings. For product 4f, the high electron-deficient effect of the trifluoromethyl groups induces the appearance of blue surfaces around the tetrahydroacridine core, the ethynyl groups and the external phenyl rings, indicating a significant decrease of their electronic densities. However, a yellow-red region is added to the electrostatic map of compound 4g, due to the positive mesomeric effect of the π-donating methoxy substituent. Accordingly, we conclude that substituents at the introduced arylethynyl groups can communicate electronically with the central tetrahydroacridine core via the ethynyl group. Consequently, they influence the electronic situation of the prepared tetrahydroacridines and are expected to change their structural proprieties. Hence, some structural parameters including gap (Eg), ionization potential (IP), electron affinity (EA) and dipole moments (µ) were deduced on the ground state from the optimized chemical structure of obtained molecules (Table 5).
Table 5: Summary of theoretical calculations.a
| Compound | EHOMO | ELUMO | IP | EA | Eg = ELUMO - EHOMO (eV) | μ (D) |
| 4a | −5.499 | −1.992 | 5.5 | 1.99 | 3.5 | 0.698 |
| 4b | −5.465 | −2.00 | 5.26 | 2 | 3.26 | 0.970 |
| 4e | −5.553 | −2.033 | 5.55 | 2.0 | 3.55 | 0.657 |
| 4f | −5.887 | −2.356 | 5.887 | 2.35 | 3.53 | 4.276 |
| 4g | −5.055 | −1.742 | 5.178 | 1.8 | 3.36 | 1.669 |
aThe DFT calculations were performed on optimized geometries with a DFT/b3lyp/6-31g(d).
The calculated permanent dipole moments µ (D) have considerably increased values for 4f and 4g, which show significant changes in their experimental emission properties. The presence of six fluorine atoms induces a large polarity difference. In fact, derivative 4f shows the highest dipole moment of 4.276 D as compared to 4a (0.698 D). As shown in Table 5, the ionization potential (IP) and electron affinity (EA) of tetrahydroacridines are almost identical. In addition, EHOMO and ELUMO do not change notably and the calculated Eg values vary only slightly from 3.26 to 3.55 eV. Although, the HOMO energy level of 4g with −5.055 eV is higher than −5.887 eV of 4e, both compounds have close band gap values of 3.36 eV and 3.53 eV, respectively.
Conclusion
In summary, we have reported a facile synthesis of 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine derivatives via a double Sonogashira cross-coupling method. The arylethynyl groups expand the π-conjugation of the tetrahydroacridine core. The substituents located at the aryl group influenced the photophysical properties of the prepared molecules. In particular, the methoxy derivative shows promising fluorescence properties.
Experimental
Materials and measurements
All reactions were carried out under an inert argon atmosphere. Anhydrous solvents and chemicals were purchased from Sigma-Aldrich and used without further purification. All reactions were monitored by thin-layer chromatography (TLC) using commercial silica-gel plate 60 coated with a fluorescence indicator and the visualization was performed by UV (254 nm). Organic compounds were purified using Merck Silica gel 60 (0.043–0.06 mm). Solvents for work-up and column chromatography were distilled before use.
NMR data were recorded on Bruker ARX 300 instruments in CDCl3 with tetramethylsilane as the internal standard (signals due to the solvent; CHCl3: δ 7.26 for 1H and δ 77.16 for 13C). The 1H NMR chemical shifts and coupling constants were determined assuming first-order behavior. Peak characterization of 1H NMR spectra: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Chemical shifts were given in ppm (δ) relative to tetramethylsilane (SiMe4). Photophysical studies were carried out in freshly prepared dichloromethane solutions with concentrations of 1 × 10−5 M. UV–vis spectra were recorded on a Shimadzu 2401 PC spectrophotometer in quartz cuvettes with a path length of 1 cm. Emission spectra were recorded on a Perkin-Elmer LS50B spectrofluorimeter.
Theoretical calculations
Theoretical studies were realized in vacuum with Gaussian 09 program [72]. The geometry of the equilibrium conformer at ground state was first found at AM1 level. Then, further optimizations through density functional theory (DFT) approach [73] at the restricted Becke3–Lee–Yang–Parr hybrid functional (B3LYP) with standard basis set 6-31G were carried out.
Experimental procedure and spectroscopic data for 2,4-dibromo-9-chloro-5,6,7,8-tetrahydroacridine (2)
3,5-Dibromoanthranilic acid (2.92 g, 10 mmol, 1 equiv) and cyclohexanone (1.07 mL, 11 mmol, 1.1 equiv) were stirred in an ice bath. Then, 15 mL of POCl3 was carefully added and the mixture was heated under reflux for 4 hours. The mixture was cooled to room temperature and concentrated to give a slurry. The residue was diluted with dichloromethane, neutralized with aqueous NaHCO3, and washed with brine. The organic layer was dried over anhydrous K2CO3 and concentrated to afford a yellow solid. It was recrystallized from acetone to give 2 as pale yellow solid (3.24 g, 87%) [65]. 1H NMR (300 MHz, CDCl3) δ 1.85–1.95 (m, 4H, CH2-CH2), 2.93 (t, 3J = 6.0 Hz, 2H, CH2), 3.11 (t,3J = 6.0 Hz, 2H, CH2), 8.05 (s, 1H, aryl-H), 8.21 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 22.40 (CH2), 22.42 (CH2), 27.63 (CH2), 34.45 (CH2), 119.73 (CAr), 125.37 (CAr), 126.00 (CAr), 127.16 (CAr), 130.78 (CAr), 135.58 (Cl-CAr), 140.36 (CAr), 142.46 (CAr), 161.25 (N =CAr).
Experimental procedure for the Sonogashira coupling and spectroscopic data for 2,4-bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine derivatives 4a–g
2,4-Dibromo-9-chloro-5,6,7,8-tetrahydroacridine (2, 372.89 mg, 1 mmol, 1 equiv), arylacetylene (2.2 mmol, 2.2 equiv), Pd(PPh3)4 (6.9 mg, 0.006 mmol, 0.6 mol %) and CuI (2.2 mg, 0.012 mmol, 1.2 mol %) were added to a dried glass pressure tube. The tube was evacuated and backfilled three times with argon, then diisopropylethylamine (4.0 mL) was added. The tube was sealed with a Teflon cap and heated to 80 °C for 3–4 hours until completion of the reaction (monitored by TLC). The mixture was then cooled to room temperature and the solvent was removed under reduced pressure. Water (10 mL) was added and the solution was extracted using CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4) and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography using heptanes/ethyl acetate 9:1 as eluent.
2,4-Bis(phenylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4a): colorless solid (375 mg, 0.9 mmol, 90%); mp 188–190 °C; 1H NMR (300 MHz, CDCl3) δ 1.81–1.92 (m, 4H, CH2-CH2), 2.94 (t, 3J = 6.0 Hz, 2H, CH2), 3.21 (t, 3J = 6.0 Hz, 2H, CH2), 7.29–7.35 (m, 6H, aryl-H), 7.49–7.53 (m, 2H, aryl-H), 7.64–7.68 (m, 2H, aryl-H), 8.01 (s, 1H, aryl-H), 8.24 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 22.4 (CH2), 22.5 (CH2), 30.9 (CH2), 34.4 (CH2), 86.5 (Csp), 88.5 (Csp), 91.1 (Csp), 96.2 (Csp), 121.3 (CAr), 122.8 (CAr), 123.0 (CAr), 123.3 (CAr), 125.4 (CAr), 127.2 (CAr), 128.3 (CAr), 128.4 (CAr), 128.5 (CAr), 128.6 (CAr), 130.2 (CAr), 131.7 (CAr), 132.0 (CAr), 136.2 (Cl-CAr), 141.5 (CAr), 145.4 (N-CAr), 161.1 (N =CAr); HRMS (ESI): [M]+ calcd for C29H20ClN, 417.1284; found, 417.1265.
2,4-Bis(o-tolylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4b): pale green solid (385 mg, 0.85 mmol, 85%); mp 119–120 °C; 1H NMR (300 MHz, CDCl3) δ 1.78–1.93 (m, 4H, 2CH2), 2.52 (s, 3H, aryl-CH3), 2.66 (s, 3H, aryl-CH3), 2.92 (t, 3J = 6.0 Hz, 2H, CH2), 3.24 (t, 3J = 6.0 Hz, 2H, CH2), 7.12–7.25 (m, 6H, aryl-H), 7.42–7.51 (m, 1H, aryl-H), 7.61–7.66 (m, 1H, aryl-H), 7.95 (s, 1H, aryl-H), 8.22 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 20.8 (CH3), 20.9 (CH3), 22.4 (CH2), 22.4 (CH2), 27.5 (CH2), 34.1 (CH2), 90.1 (Csp), 90.2 (Csp), 92.3 (Csp), 95.8 (Csp), 119.7 (CAr), 121.80 (CAr), 122.9 (CAr), 123.1 (CAr), 125.5 (CAr), 126.8 (CAr), 128.6 (CAr), 128.8 (CAr), 129.4 (CAr), 129.6 (CAr), 130.3 (CAr), 130.4 (CAr), 132.1 (CAr), 135.8 (Cl-CAr), 141.1 (CAr), 145.0 (N-CAr), 160.8 (N =CAr); HRMS (ESI): [M]+ calcd for C31H24ClN, 445.1597; found, 445.1576.
2,4-Bis(m-tolylethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4c): pale black solid (364 mg, 0.82 mmol, 82%); mp 170–172 °C; 1H NMR (300 MHz, CDCl3) δ 1.80–1.94 (m, 4H, 2CH2), 2.31 (s, 3H, aryl-CH3), 2.33 (s, 3H, aryl-CH3), 2.92 (t, 3J = 6.0 Hz, 2H, CH2), 3.21 (t, 3J = 6.0 Hz, 2H, CH2), 7.05–7.36 (m, 4H, aryl-H), 7.40–7.51 (m, 2H, aryl-H), 7.58–7.64 (m, 2H, aryl-H), 7.91 (s, 1H, aryl-H), 8.21 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3)δ 21.27 (CH3), 21.29 (CH3), 22.41 (CH2), 22.49 (CH2), 27.56 (CH2), 34.36 (CH2), 86.10 (Csp), 88.17 (Csp), 91.42 (Csp), 96.58 (Csp), 121.53 (CAr), 122.58 (CAr), 122.96 (CAr), 123.15 (CAr), 125.51 (CAr), 127.08 (CAr), 128.19 (CAr), 128.36 (CAr), 129.15 (CAr), 129.62 (CAr), 130.25 (CAr), 132.60 (CAr), 136.37 (CAr), 138.16 (Cl-CAr), 141.79 (CAr), 145.15 (N-CAr), 160.99 (N =CAr); HRMS (ESI): [M]+ calcd for C31H24ClN, 445.1597; found, 445.1584.
2,4-Bis((p-ethylphenyl)ethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4d): pale black solid (392 mg, 0.83 mmol, 83%); mp 189–191 °C; 1H NMR (300 MHz, CDCl3) δ 1.19 (t, 6H, 2CH3), 1.82–1.93 (m, 4H, 2CH2), 2.54 (q, 4H, 2CH2), 2.93 (t, 3J = 6.0 Hz, 2H, CH2), 3.28 (t, 3J = 6.0 Hz, 2H, CH2), 7.11–7.19 (m, 4H, aryl-H), 7.35–7.42 (m, 2H, aryl-H), 7.62–7.69 (m, 2H, aryl-H), 7.96 (s, 1H, aryl-H), 8.23 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 15.31 (CH3), 15.32 (CH3), 22.26 (CH2), 22.37 (CH2), 27.56 (CH2), 28.89 (CH2), 28.91 (CH2), 33.99 (CH2), 85.45 (Csp), 87.75 (Csp), 91.77. (Csp), 97.24 (Csp), 119.84 (CAr), 120.39 (CAr), 122.03 (CAr), 122.62 (CAr), 125.60 (CAr), 126.73 (CAr), 127.86 (CAr), 128.05 (CAr), 130.41 (CAr), 131.78 (CAr), 132.14 (CAr), 136.72 (Cl-CAr), 142.60 (CAr), 144.24 (CAr), 145.08 (CAr), 145.30 (N-CAr), 160.84 (N=CAr); HRMS (ESI): [M]+ calcd for C33H28ClN, 473.1910; found, 473.1924.
2,4-Bis((o-fluorophenyl)ethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4e): pale red solid (375 mg, 0.8 mmol, 80%); mp 179–181 °C; 1H NMR (300 MHz, CDCl3) δ 1.81–1.96 (m, 4H, 2CH2), 2.93 (t, 3J = 6.0 Hz, 2H, CH2), 3.25 (t, 3J = 6.0 Hz, 2H, CH2), 7.06–7.15 (m, 4H, aryl-H), 7.21–7.31 (m, 2H, aryl-H), 7.45–7.53 (m, 1H, aryl-H), 7.60–7.65 (m, 1H, aryl-H), 8.03 (s, 1H, aryl-H), 8.27 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 21.3 (CH2), 21.4 (CH2), 26.5 (CH2), 33.4 (CH2), 83.5 (Csp), 88.6 (Csp), 90.3. (Csp), 92.3 (Csp), 110.3 (CAr), 110.9 (CAr), 114.3 (CAr), 114.8 (CAr), 120.0 (CAr), 121.6 (CAr), 122.9 (CAr), 123.0 (CAr), 124.4 (CAr), 126.7 (CAr), 129.2 (CAr), 128.8 (CAr), 129.4 (CAr), 129.6 (CAr), 132.9 (CAr), 135.3 (Cl-CAr), 140.8 (CAr), 144.3 (N-CAr), 159.8 (F-CAr), 160.04 (F-CAr). 163.8 (N=CAr); HRMS (ESI): [M]+ calcd for C29H18ClF2N, 453.1095; found, 453.1081.
2,4-Bis((p-trifluoromethylphenyl)ethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4f): brown solid (414 mg, 0.75 mmol, 75%); mp 170–172 °C; 1H NMR (300 MHz, CDCl3) δ 1.81–1.95 (m, 4H, 2CH2), 2.95 (t, 3J = 6.0 Hz, 2H, CH2), 3.23 (t, 3J = 6.0 Hz, 2H, CH2), 7.51–7.75 (m, 8H, aryl-H), 8.05 (s, 1H, aryl-H), 8.31 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 22.3 (CH2), 22.4 (CH2), 27.6 (CH2), 34.5 (CH2), 86.5 (Csp), 89.7 (Csp), 90.5 (Csp), 93.2 (Csp), 120.6 (CAr), 122.4 (CAr), 125.2 (F3C-CAr), 125.3 (F3C-CAr), 125.4 (CAr), 125.5 (CAr), 126.5 (CAr), 127.1 (CAr), 128.2 (CAr), 129.9 (CAr), 130.4 (CAr), 130.6 (CAr), 131.9 (CAr), 132.2 (CAr), 136.4 (Cl-CAr), 141.8 (CAr), 145.5 (N-CAr), 161.7 (N =CAr); HRMS (ESI): [M]+ calcd for C31H18ClF6N, 553.1032; found, 553.1015.
2,4-Bis((p-methoxyphenyl)ethynyl)-9-chloro-5,6,7,8-tetrahydroacridine (4g): yellow solid (536 mg, 0.93 mmol, 93%); mp 125–127 °C; NMR (300 MHz, CDCl3) δ 1.83–1.92 (m, 4H, 2CH2), 2.94 (t, 3J = 6.0 Hz, 2H, CH2), 3.33 (t, 3J = 6.0 Hz, 2H, CH2), 3.79 (s, 6H, 2 OCH3), 7.75–7.89 (m, 4H, aryl-H), 7.47 (d, 2H, aryl-H), 8.62 (d, 2H, aryl-H), 7.93 (s, 1H, aryl-H), 8.21 (s, 1H, aryl-H); 13C NMR (75 MHz, CDCl3) δ 22.2 (CH2), 22.3 (CH2), 27.5 (CH2), 33.9 (CH2), 55.3 (OCH3), 55.3 (OCH3), 87.2 (Csp), 88.7 (Csp), 90.2 (Csp), 92.2 (Csp), 113.9 (CAr), 114.1 (CAr), 114.7 (CAr), 115.3 (CAr), 122.2 (CAr), 122.6 (CAr), 125.6 (CAr), 126.3 (CAr), 130.0 (CAr), 130.4 (CAr), 132.9 (CAr), 133.3 (CAr), 133.7 (CAr), 136.5 (Cl-CAr), 143.1 (N-CAr), 159.9 (OCH3-CAr), 160.0 (OCH3-CAr), 160.9 (N =CAr); HRMS (ESI): [M]+ calcd for C31H18ClF6N, 577.1495; found, 577.1479.
Supporting Information
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Hong, T. R.; Shin, J.; Um, H. A.; Lee, T. W.; Cho, M. J.; Kim, G. W.; Kwon, J. H.; Choi, D. H. Dyes Pigm. 2014, 108, 7–14. doi:10.1016/j.dyepig.2014.04.015
Return to citation in text: [1] -
Reddy, G.; Basak, P.; Jones, L. A.; Della Gaspera, E.; Islavath, N.; Giribabu, L. Sol. Energy 2020, 206, 539–547. doi:10.1016/j.solener.2020.06.040
Return to citation in text: [1] -
Sun, Z.-Z.; Sun, P.-P.; Feng, S.; Xu, Y.-L.; Liu, J.-F. Synth. Met. 2019, 254, 34–41. doi:10.1016/j.synthmet.2019.05.014
Return to citation in text: [1] -
Payne, A.-J.; McCahill, J. S. J.; Welch, G. C. Dyes Pigm. 2015, 123, 139–146. doi:10.1016/j.dyepig.2015.07.035
Return to citation in text: [1] -
Hoang, M. H.; Cho, M. J.; Kim, K. H.; Cho, M. Y.; Joo, J.-s.; Choi, D. H. Thin Solid Films 2009, 518, 501–506. doi:10.1016/j.tsf.2009.07.030
Return to citation in text: [1] -
Pham, H. T.; Lee, D.-S.; Dao, T. D.; Jeong, H.-D. J. Ind. Eng. Chem. (Amsterdam, Neth.) 2018, 57, 22–27. doi:10.1016/j.jiec.2017.08.003
Return to citation in text: [1] -
Ma, L.; Yu, Y.; Li, L.; Lei, T.; Jiao, B.; Hou, X.; Wu, Z. Org. Electron. 2018, 57, 123–132. doi:10.1016/j.orgel.2018.02.042
Return to citation in text: [1] -
Fujita, T.; Haketa, Y.; Maeda, H.; Yamamoto, T. Org. Electron. 2017, 49, 53–63. doi:10.1016/j.orgel.2017.06.028
Return to citation in text: [1] -
Yan, Z.; Wang, C.; Tang, Y.; Zhu, Y.; Cao, Q.; Yang, T.; Hu, L. Spectrochim. Acta, Part A 2020, 224, 117451. doi:10.1016/j.saa.2019.117451
Return to citation in text: [1] -
Zhao, Q.; Yuan, H.; Xu, X.; Hu, L.; Gong, P.; Yan, Z. Dyes Pigm. 2019, 165, 217–222. doi:10.1016/j.dyepig.2019.02.030
Return to citation in text: [1] -
Graebe, C.; Caro, H. Ann. Chem. Pharm. 1871, 158, 265–281. doi:10.1002/jlac.18711580302
Return to citation in text: [1] -
Kumar, R.; Kaur, M.; Kumari, M. Acta Pol. Pharm. 2012, 69, 3–9.
Return to citation in text: [1] -
Campbell, N. H.; Parkinson, G. N.; Reszka, A. P.; Neidle, S. J. Am. Chem. Soc. 2008, 130, 6722–6724. doi:10.1021/ja8016973
Return to citation in text: [1] -
Albert, A. The Acridines. Their Preparation, Physical, Chemical, and Biological Properties and Uses, 2nd ed.; Edward Arnold Ltd.: London, U.K., 1966.
Return to citation in text: [1] -
Goodell, J. R.; Ougolkov, A. V.; Hiasa, H.; Kaur, H.; Remmel, R.; Billadeau, D. D.; Ferguson, D. M. J. Med. Chem. 2008, 51, 179–182. doi:10.1021/jm701228e
Return to citation in text: [1] -
Fernández-Calienes Valdés, A. Open Med. Chem. J. 2011, 5, 11–20. doi:10.2174/1874104501105010011
Return to citation in text: [1] -
Nowak, K. J. Mol. Struct. 2017, 1146, 562–570. doi:10.1016/j.molstruc.2017.05.042
Return to citation in text: [1] -
Budiman, M. E.; Bierbach, U.; Alexander, R. W. Biochemistry 2005, 44, 11262–11268. doi:10.1021/bi050745n
Return to citation in text: [1] -
Sondhi, S. M.; Singh, J.; Rani, R.; Gupta, P. P.; Agrawal, S. K.; Saxena, A. K. Eur. J. Med. Chem. 2010, 45, 555–563. doi:10.1016/j.ejmech.2009.10.042
Return to citation in text: [1] -
Benoit, A. R.; Schiaffo, C.; Salomon, C. E.; Goodell, J. R.; Hiasa, H.; Ferguson, D. M. Bioorg. Med. Chem. Lett. 2014, 24, 3014–3017. doi:10.1016/j.bmcl.2014.05.037
Return to citation in text: [1] -
Denny, W. A. Curr. Med. Chem. 2002, 9, 1655–1665. doi:10.2174/0929867023369277
Return to citation in text: [1] -
Demeunynck, M.; Charmantray, F.; Martelli, A. Curr. Pharm. Des. 2001, 7, 1703–1724. doi:10.2174/1381612013397131
Return to citation in text: [1] -
Demeunynck, M. Expert Opin. Ther. Pat. 2004, 14, 55e70.
Return to citation in text: [1] -
Denny, W. A. Med. Chem. Rev.–Online 2004, 1, 257–266. doi:10.2174/1567203043401923
Return to citation in text: [1] -
Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Anti-Cancer Agents Med. Chem. 2007, 7, 139–169. doi:10.2174/187152007780058669
Return to citation in text: [1] -
Cholewiński, G.; Dzierzbicka, K.; Kołodziejczyk, A. M. Pharmacol. Rep. 2011, 63, 305–336. doi:10.1016/s1734-1140(11)70499-6
Return to citation in text: [1] -
Dopierała, A.; Wrosz, P.; Mazerski, J. Postepy Hig. Med. Dosw. 2011, 65, 263–269. doi:10.5604/17322693.941521
Return to citation in text: [1] -
Pérez, S. A.; de Haro, C.; Vicente, C.; Donaire, A.; Zamora, A.; Zajac, J.; Kostrhunova, H.; Brabec, V.; Bautista, D.; Ruiz, J. ACS Chem. Biol. 2017, 12, 1524–1537. doi:10.1021/acschembio.7b00090
Return to citation in text: [1] -
Galdino-Pitta, M. R.; Pitta, M. G. R.; Lima, M. C. A.; Galdino, L. S.; Pitta, R. I. Mini-Rev. Med. Chem. 2013, 13, 1256–1271. doi:10.2174/1389557511313090002
Return to citation in text: [1] -
Singh, H.; Singh, H.; Sharma, S.; Singh Bedi, P. M. Heterocycles 2015, 91, 2043–2085. doi:10.3987/rev-15-826
Return to citation in text: [1] -
Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Expert Opin. Ther. Pat. 2014, 24, 647–664. doi:10.1517/13543776.2014.902052
Return to citation in text: [1] -
Wiseman, A.; Sims, L. A.; Snead, R.; Gronert, S.; Maclagan, R. G. A. R.; Meot-Ner (Mautner), M. J. Phys. Chem. A 2015, 119, 118–126. doi:10.1021/jp506913r
Return to citation in text: [1] -
Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076
Return to citation in text: [1] -
Yang, W.; Monteiro, J. H. S. K.; de Bettencourt-Dias, A.; Catalano, V. J.; Chalifoux, W. A. Angew. Chem., Int. Ed. 2016, 55, 10427–10430. doi:10.1002/anie.201604741
Return to citation in text: [1] -
Zhang, J.; Lakowicz, J. R. J. Phys. Chem. B 2005, 109, 8701–8706. doi:10.1021/jp046016j
Return to citation in text: [1] -
Magnan, F.; Gabidullin, B.; Brusso, J. L. RSC Adv. 2016, 6, 97420–97429. doi:10.1039/c6ra18897d
Return to citation in text: [1] -
Yamashita, Y. Sci. Technol. Adv. Mater. 2009, 10, 024313. doi:10.1088/1468-6996/10/2/024313
Return to citation in text: [1] -
Boudreault, P.-L. T.; Alleyne, B.; Xia, C. Organic electroluminescent materials and devices. U.S. Pat. Appl. US20180013077A1, Jan 11, 2018.
Return to citation in text: [1] -
Takimiya, K.; Yamamoto, T.; Ebata, H.; Izawa, T. Sci. Technol. Adv. Mater. 2007, 8, 273–276. doi:10.1016/j.stam.2007.02.010
Return to citation in text: [1] -
Goel, A.; Kumar, V.; Singh, S. P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R. S. J. Mater. Chem. 2012, 22, 14880. doi:10.1039/c2jm31052j
Return to citation in text: [1] -
Lian, Y.; Hummel, J. R.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2013, 135, 12548–12551. doi:10.1021/ja406131a
Return to citation in text: [1] -
Wang, T.-J.; Chen, W.-W.; Li, Y.; Xu, M.-H. Org. Biomol. Chem. 2015, 13, 6580–6586. doi:10.1039/c5ob00755k
Return to citation in text: [1] -
Tsvelikhovsky, D.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 14048–14051. doi:10.1021/ja107511g
Return to citation in text: [1] -
Mohammadi-Khanaposhtani, M.; Rezaei, S.; Khalifeh, R.; Imanparast, S.; Faramarzi, M. A.; Bahadorikhalili, S.; Safavi, M.; Bandarian, F.; Nasli Esfahani, E.; Mahdavi, M.; Larijani, B. Bioorg. Chem. 2018, 80, 288–295. doi:10.1016/j.bioorg.2018.06.035
Return to citation in text: [1] -
Pintori, D. G.; Greaney, M. F. Org. Lett. 2010, 12, 168–171. doi:10.1021/ol902568x
Return to citation in text: [1] -
Rogness, D. C.; Larock, R. C. J. Org. Chem. 2010, 75, 2289–2295. doi:10.1021/jo1000687
Return to citation in text: [1] -
Huang, X.; Zhang, T. J. Org. Chem. 2010, 75, 506–509. doi:10.1021/jo902311a
Return to citation in text: [1] -
Olszewska, P.; Mikiciuk-Olasik, E.; Błaszczak-Świątkiewicz, K.; Szymański, J.; Szymański, P. Biomed. Pharmacother. 2014, 68, 959–967. doi:10.1016/j.biopha.2014.10.018
Return to citation in text: [1] -
Pirrung, M. C.; Chau, J. H.-L.; Chen, J. Chem. Biol. 1995, 2, 621–626. doi:10.1016/1074-5521(95)90127-2
Return to citation in text: [1] -
Tripathi, R. P.; Verma, S. S.; Pandey, J.; Agarwal, K. C.; Chaturvedi, V.; Manju, Y. K.; Srivastva, A. K.; Gaikwad, A.; Sinha, S. Bioorg. Med. Chem. Lett. 2006, 16, 5144–5147. doi:10.1016/j.bmcl.2006.07.025
Return to citation in text: [1] -
Czarnecka, K.; Chufarova, N.; Halczuk, K.; Maciejewska, K.; Girek, M.; Skibiński, R.; Jończyk, J.; Bajda, M.; Kabziński, J.; Majsterek, I.; Szymański, P. Eur. J. Med. Chem. 2018, 145, 760–769. doi:10.1016/j.ejmech.2018.01.014
Return to citation in text: [1] -
Bajda, M.; Jończyk, J.; Malawska, B.; Czarnecka, K.; Girek, M.; Olszewska, P.; Sikora, J.; Mikiciuk-Olasik, E.; Skibiński, R.; Gumieniczek, A.; Szymański, P. Bioorg. Med. Chem. 2015, 23, 5610–5618. doi:10.1016/j.bmc.2015.07.029
Return to citation in text: [1] -
Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Bioorg. Chem. 2011, 39, 138–142. doi:10.1016/j.bioorg.2011.05.001
Return to citation in text: [1] -
Nishibori, M.; Oishi, R.; Itoh, Y.; Saeki, K. Jpn. J. Pharmacol. 1991, 55, 539–546. doi:10.1016/s0021-5198(19)39924-x
Return to citation in text: [1] -
Romero, A.; Cacabelos, R.; Oset-Gasque, M. J.; Samadi, A.; Marco-Contelles, J. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. doi:10.1016/j.bmcl.2013.02.017
Return to citation in text: [1] -
Liu, Z.; Fang, L.; Zhang, H.; Gou, S.; Chen, L. Bioorg. Med. Chem. 2017, 25, 2387–2398. doi:10.1016/j.bmc.2017.02.049
Return to citation in text: [1] -
Martins, C.; Carreiras, M. C.; León, R.; de los Ríos, C.; Bartolini, M.; Andrisano, V.; Iriepa, I.; Moraleda, I.; Gálvez, E.; García, M.; Egea, J.; Samadi, A.; Chioua, M.; Marco-Contelles, J. Eur. J. Med. Chem. 2011, 46, 6119–6130. doi:10.1016/j.ejmech.2011.09.038
Return to citation in text: [1] -
Flader, A.; Ohlendorf, L.; Ehlers, P.; Ammon, E.; Villinger, A.; Langer, P. Adv. Synth. Catal. 2019, 361, 2981–2991. doi:10.1002/adsc.201900034
Return to citation in text: [1] -
Janke, S.; Boldt, S.; Ghazargan, K.; Ehlers, P.; Villinger, A.; Langer, P. Eur. J. Org. Chem. 2019, 6177–6197. doi:10.1002/ejoc.201900913
Return to citation in text: [1] -
Janke, J.; Villinger, A.; Ehlers, P.; Langer, P. Synlett 2019, 30, 817–820. doi:10.1055/s-0037-1612256
Return to citation in text: [1] -
Tka, N.; Jabli, M.; Saleh, T. A.; Salman, G. A. J. Mol. Liq. 2018, 250, 423–432. doi:10.1016/j.molliq.2017.12.026
Return to citation in text: [1] -
Jabli, M.; Tka, N.; Ramzi, K.; Saleh, T. A. J. Mol. Liq. 2018, 249, 1138–1144. doi:10.1016/j.molliq.2017.11.126
Return to citation in text: [1] -
Jabli, M.; Tka, N.; Salman, G. A.; Elaissi, A.; Sebeia, N.; Hamdaoui, M. J. Mol. Liq. 2017, 242, 272–283. doi:10.1016/j.molliq.2017.07.018
Return to citation in text: [1] -
Modh, R. P.; De Clercq, E.; Pannecouque, C.; Chikhalia, K. H. J. Enzyme Inhib. Med. Chem. 2014, 29, 100–108. doi:10.3109/14756366.2012.755622
Return to citation in text: [1] -
Keri, R. S.; Quintanova, C.; Marques, S. M.; Esteves, A. R.; Cardoso, S. M.; Santos, M. A. Bioorg. Med. Chem. 2013, 21, 4559–4569. doi:10.1016/j.bmc.2013.05.028
Return to citation in text: [1] [2] -
Dehbanipour, Z.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor–Baltork, I. J. Organomet. Chem. 2017, 853, 5–12. doi:10.1016/j.jorganchem.2017.10.006
Return to citation in text: [1] -
Gogoi, R.; Saikia, R.; Borah, G. J. Organomet. Chem. 2019, 897, 80–88. doi:10.1016/j.jorganchem.2019.06.015
Return to citation in text: [1] -
Modak, A.; Mondal, J.; Bhaumik, A. Green Chem. 2012, 14, 2840. doi:10.1039/c2gc35820d
Return to citation in text: [1] -
Gogoi, A.; Dewan, A.; Borah, G.; Bora, U. New J. Chem. 2015, 39, 3341–3344. doi:10.1039/c4nj01822b
Return to citation in text: [1] -
Melhuish, W. H. J. Phys. Chem. 1961, 65, 229–235. doi:10.1021/j100820a009
Return to citation in text: [1] [2] -
Qin, Y.; Gu, H.; Liu, S.; Dai, W.; Luo, X. Synth. Met. 2018, 245, 42–50. doi:10.1016/j.synthmet.2018.08.008
Return to citation in text: [1] -
Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
Return to citation in text: [1] -
Koch, W.; Holthausen, M. C. A. Chemist's Guide to Density Functional Theory; Wiley-VCH: Weinheim, Germany, 2000. doi:10.1002/3527600043
Return to citation in text: [1]
| 65. | Keri, R. S.; Quintanova, C.; Marques, S. M.; Esteves, A. R.; Cardoso, S. M.; Santos, M. A. Bioorg. Med. Chem. 2013, 21, 4559–4569. doi:10.1016/j.bmc.2013.05.028 |
| 66. | Dehbanipour, Z.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor–Baltork, I. J. Organomet. Chem. 2017, 853, 5–12. doi:10.1016/j.jorganchem.2017.10.006 |
| 67. | Gogoi, R.; Saikia, R.; Borah, G. J. Organomet. Chem. 2019, 897, 80–88. doi:10.1016/j.jorganchem.2019.06.015 |
| 68. | Modak, A.; Mondal, J.; Bhaumik, A. Green Chem. 2012, 14, 2840. doi:10.1039/c2gc35820d |
| 69. | Gogoi, A.; Dewan, A.; Borah, G.; Bora, U. New J. Chem. 2015, 39, 3341–3344. doi:10.1039/c4nj01822b |
| 1. | Hong, T. R.; Shin, J.; Um, H. A.; Lee, T. W.; Cho, M. J.; Kim, G. W.; Kwon, J. H.; Choi, D. H. Dyes Pigm. 2014, 108, 7–14. doi:10.1016/j.dyepig.2014.04.015 |
| 2. | Reddy, G.; Basak, P.; Jones, L. A.; Della Gaspera, E.; Islavath, N.; Giribabu, L. Sol. Energy 2020, 206, 539–547. doi:10.1016/j.solener.2020.06.040 |
| 3. | Sun, Z.-Z.; Sun, P.-P.; Feng, S.; Xu, Y.-L.; Liu, J.-F. Synth. Met. 2019, 254, 34–41. doi:10.1016/j.synthmet.2019.05.014 |
| 12. | Kumar, R.; Kaur, M.; Kumari, M. Acta Pol. Pharm. 2012, 69, 3–9. |
| 13. | Campbell, N. H.; Parkinson, G. N.; Reszka, A. P.; Neidle, S. J. Am. Chem. Soc. 2008, 130, 6722–6724. doi:10.1021/ja8016973 |
| 14. | Albert, A. The Acridines. Their Preparation, Physical, Chemical, and Biological Properties and Uses, 2nd ed.; Edward Arnold Ltd.: London, U.K., 1966. |
| 61. | Tka, N.; Jabli, M.; Saleh, T. A.; Salman, G. A. J. Mol. Liq. 2018, 250, 423–432. doi:10.1016/j.molliq.2017.12.026 |
| 62. | Jabli, M.; Tka, N.; Ramzi, K.; Saleh, T. A. J. Mol. Liq. 2018, 249, 1138–1144. doi:10.1016/j.molliq.2017.11.126 |
| 63. | Jabli, M.; Tka, N.; Salman, G. A.; Elaissi, A.; Sebeia, N.; Hamdaoui, M. J. Mol. Liq. 2017, 242, 272–283. doi:10.1016/j.molliq.2017.07.018 |
| 11. | Graebe, C.; Caro, H. Ann. Chem. Pharm. 1871, 158, 265–281. doi:10.1002/jlac.18711580302 |
| 64. | Modh, R. P.; De Clercq, E.; Pannecouque, C.; Chikhalia, K. H. J. Enzyme Inhib. Med. Chem. 2014, 29, 100–108. doi:10.3109/14756366.2012.755622 |
| 9. | Yan, Z.; Wang, C.; Tang, Y.; Zhu, Y.; Cao, Q.; Yang, T.; Hu, L. Spectrochim. Acta, Part A 2020, 224, 117451. doi:10.1016/j.saa.2019.117451 |
| 10. | Zhao, Q.; Yuan, H.; Xu, X.; Hu, L.; Gong, P.; Yan, Z. Dyes Pigm. 2019, 165, 217–222. doi:10.1016/j.dyepig.2019.02.030 |
| 55. | Romero, A.; Cacabelos, R.; Oset-Gasque, M. J.; Samadi, A.; Marco-Contelles, J. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. doi:10.1016/j.bmcl.2013.02.017 |
| 56. | Liu, Z.; Fang, L.; Zhang, H.; Gou, S.; Chen, L. Bioorg. Med. Chem. 2017, 25, 2387–2398. doi:10.1016/j.bmc.2017.02.049 |
| 57. | Martins, C.; Carreiras, M. C.; León, R.; de los Ríos, C.; Bartolini, M.; Andrisano, V.; Iriepa, I.; Moraleda, I.; Gálvez, E.; García, M.; Egea, J.; Samadi, A.; Chioua, M.; Marco-Contelles, J. Eur. J. Med. Chem. 2011, 46, 6119–6130. doi:10.1016/j.ejmech.2011.09.038 |
| 65. | Keri, R. S.; Quintanova, C.; Marques, S. M.; Esteves, A. R.; Cardoso, S. M.; Santos, M. A. Bioorg. Med. Chem. 2013, 21, 4559–4569. doi:10.1016/j.bmc.2013.05.028 |
| 4. | Payne, A.-J.; McCahill, J. S. J.; Welch, G. C. Dyes Pigm. 2015, 123, 139–146. doi:10.1016/j.dyepig.2015.07.035 |
| 5. | Hoang, M. H.; Cho, M. J.; Kim, K. H.; Cho, M. Y.; Joo, J.-s.; Choi, D. H. Thin Solid Films 2009, 518, 501–506. doi:10.1016/j.tsf.2009.07.030 |
| 6. | Pham, H. T.; Lee, D.-S.; Dao, T. D.; Jeong, H.-D. J. Ind. Eng. Chem. (Amsterdam, Neth.) 2018, 57, 22–27. doi:10.1016/j.jiec.2017.08.003 |
| 7. | Ma, L.; Yu, Y.; Li, L.; Lei, T.; Jiao, B.; Hou, X.; Wu, Z. Org. Electron. 2018, 57, 123–132. doi:10.1016/j.orgel.2018.02.042 |
| 8. | Fujita, T.; Haketa, Y.; Maeda, H.; Yamamoto, T. Org. Electron. 2017, 49, 53–63. doi:10.1016/j.orgel.2017.06.028 |
| 58. | Flader, A.; Ohlendorf, L.; Ehlers, P.; Ammon, E.; Villinger, A.; Langer, P. Adv. Synth. Catal. 2019, 361, 2981–2991. doi:10.1002/adsc.201900034 |
| 59. | Janke, S.; Boldt, S.; Ghazargan, K.; Ehlers, P.; Villinger, A.; Langer, P. Eur. J. Org. Chem. 2019, 6177–6197. doi:10.1002/ejoc.201900913 |
| 60. | Janke, J.; Villinger, A.; Ehlers, P.; Langer, P. Synlett 2019, 30, 817–820. doi:10.1055/s-0037-1612256 |
| 36. | Magnan, F.; Gabidullin, B.; Brusso, J. L. RSC Adv. 2016, 6, 97420–97429. doi:10.1039/c6ra18897d |
| 37. | Yamashita, Y. Sci. Technol. Adv. Mater. 2009, 10, 024313. doi:10.1088/1468-6996/10/2/024313 |
| 38. | Boudreault, P.-L. T.; Alleyne, B.; Xia, C. Organic electroluminescent materials and devices. U.S. Pat. Appl. US20180013077A1, Jan 11, 2018. |
| 39. | Takimiya, K.; Yamamoto, T.; Ebata, H.; Izawa, T. Sci. Technol. Adv. Mater. 2007, 8, 273–276. doi:10.1016/j.stam.2007.02.010 |
| 41. | Lian, Y.; Hummel, J. R.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2013, 135, 12548–12551. doi:10.1021/ja406131a |
| 42. | Wang, T.-J.; Chen, W.-W.; Li, Y.; Xu, M.-H. Org. Biomol. Chem. 2015, 13, 6580–6586. doi:10.1039/c5ob00755k |
| 43. | Tsvelikhovsky, D.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 14048–14051. doi:10.1021/ja107511g |
| 44. | Mohammadi-Khanaposhtani, M.; Rezaei, S.; Khalifeh, R.; Imanparast, S.; Faramarzi, M. A.; Bahadorikhalili, S.; Safavi, M.; Bandarian, F.; Nasli Esfahani, E.; Mahdavi, M.; Larijani, B. Bioorg. Chem. 2018, 80, 288–295. doi:10.1016/j.bioorg.2018.06.035 |
| 45. | Pintori, D. G.; Greaney, M. F. Org. Lett. 2010, 12, 168–171. doi:10.1021/ol902568x |
| 46. | Rogness, D. C.; Larock, R. C. J. Org. Chem. 2010, 75, 2289–2295. doi:10.1021/jo1000687 |
| 47. | Huang, X.; Zhang, T. J. Org. Chem. 2010, 75, 506–509. doi:10.1021/jo902311a |
| 32. | Wiseman, A.; Sims, L. A.; Snead, R.; Gronert, S.; Maclagan, R. G. A. R.; Meot-Ner (Mautner), M. J. Phys. Chem. A 2015, 119, 118–126. doi:10.1021/jp506913r |
| 33. | Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076 |
| 34. | Yang, W.; Monteiro, J. H. S. K.; de Bettencourt-Dias, A.; Catalano, V. J.; Chalifoux, W. A. Angew. Chem., Int. Ed. 2016, 55, 10427–10430. doi:10.1002/anie.201604741 |
| 35. | Zhang, J.; Lakowicz, J. R. J. Phys. Chem. B 2005, 109, 8701–8706. doi:10.1021/jp046016j |
| 48. | Olszewska, P.; Mikiciuk-Olasik, E.; Błaszczak-Świątkiewicz, K.; Szymański, J.; Szymański, P. Biomed. Pharmacother. 2014, 68, 959–967. doi:10.1016/j.biopha.2014.10.018 |
| 49. | Pirrung, M. C.; Chau, J. H.-L.; Chen, J. Chem. Biol. 1995, 2, 621–626. doi:10.1016/1074-5521(95)90127-2 |
| 50. | Tripathi, R. P.; Verma, S. S.; Pandey, J.; Agarwal, K. C.; Chaturvedi, V.; Manju, Y. K.; Srivastva, A. K.; Gaikwad, A.; Sinha, S. Bioorg. Med. Chem. Lett. 2006, 16, 5144–5147. doi:10.1016/j.bmcl.2006.07.025 |
| 51. | Czarnecka, K.; Chufarova, N.; Halczuk, K.; Maciejewska, K.; Girek, M.; Skibiński, R.; Jończyk, J.; Bajda, M.; Kabziński, J.; Majsterek, I.; Szymański, P. Eur. J. Med. Chem. 2018, 145, 760–769. doi:10.1016/j.ejmech.2018.01.014 |
| 52. | Bajda, M.; Jończyk, J.; Malawska, B.; Czarnecka, K.; Girek, M.; Olszewska, P.; Sikora, J.; Mikiciuk-Olasik, E.; Skibiński, R.; Gumieniczek, A.; Szymański, P. Bioorg. Med. Chem. 2015, 23, 5610–5618. doi:10.1016/j.bmc.2015.07.029 |
| 53. | Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Bioorg. Chem. 2011, 39, 138–142. doi:10.1016/j.bioorg.2011.05.001 |
| 54. | Nishibori, M.; Oishi, R.; Itoh, Y.; Saeki, K. Jpn. J. Pharmacol. 1991, 55, 539–546. doi:10.1016/s0021-5198(19)39924-x |
| 73. | Koch, W.; Holthausen, M. C. A. Chemist's Guide to Density Functional Theory; Wiley-VCH: Weinheim, Germany, 2000. doi:10.1002/3527600043 |
| 21. | Denny, W. A. Curr. Med. Chem. 2002, 9, 1655–1665. doi:10.2174/0929867023369277 |
| 22. | Demeunynck, M.; Charmantray, F.; Martelli, A. Curr. Pharm. Des. 2001, 7, 1703–1724. doi:10.2174/1381612013397131 |
| 23. | Demeunynck, M. Expert Opin. Ther. Pat. 2004, 14, 55e70. |
| 24. | Denny, W. A. Med. Chem. Rev.–Online 2004, 1, 257–266. doi:10.2174/1567203043401923 |
| 25. | Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Anti-Cancer Agents Med. Chem. 2007, 7, 139–169. doi:10.2174/187152007780058669 |
| 26. | Cholewiński, G.; Dzierzbicka, K.; Kołodziejczyk, A. M. Pharmacol. Rep. 2011, 63, 305–336. doi:10.1016/s1734-1140(11)70499-6 |
| 27. | Dopierała, A.; Wrosz, P.; Mazerski, J. Postepy Hig. Med. Dosw. 2011, 65, 263–269. doi:10.5604/17322693.941521 |
| 28. | Pérez, S. A.; de Haro, C.; Vicente, C.; Donaire, A.; Zamora, A.; Zajac, J.; Kostrhunova, H.; Brabec, V.; Bautista, D.; Ruiz, J. ACS Chem. Biol. 2017, 12, 1524–1537. doi:10.1021/acschembio.7b00090 |
| 29. | Galdino-Pitta, M. R.; Pitta, M. G. R.; Lima, M. C. A.; Galdino, L. S.; Pitta, R. I. Mini-Rev. Med. Chem. 2013, 13, 1256–1271. doi:10.2174/1389557511313090002 |
| 30. | Singh, H.; Singh, H.; Sharma, S.; Singh Bedi, P. M. Heterocycles 2015, 91, 2043–2085. doi:10.3987/rev-15-826 |
| 31. | Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Expert Opin. Ther. Pat. 2014, 24, 647–664. doi:10.1517/13543776.2014.902052 |
| 71. | Qin, Y.; Gu, H.; Liu, S.; Dai, W.; Luo, X. Synth. Met. 2018, 245, 42–50. doi:10.1016/j.synthmet.2018.08.008 |
| 15. | Goodell, J. R.; Ougolkov, A. V.; Hiasa, H.; Kaur, H.; Remmel, R.; Billadeau, D. D.; Ferguson, D. M. J. Med. Chem. 2008, 51, 179–182. doi:10.1021/jm701228e |
| 16. | Fernández-Calienes Valdés, A. Open Med. Chem. J. 2011, 5, 11–20. doi:10.2174/1874104501105010011 |
| 17. | Nowak, K. J. Mol. Struct. 2017, 1146, 562–570. doi:10.1016/j.molstruc.2017.05.042 |
| 18. | Budiman, M. E.; Bierbach, U.; Alexander, R. W. Biochemistry 2005, 44, 11262–11268. doi:10.1021/bi050745n |
| 19. | Sondhi, S. M.; Singh, J.; Rani, R.; Gupta, P. P.; Agrawal, S. K.; Saxena, A. K. Eur. J. Med. Chem. 2010, 45, 555–563. doi:10.1016/j.ejmech.2009.10.042 |
| 20. | Benoit, A. R.; Schiaffo, C.; Salomon, C. E.; Goodell, J. R.; Hiasa, H.; Ferguson, D. M. Bioorg. Med. Chem. Lett. 2014, 24, 3014–3017. doi:10.1016/j.bmcl.2014.05.037 |
| 40. | Goel, A.; Kumar, V.; Singh, S. P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R. S. J. Mater. Chem. 2012, 22, 14880. doi:10.1039/c2jm31052j |
© 2021 Tka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)