Abstract
(1,1,2,2-Tetrafluorobut-3-en-1-yl)zinc bromide was prepared by insertion of the zinc–silver couple into the CF2–Br bond of commercially available 4-bromo-3,3,4,4-tetrafluorobut-1-ene in DMF at 0 °C for 0.5 h, The resultant polyfluorinated zinc reagent was found to be thermally stable at ambient temperature and storable for at least 1.5 years in the refrigerator. This CF2CF2-containing organozinc reagent could be easily transmetallated to copper species, which underwent cross-coupling reactions with various aromatic iodides or acyl chlorides to produce a broad range of CF2CF2-containing organic molecules in good-to-excellent yields. Therefore, the zinc reagent could become a new and practical synthetic tool for producing functional molecules with a CF2CF2 fragment.

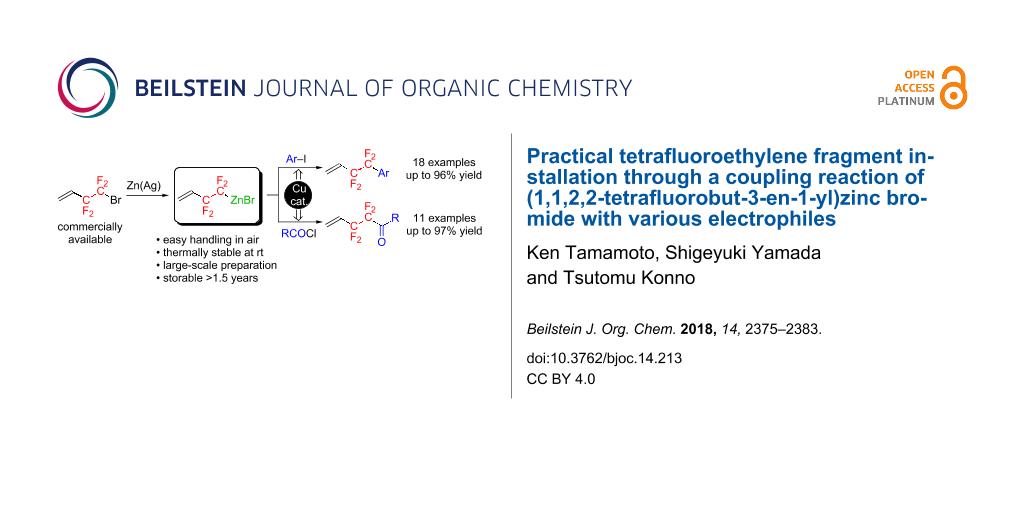
Graphical Abstract
Introduction
Recently, much attention has been paid to organic compounds containing a perfluoroalkylene unit, e.g., –(CF2)n–, in various fields, such as medicine and materials sciences [1-4], because incorporation of multiple fluorine atoms into organic substances causes dramatic alterations in the chemical and physical properties of substances, which may significantly enhance their potential functionality [5,6]. Notably, organofluorine compounds bearing a tetrafluoroethylene (–CF2CF2–) unit have attracted significant interest as a promising framework for various functional molecules. In the medicinal field, for example, Linclau and co-workers reported the first enantioselective synthesis and the intriguing biological activities of CF2CF2-containing pyranose and furanose derivatives (Figure 1a) [7-9]. Subsequently, Gouverneur et al. also developed novel CF2CF2-containing C-nucleosides (Figure 1b) [10]. Meanwhile, in the field of materials sciences, Kirsch et al. revealed that the incorporation of the CF2CF2 unit between two cyclohexane rings caused significant enhancement in thermal stability in a liquid crystalline phase (Figure 1c) [11]. In addition to the development of a convenient access to CF2CF2-containing pyranoses [12], our group also showed that tricyclic mesogens with a CF2CF2-containing carbocycle exhibited large negative dielectric anisotropies (Figure 1d), which would be promising for candidates for vertically aligned (VA)-type display materials [13-17]. Owing to such valuable applications, the development of a much more efficient synthetic protocol for CF2CF2-containing organic molecules has a high priority in various research areas.
![[1860-5397-14-213-1]](/bjoc/content/figures/1860-5397-14-213-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Functional molecules with CF2CF2-fragment.
Figure 1: Functional molecules with CF2CF2-fragment.
A number of synthetic protocols for CF2CF2-containing molecules have been developed in the last half-decade. Ogoshi and co-workers reported an efficient synthetic protocol for CF2CF2-containing compounds via carbocupration or oxycupration of tetrafluoroethylene (TFE) [18-20]. Gouverneur et al. disclosed a Cu(I)-mediated cross-coupling reaction using ArCF2CF2SiMe3 derivatives that produced the corresponding tetrafluoroethylenated compounds [21]. Beier’s group also described a broad range of tetrafluoroethylene compounds generated by the reductive coupling of in situ-formed RCF2CF2MgCl·LiCl with various electrophiles [22,23]. Our group revealed that commercially available 4-bromo-3,3,4,4-tetrafluorobut-1-ene (1) underwent a Cu(0)-mediated cross-coupling reaction with aromatic iodides in DMSO at 160 °C in a sealed-tube apparatus [24] or reductive couplings with various carbonyl compounds [25], leading to good yields of versatile CF2CF2-containing substances. However, many obstacles to practical synthesis remain, e.g., the use of 1,2-dibromotetrafluoroethane (Halon-2402) with ozone depletion and global warming potentials as a starting material [26], explosive and difficult-to-handle gaseous tetrafluoroethylene [18-20], thermally unstable polyfluoroalkylmetal species [8,27,28], etc. Therefore, the development of a more efficient synthetic protocol featuring easy-handling, simple preparation, and thermal stability is highly desirable.
Our strategy focused on the preparation of a thermally stable CF2CF2-containing metal species for which the “unreactive” form can be easily changed to the “reactive” form through chemical transformation. Out of a variety of organometallics, we selected an organozinc reagent that possesses higher thermal stability than the corresponding organolithium or -magnesium species due to the almost covalent C–Zn bond [29,30]. Moreover, organozinc reagents can be easily transformed to the “reactive” species, through a transmetallation process with a transition metal (e.g., Pd or Cu), which can efficiently construct a new C–C bond [30-32]. Thus, we aimed at the development of an efficient preparation of a thermally stable organozinc reagent, possessing a CF2CF2 fragment as a thermally stable tetrafluoroethylenating agent, and successive C–C bond formation to produce a wide variety of CF2CF2-containing molecules, and the results are described in this article (Scheme 1).
![[1860-5397-14-213-i1]](/bjoc/content/inline/1860-5397-14-213-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation and synthetic applications of 2-Zn.
Scheme 1: Preparation and synthetic applications of 2-Zn.
Results and Discussion
We carried out the optimization of the reaction conditions for the preparation of 2-Zn by direct zinc insertion into the CF2–Br bond of commercially available fluorinated substance 1. The results are summarized in Table 1.
Table 1: Optimization of the reaction conditions for the preparation of 2-Zn.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-213-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | Zn(Ag) (equiv) | solvent | temp (°C) | yielda (%) | recoverya (% of 1) | |
| 2-Zn | 2-H | |||||
| 1b | 2.0 | DMF | 0 | 0 | 0 | quant |
| 2 | 2.0 | DMF | 0 | 86 | 9 | 0 |
| 3 | 1.2 | DMF | 0 | 50 | 12 | 0 |
| 4 | 2.0 | DMF | −30 | 0 | 0 | quant |
| 5 | 2.0 | DMA | 0 | 58 | 16 | 0 |
| 6 | 2.0 | DMPU | 0 | 22 | 28 | 0 |
| 7 | 2.0 | NMP | 0 | 38 | 20 | 0 |
| 8 | 2.0 | THF | 0 | 0 | 0 | quant |
| 9c | 2.0 | THF | 40 | 75 | 0 | trace |
aDetermined by 19F NMR. bWith zinc powder pre-activated by dilute HCl aq, instead of Zn(Ag). cCarried out for 4 h.
Initially, we attempted zinc insertion using a typical protocol [33], i.e., treating 1 with 2.0 equiv of zinc powder, pre-activated with dilute HCl solution, in DMF at 0 °C for 0.5 h. Unfortunately, no desired zinc insertion occurred and substrate 1 was quantitatively recovered (Table 1, entry 1). Interestingly, when a zinc–silver couple Zn(Ag) [34] was employed instead of zinc powder, the desired zinc insertion took place very smoothly to form the desired (1,1,2,2-tetrafluorobut-3-en-1-yl)zinc bromide (2-Zn) in 86% NMR yield, along with a small amount of a reduction byproduct (2-H) (Table 1, entry 2). Reducing the amount of Zn(Ag) slightly retarded the zinc insertion, with only 50% NMR yield (Table 1, entry 3). Lowering the temperature to –30 °C also inhibited the zinc insertion, with quantitative recovery of 1 (Table 1, entry 4). The reaction was also attempted in various solvents, namely N,N-dimethylacetamide (DMA), N,N’-dimethylpropyleneurea (DMPU), N-methylpyrrolidin-2-one (NMP), and THF. In polar solvents (DMA, DMPU, and NMP), the reaction successfully produced the desired 2-Zn, although the yield was still unsatisfactory (22–58%, Table 1, entries 5–7). In the less polar THF, in contrast, complete recovery of 1 was observed (Table 1, entry 8). Nevertheless, after raising the temperature to 40 °C, directed zinc insertion into the CF2–Br bond was achieved in THF, producing 2-Zn in 75% NMR yield (Table 1, entry 9). Eventually, the optimal conditions for the preparation of 2-Zn were determined to be that given in entry 2 (Table 1). It is noteworthy that 2-Zn can be prepared in DMF or THF on a large scale (approx. 40–50 mmol scale) without any problem. More remarkably, the prepared 2-Zn can be stored in DMF solution (ca. 0.70 M) for at least 1.5 years in the refrigerator [35].
In order to examine the stability of 2-Zn in more detail, we quantitatively evaluated the thermal stability of 2-Zn under various temperature conditions (Figure 2). After a given duration, the recovery yield of 2-Zn was determined by 19F NMR analysis using an internal reference.
![[1860-5397-14-213-2]](/bjoc/content/figures/1860-5397-14-213-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Recovery yield of 2-Zn in DMF (ca. 0.70 M) after stirring at various temperature conditions.
Figure 2: Recovery yield of 2-Zn in DMF (ca. 0.70 M) after stirring at various temperature conditions.
Complete recovery of 2-Zn (0.70 M in DMF) was observed below 50 °C and gradual degradation occurred at 80 °C. It was revealed that 50% decomposition of 2-Zn was observed after 4 h stirring, and almost all 2-Zn was decomposed after 12 h stirring (96%). At 100 °C, 2-Zn was almost completely decomposed after being stirred for 2 h, with a recovery yield of only 8%. According to Beier’s report [22,23], tetrafluoroethylmagnesium species fully decompose within 50 min at −40 °C; CH2=CHCF2CF2ZnBr (2-Zn) used in the present study was shown to be much more thermally stable than tetrafluoroethyllithium [12,25] and -magnesium species [22,23].
The synthetic uses of 2-Zn as a promising tetrafluorinating agent were tested in several reactions. First, we demonstrated a typical C–C bond formation reaction. Treating freshly prepared 2-Zn (ca. 0.70 M in DMF) with 5.0 equiv of iodobenzene (3a) in the presence of 10 mol % of CuI in DMF at 50 °C for 24 h resulted in limited formation (11% yield) of the cross-coupling product 4a (Table 2, entry 1). The Cu(I)-catalyzed cross-coupling reaction with 3a was proposed to take place via the following three key reaction steps [36]: (i) transmetallation from 2-Zn to the corresponding Cu(I) species, (ii) oxidative addition of a CAr–I bond to the Cu(I) atomic center to generate Cu(III) species, and (iii) reductive elimination of the product 4a along with the regeneration of the Cu(I) salt. When the initial transmetallation from 2-Zn to the reactive Cu(I) species proceeds much faster than the thermal decomposition of 2-Zn, the yield of 4a should be improved. Indeed, in the cross-coupling reaction carried out at 80 °C for 24 h, an enhanced yield of 4a was observed (Table 2, entry 2), while the reaction in THF did not give any trace of the product (Table 2, entry 3). Catalyst optimization (Table 2, entries 4–6) showed that Cu2O led to the highest product yield (46%, Table 2, entry 6) [37]. After further exploring the reaction conditions (Table 2, entries 7–13) including catalyst loading, equivalents of 3a, additives, and reaction temperature, the best result (64% NMR yield) was observed with 6.0 equiv of 3a in the presence of 30 mol % of Cu2O in DMF at 100 °C for 24 h (Table 2, entry 12) [38].
Table 2: Optimization of reaction conditions for Cu(I)-catalyzed cross-coupling reaction of 2-Zn with iodobenzene (3a).
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-213-i5.svg?max-width=637&scale=1.0)
|
||||
| entrya | Cu(I) salt (mol %) | 3a (equiv) | temp (°C)b | yieldc (% of 4a) |
| 1 | CuI (10) | 5.0 | 50 | 11 |
| 2 | CuI (10) | 5.0 | 80 | 37 |
| 3d | CuI (10) | 5.0 | 80 | 0 |
| 4 | CuBr (10) | 5.0 | 80 | 37 |
| 5 | CuCl (10) | 5.0 | 80 | 34 |
| 6 | Cu2O (10) | 5.0 | 80 | 46 |
| 7 | Cu2O (30) | 5.0 | 80 | 53 |
| 8 | Cu2O (50) | 5.0 | 80 | 54 |
| 9e | Cu2O (30) | 5.0 | 80 | 50 |
| 10f | Cu2O (30) | 5.0 | 80 | 35 |
| 11 | Cu2O (30) | 6.0 | 80 | 59 |
| 12 | Cu2O (30) | 6.0 | 100 | 64 (29)g |
| 13 | Cu2O (30) | 6.0 | 120 | 57 |
aCarried out using ca. 0.6 mmol of 2-Zn in DMF solution (ca. 0.70 M). bBath temperature. cDetermined by 19F NMR. Values in parentheses are isolated yields. dThe reaction was carried out using 2-Zn in THF instead of 2-Zn in DMF. eN,N,N’,N’-Tetramethylethylenediamine (TMEDA) was used as an additive. f1,10-Phenanthroline (phen) was used as an additive. gThe low isolated yield was due to the low boiling point and/or the high volatility of the product.
Using the optimal conditions for the reaction in entry 12, Table 2, various iodoarenes or -heteroarenes (3b–r) could be converted into the corresponding CF2CF2-substituted aromatic compounds 4b–r (Figure 3).
![[1860-5397-14-213-3]](/bjoc/content/figures/1860-5397-14-213-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Copper(I)-catalyzed cross-coupling reaction of 2-Zn with various iodoarene derivatives. NMR (isolated) yields are indicated. aLow yields are due to the low boiling point and/or the high volatility of the products. bReaction was carried out using 1.0 equiv of 3 and 2.0 equiv of 2-Zn in the presence of 60 mol % of Cu2O.
Figure 3: Copper(I)-catalyzed cross-coupling reaction of 2-Zn with various iodoarene derivatives. NMR (isolat...
Aromatic iodides with an electron-donating group, such as OMe (3b) and Me (3c), at the para position on the benzene ring were successfully coupled with 2-Zn under the optimized conditions to form 4b and 4c in 43% and 36% isolated yields, respectively. p-Chloro- (3d) or p-trifluoromethyl-substituted iodobenzene (3e) could also participate in the coupling reaction, leading to the corresponding products 4d and 4e in 53% and 75% NMR (49% and 13% isolated) yields, respectively. Notably, iodoarenes with an ethoxycarbonyl (3f) or a nitro group (3g) at the para-position were successfully converted to the corresponding products 4f and 4g with good yields. Differences in the position of the substituents on the benzene ring had no effect on the coupling reaction; m-chloro- (3h) and o-chloroiodobenzene (3i) were transformed into their corresponding products 4h and 4i in 60% and 64% yields, respectively. Interestingly, the reaction using ethyl o-iodobenzoate (3j) quite efficiently proceeded to give the coupling product 4j almost quantitatively. Comparing the result from the reaction with the para-substituted analog 3f, the ester functionality at the ortho-position seems to significantly facilitate the formation of the coupling product. According to the previous reports by Jiang and co-workers [36], the oxygen atom in the ethoxycarbonyl group substituted at the ortho-position coordinates with the copper center to stabilize the Cu(III) intermediate, which facilitates the subsequent reductive elimination to form the desired coupling product. This acceleration effect of the ester functional group at the ortho-position led to a reduction in the chemical substances used. That is to say, the product 4j was obtained in a quantitative manner even when the reaction of 2-Zn was carried out with only a half equivalent of 3j. The same effect was also noted for iodoarenes having a labile functional group, such as formyl (3k), acetyl (3l), methoxymethyl (3m), or a nitro group (3n–q) at the ortho-position, giving rise to the corresponding products 4k–q with excellent efficiency. These results strongly suggest that 2-Zn can be successfully employed for the coupling reaction with an electrophile bearing a reactive functional group. Lastly, this synthetic protocol could be applied to prepare the CF2CF2-substituted heteroaromatic compound 4r from 3-iodopyridine (3r), demonstrating a promising pathway for constructing CF2CF2-containing heterocyclic compounds.
We also demonstrated the multigram preparation of CF2CF2-containing arenes through the present cross-coupling reaction, as shown in Scheme 2. Thus, treatment of 1.38 g (5.00 mmol) of ethyl o-iodobenzoate (3j) with 10.1 mmol of 2-Zn in the presence of 3 mmol of Cu2O in DMF at 100 °C for 24 h gave 1.32 g (4.78 mmol) of the corresponding coupling product 4j (96% yield). Similarly, the reaction of 2-Zn with 1.49 g of o-iodonitrobenzene (3n) afforded 1.25 g (5.02 mmol) of the adduct 4n (84% yield). This achievement may lead to a significant contribution as a first scalable CF2CF2 fragment installation method.
![[1860-5397-14-213-i2]](/bjoc/content/inline/1860-5397-14-213-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Multigram-scale cross-coupling of 2-Zn with iodoarenes.
Scheme 2: Multigram-scale cross-coupling of 2-Zn with iodoarenes.
To be further convinced of the importance of the present reaction, we carried out an additional transformation of the coupling adduct 4n to a CF2CF2-containing π-conjugates molecule, a fluorinated tolane derivative, applicable to promising functional materials, such as light-emitting and liquid-crystalline materials (Scheme 3) [39-44].
![[1860-5397-14-213-i3]](/bjoc/content/inline/1860-5397-14-213-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of a CF2CF2 group containing tolane derivative.
Scheme 3: Synthesis of a CF2CF2 group containing tolane derivative.
Thus, CF2CF2 group containing 4n with a nitro group at the ortho-position of the aromatic ring was effectively prepared from 2-Zn with the present protocol. 4n was smoothly converted to the corresponding aniline derivative 4s in 87% isolated yield, after exposure to a reductive environment using Fe powder and NH4Cl. Subsequent Sandmeyer reaction of 4s took place very nicely to afford the corresponding CF2CF2-substituted iodobenzene derivative 4t in 67% isolated yield. Then, 4t underwent Pd(0)-catalyzed Sonogashira cross-coupling reaction with phenylacetylene, producing the corresponding tolane derivative 4u with a CF2CF2 fragment in good yield (30% overall yield from 2-Zn). Consequently, 2-Zn is found to be a powerful tetrafluoroethylenating agent for producing a broad range of organic molecules with a CF2CF2 unit.
Next, the Cu(I)-catalyzed cross-coupling reaction of 2-Zn with benzoyl chloride (5a) as a coupling partner was investigated (Table 3).
Table 3: Optimization of reaction conditions for Cu(I)-catalyzed cross-coupling reaction of 2-Zn with benzoyl chloride (5a).
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-213-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | Cu(I) salt (mol %) | 5a (equiv) | additive | tempa (°C) | yieldb (% of 4a) |
| 1 | CuI (30) | 2.4 | – | 100 | 67 |
| 2 | CuBr (30) | 2.4 | – | 100 | 51 |
| 3 | CuCN (30) | 2.4 | – | 100 | 46 |
| 4 | Cu2O (30) | 2.4 | – | 100 | 81 |
| 5 | Cu2O (10) | 2.4 | TMEDA | 100 | ndc |
| 6 | Cu2O (30) | 2.4 | phen | 100 | 69 |
| 7 | Cu2O (50) | 2.4 | bpy | 100 | quant |
| 8 | Cu2O (30) | 2.4 | bpy | rt | quant (92) |
| 9 | Cu2O (30) | 2.0 | bpy | rt | 83 |
aBath temperature. bDetermined by 19F NMR. Values in parentheses are isolated yield. cNot detected.
Treating 2-Zn (ca. 0.70 M DMF solution) with 2.4 equiv of 5a in the presence of CuI (30 mol %) in DMF at 100 °C for 20 h enabled the formation of the corresponding cross-coupling reaction product 6a in 67% yield (Table 3, entry 1). Optimization of the Cu(I) catalyst for the benzoylation reaction (Table 3, entries 2–4) revealed that Cu2O was the most efficient for producing 6a (81% by NMR, Table 3, entry 4). To further improve the reaction efficiency, three different additives (TMEDA, phen, and 2,2’-bipyridyl (bpy)) were tested. The first two additives were found to be ineffective for the present reaction (Table 3, entries 5 and 6), whereas bpy led to the quantitative formation of 6a (Table 3, entry 7). The facilitative effect of bpy as an additive made it possible to convert 5a to 6a even at ambient temperature (Table 3, entry 8). Additionally, to utilize the present reaction in an environmentally benign protocol, we conducted the reaction with a decreased amount of 5a. Unfortunately, this slightly lowered the yield of 6a (Table 3, entry 9).
With the optimized conditions (Table 3, entry 8), various kinds of acid chlorides 5b–k could be converted to the corresponding CF2CF2-substituted products 6b–k (Figure 4).
![[1860-5397-14-213-4]](/bjoc/content/figures/1860-5397-14-213-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Copper(I)-catalyzed cross-coupling reaction of 2-Zn with various acid chlorides. NMR yields (isolated yields) are indicated. aCarried out at 100 °C. bWith 4.0 equiv of 5g.
Figure 4: Copper(I)-catalyzed cross-coupling reaction of 2-Zn with various acid chlorides. NMR yields (isolat...
Benzoyl chloride derivatives with an electron-donating (5b–d) and an electron-withdrawing group (5e,f) gave rise to the corresponding coupling products 6b–f in 50–98% isolated yields. However, the reaction using nitro-substituted substrate 5g was quite slow under the same conditions. Slight modification of the reaction conditions, namely increasing the amount of 5g and raising the reaction temperature, significantly improved the yield of 6g. Acid chlorides with a heteroaromatic moiety, e.g., 5h and 5i, could also readily participate in the coupling reaction, leading to 6h and 6i in 83% and 87% isolated yields, respectively. Additionally, cinnamoyl chloride (5j) and n-octanoyl chloride (5k) were also suitable electrophiles to form the respective products 6j and 6k in high-to-excellent yields.
Conclusion
We developed a novel and practical tetrafluoroethylenating agent, viz. CH2=CHCF2CF2ZnBr (2-Zn), which can be prepared in large-scale and stored for at least 1.5 years in the refrigerator without decomposition. 2-Zn could be successfully transformed into a broad range of CF2CF2-containing molecules with good-to-excellent efficiency. Considering that our previous study found that the vinyl moiety in the coupling product could be a useful molecular building block [14,45], 2-Zn presented here should be a suitable and valuable tetrafluoroethylenating agent for preparing various CF2CF2-containing molecules, thereby providing a powerful and practical synthetic tool in organofluorine chemistry.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data (1H, 13C, 19F NMR, IR and HRMS), copies of 1H, 13C and 19F NMR spectra. | ||
| Format: PDF | Size: 10.4 MB | Download |
References
-
Kaplan, P. T.; Vicic, D. A. Org. Lett. 2016, 18, 884–886. doi:10.1021/acs.orglett.6b00032
Return to citation in text: [1] -
Irie, M. Pure Appl. Chem. 2015, 87, 617–626. doi:10.1515/pac-2015-0208
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] -
Kim, H. W.; Rossi, P.; Shoemaker, R. K.; DiMagno, S. G. J. Am. Chem. Soc. 1998, 120, 9082–9083. doi:10.1021/ja9803714
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
van Straaten, K. E.; Kuttiyatveetil, J. R. A.; Sevrain, C. M.; Villaume, S. A.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S. P.; Sanders, D. A. R. J. Am. Chem. Soc. 2015, 137, 1230–1244. doi:10.1021/ja511204p
Return to citation in text: [1] -
Timofte, R. S.; Linclau, B. Org. Lett. 2008, 10, 3673–3676. doi:10.1021/ol801272e
Return to citation in text: [1] [2] -
Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746
Return to citation in text: [1] -
Bonnac, L.; Lee, S. E.; Giuffredi, G. T.; Elphick, L. M.; Anderson, A. A.; Child, E. S.; Mann, D. J.; Gouverneur, V. Org. Biomol. Chem. 2010, 8, 1445–1454. doi:10.1039/b922442d
Return to citation in text: [1] -
Kirsch, P.; Bremer, M.; Huber, F.; Lannert, H.; Ruhl, A.; Lieb, M.; Wallmichrath, T. J. Am. Chem. Soc. 2001, 123, 5414–5417. doi:10.1021/ja010024l
Return to citation in text: [1] -
Konno, T.; Hoshino, T.; Kida, T.; Takano, S.; Ishihara, T. J. Fluorine Chem. 2013, 152, 106–113. doi:10.1016/j.jfluchem.2013.02.013
Return to citation in text: [1] [2] -
Kumon, T.; Hashishita, S.; Kida, T.; Yamada, S.; Ishihara, T.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 148–154. doi:10.3762/bjoc.14.10
Return to citation in text: [1] -
Yamashika, K.; Morishitabara, S.; Yamada, S.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 207, 24–37. doi:10.1016/j.jfluchem.2017.12.013
Return to citation in text: [1] [2] -
Yamada, S.; Tamamoto, K.; Kida, T.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 9442–9454. doi:10.1039/C7OB02399E
Return to citation in text: [1] -
Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013
Return to citation in text: [1] -
Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A
Return to citation in text: [1] -
Ohashi, M.; Adachi, T.; Ishida, N.; Kikushima, K.; Ogoshi, S. Angew. Chem., Int. Ed. 2017, 56, 11911–11915. doi:10.1002/anie.201703923
Return to citation in text: [1] [2] -
Kikushima, K.; Sakaguchi, H.; Saijo, H.; Ohashi, M.; Ogoshi, S. Chem. Lett. 2015, 44, 1019–1021. doi:10.1246/cl.150322
Return to citation in text: [1] [2] -
Saijo, H.; Ohashi, M.; Ogoshi, S. J. Am. Chem. Soc. 2014, 136, 15158–15161. doi:10.1021/ja5093776
Return to citation in text: [1] [2] -
O’Duill, M.; Dubost, E.; Pfeifer, L.; Gouverneur, V. Org. Lett. 2015, 17, 3466–3469. doi:10.1021/acs.orglett.5b01510
Return to citation in text: [1] -
Voltrová, S.; Muselli, M.; Filgas, J.; Matoušek, V.; Klepetářová, B.; Beier, P. Org. Biomol. Chem. 2017, 15, 4962–4965. doi:10.1039/c7ob01151b
Return to citation in text: [1] [2] [3] -
Budinská, A.; Václavík, J.; Matoušek, V.; Beier, P. Org. Lett. 2016, 18, 5844–5847. doi:10.1021/acs.orglett.6b02890
Return to citation in text: [1] [2] [3] -
Watanabe, Y.; Konno, T. J. Fluorine Chem. 2015, 174, 102–107. doi:10.1016/j.jfluchem.2014.12.008
Return to citation in text: [1] -
Konno, T.; Takano, S.; Takahashi, Y.; Konishi, H.; Tanaka, Y.; Ishihara, T. Synthesis 2011, 33–44. doi:10.1055/s-0030-1258330
Return to citation in text: [1] [2] -
Dmowski, W. J. Fluorine Chem. 2012, 142, 6–13. doi:10.1016/j.jfluchem.2012.06.018
Return to citation in text: [1] -
Chambers, R. D. Chapter 10. In Fluorine in Organic Chemistry; Chambers, R. D., Ed.; Blackwell Publishing Ltd.: Hoboken, NJ, U.S.A., 2004. doi:10.1002/9781444305371
Return to citation in text: [1] -
Uno, H.; Suzukib, H. Synlett 1993, 91–96. doi:10.1055/s-1993-22361
Return to citation in text: [1] -
Knochel, P.; Leuser, H.; Gong, L.-Z.; Perrone, S. Chapter 7. In Handbook of Functionalized Organometallics, Applications in Synthesis; Knochel, P., Ed.; Wiley-VCH Verlag GmbH&Co. KGaA: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Kato, H.; Hirano, K.; Kurauchi, D.; Toriumi, N.; Uchiyama, M. Chem. – Eur. J. 2015, 21, 3895–3900. doi:10.1002/chem.201406292
Return to citation in text: [1] [2] -
Aikawa, K.; Nakamura, Y.; Yokota, Y.; Toya, W.; Mikami, K. Chem. – Eur. J. 2015, 21, 96–100. doi:10.1002/chem.201405677
Return to citation in text: [1] -
Popov, I.; Lindeman, S.; Daugulis, O. J. Am. Chem. Soc. 2011, 133, 9286–9289. doi:10.1021/ja2041942
Return to citation in text: [1] -
Burton, D. J.; Jairaj, V. J. Fluorine Chem. 2004, 125, 673–680. doi:10.1016/j.jfluchem.2003.11.010
Return to citation in text: [1] -
Jiang, B.; Xu, Y. J. Org. Chem. 1991, 56, 7336–7340. doi:10.1021/jo00026a028
Return to citation in text: [1] -
After storage in the refrigerator for 1.5 years, the 2-Zn solution was found to have the same concentration as the freshly prepared one, as measured by quantitative titration. Furthermore, the one and a half-year-old reagent was also quantitatively coupled with ethyl 2-iodobenzoate (3j) to give the corresponding 4j. The details are described in Scheme S1 in Supporting Information File 1.
Return to citation in text: [1] -
Jiang, H.; Lu, W.; Yang, K.; Ma, G.; Xu, M.; Li, J.; Yao, J.; Wan, W.; Deng, H.; Wu, S.; Zhu, S.; Hao, J. Chem. – Eur. J. 2014, 20, 10084–10092. doi:10.1002/chem.201402205
Return to citation in text: [1] [2] -
Li, J.-H.; Tang, B.-X.; Tao, L.-M.; Xie, Y.-X.; Liang, Y.; Zhang, M.-B. J. Org. Chem. 2006, 71, 7488–7490. doi:10.1021/jo061220j
Return to citation in text: [1] -
We also carried out the reaction of 2-Zn with 3a under the influence of 100 mol % of Cu2O in DMF at 100 °C for 1 h. As a result, it was found that 4a could be obtained in 58% yield. This result indicates that this catalytic cycle proceeds quickly.
Return to citation in text: [1] -
Peng, Z.; Wang, Q.; Liu, Y.; Mu, Q.; Cao, Z.; Xu, H.; Zhang, P.; Yang, C.; Yao, L.; Xuan, L.; Zhang, Z. Liq. Cryst. 2016, 43, 276–284. doi:10.1080/02678292.2015.1105311
Return to citation in text: [1] -
Li, B.; He, W.; Wang, L.; Xiao, X.; Yang, H. Soft Matter 2013, 9, 1172–1177. doi:10.1039/c2sm26807h
Return to citation in text: [1] -
Hird, M.; Toyne, K. J.; Goodby, J. W.; Gray, G. W.; Minter, V.; Tuffin, R. P.; McDonnell, D. G. J. Mater. Chem. 2004, 14, 1731–1743. doi:10.1039/b400630e
Return to citation in text: [1] -
Yamada, S.; Morita, M.; Konno, T. J. Fluorine Chem. 2017, 202, 54–64. doi:10.1016/j.jfluchem.2017.09.003
Return to citation in text: [1] -
Yamada, S.; Miyano, K.; Konno, T.; Agou, T.; Kubota, T.; Hosokai, T. Org. Biomol. Chem. 2017, 15, 5949–5958. doi:10.1039/C7OB01369H
Return to citation in text: [1] -
Reimann, S.; Wittler, K.; Schmode, S.; Sharif, M.; Fahrenwaldt, T.; Ludwig, R.; Spannenberg, A.; Langer, P. Eur. J. Org. Chem. 2013, 8115–8134. doi:10.1002/ejoc.201300607
Return to citation in text: [1] -
Sakaguchi, Y.; Yamada, S.; Konno, T.; Agou, T.; Kubota, T. J. Org. Chem. 2017, 82, 1618–1631. doi:10.1021/acs.joc.6b02793
Return to citation in text: [1]
| 39. | Peng, Z.; Wang, Q.; Liu, Y.; Mu, Q.; Cao, Z.; Xu, H.; Zhang, P.; Yang, C.; Yao, L.; Xuan, L.; Zhang, Z. Liq. Cryst. 2016, 43, 276–284. doi:10.1080/02678292.2015.1105311 |
| 40. | Li, B.; He, W.; Wang, L.; Xiao, X.; Yang, H. Soft Matter 2013, 9, 1172–1177. doi:10.1039/c2sm26807h |
| 41. | Hird, M.; Toyne, K. J.; Goodby, J. W.; Gray, G. W.; Minter, V.; Tuffin, R. P.; McDonnell, D. G. J. Mater. Chem. 2004, 14, 1731–1743. doi:10.1039/b400630e |
| 42. | Yamada, S.; Morita, M.; Konno, T. J. Fluorine Chem. 2017, 202, 54–64. doi:10.1016/j.jfluchem.2017.09.003 |
| 43. | Yamada, S.; Miyano, K.; Konno, T.; Agou, T.; Kubota, T.; Hosokai, T. Org. Biomol. Chem. 2017, 15, 5949–5958. doi:10.1039/C7OB01369H |
| 44. | Reimann, S.; Wittler, K.; Schmode, S.; Sharif, M.; Fahrenwaldt, T.; Ludwig, R.; Spannenberg, A.; Langer, P. Eur. J. Org. Chem. 2013, 8115–8134. doi:10.1002/ejoc.201300607 |
| 14. | Yamashika, K.; Morishitabara, S.; Yamada, S.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 207, 24–37. doi:10.1016/j.jfluchem.2017.12.013 |
| 45. | Sakaguchi, Y.; Yamada, S.; Konno, T.; Agou, T.; Kubota, T. J. Org. Chem. 2017, 82, 1618–1631. doi:10.1021/acs.joc.6b02793 |
| 1. | Kaplan, P. T.; Vicic, D. A. Org. Lett. 2016, 18, 884–886. doi:10.1021/acs.orglett.6b00032 |
| 2. | Irie, M. Pure Appl. Chem. 2015, 87, 617–626. doi:10.1515/pac-2015-0208 |
| 3. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 4. | Kim, H. W.; Rossi, P.; Shoemaker, R. K.; DiMagno, S. G. J. Am. Chem. Soc. 1998, 120, 9082–9083. doi:10.1021/ja9803714 |
| 11. | Kirsch, P.; Bremer, M.; Huber, F.; Lannert, H.; Ruhl, A.; Lieb, M.; Wallmichrath, T. J. Am. Chem. Soc. 2001, 123, 5414–5417. doi:10.1021/ja010024l |
| 8. | Timofte, R. S.; Linclau, B. Org. Lett. 2008, 10, 3673–3676. doi:10.1021/ol801272e |
| 27. | Chambers, R. D. Chapter 10. In Fluorine in Organic Chemistry; Chambers, R. D., Ed.; Blackwell Publishing Ltd.: Hoboken, NJ, U.S.A., 2004. doi:10.1002/9781444305371 |
| 28. | Uno, H.; Suzukib, H. Synlett 1993, 91–96. doi:10.1055/s-1993-22361 |
| 10. | Bonnac, L.; Lee, S. E.; Giuffredi, G. T.; Elphick, L. M.; Anderson, A. A.; Child, E. S.; Mann, D. J.; Gouverneur, V. Org. Biomol. Chem. 2010, 8, 1445–1454. doi:10.1039/b922442d |
| 29. | Knochel, P.; Leuser, H.; Gong, L.-Z.; Perrone, S. Chapter 7. In Handbook of Functionalized Organometallics, Applications in Synthesis; Knochel, P., Ed.; Wiley-VCH Verlag GmbH&Co. KGaA: Weinheim, Germany, 2005. |
| 30. | Kato, H.; Hirano, K.; Kurauchi, D.; Toriumi, N.; Uchiyama, M. Chem. – Eur. J. 2015, 21, 3895–3900. doi:10.1002/chem.201406292 |
| 7. | van Straaten, K. E.; Kuttiyatveetil, J. R. A.; Sevrain, C. M.; Villaume, S. A.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S. P.; Sanders, D. A. R. J. Am. Chem. Soc. 2015, 137, 1230–1244. doi:10.1021/ja511204p |
| 8. | Timofte, R. S.; Linclau, B. Org. Lett. 2008, 10, 3673–3676. doi:10.1021/ol801272e |
| 9. | Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746 |
| 26. | Dmowski, W. J. Fluorine Chem. 2012, 142, 6–13. doi:10.1016/j.jfluchem.2012.06.018 |
| 5. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 6. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 18. | Ohashi, M.; Adachi, T.; Ishida, N.; Kikushima, K.; Ogoshi, S. Angew. Chem., Int. Ed. 2017, 56, 11911–11915. doi:10.1002/anie.201703923 |
| 19. | Kikushima, K.; Sakaguchi, H.; Saijo, H.; Ohashi, M.; Ogoshi, S. Chem. Lett. 2015, 44, 1019–1021. doi:10.1246/cl.150322 |
| 20. | Saijo, H.; Ohashi, M.; Ogoshi, S. J. Am. Chem. Soc. 2014, 136, 15158–15161. doi:10.1021/ja5093776 |
| 21. | O’Duill, M.; Dubost, E.; Pfeifer, L.; Gouverneur, V. Org. Lett. 2015, 17, 3466–3469. doi:10.1021/acs.orglett.5b01510 |
| 24. | Watanabe, Y.; Konno, T. J. Fluorine Chem. 2015, 174, 102–107. doi:10.1016/j.jfluchem.2014.12.008 |
| 18. | Ohashi, M.; Adachi, T.; Ishida, N.; Kikushima, K.; Ogoshi, S. Angew. Chem., Int. Ed. 2017, 56, 11911–11915. doi:10.1002/anie.201703923 |
| 19. | Kikushima, K.; Sakaguchi, H.; Saijo, H.; Ohashi, M.; Ogoshi, S. Chem. Lett. 2015, 44, 1019–1021. doi:10.1246/cl.150322 |
| 20. | Saijo, H.; Ohashi, M.; Ogoshi, S. J. Am. Chem. Soc. 2014, 136, 15158–15161. doi:10.1021/ja5093776 |
| 25. | Konno, T.; Takano, S.; Takahashi, Y.; Konishi, H.; Tanaka, Y.; Ishihara, T. Synthesis 2011, 33–44. doi:10.1055/s-0030-1258330 |
| 13. | Kumon, T.; Hashishita, S.; Kida, T.; Yamada, S.; Ishihara, T.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 148–154. doi:10.3762/bjoc.14.10 |
| 14. | Yamashika, K.; Morishitabara, S.; Yamada, S.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 207, 24–37. doi:10.1016/j.jfluchem.2017.12.013 |
| 15. | Yamada, S.; Tamamoto, K.; Kida, T.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 9442–9454. doi:10.1039/C7OB02399E |
| 16. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 17. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 12. | Konno, T.; Hoshino, T.; Kida, T.; Takano, S.; Ishihara, T. J. Fluorine Chem. 2013, 152, 106–113. doi:10.1016/j.jfluchem.2013.02.013 |
| 22. | Voltrová, S.; Muselli, M.; Filgas, J.; Matoušek, V.; Klepetářová, B.; Beier, P. Org. Biomol. Chem. 2017, 15, 4962–4965. doi:10.1039/c7ob01151b |
| 23. | Budinská, A.; Václavík, J.; Matoušek, V.; Beier, P. Org. Lett. 2016, 18, 5844–5847. doi:10.1021/acs.orglett.6b02890 |
| 30. | Kato, H.; Hirano, K.; Kurauchi, D.; Toriumi, N.; Uchiyama, M. Chem. – Eur. J. 2015, 21, 3895–3900. doi:10.1002/chem.201406292 |
| 31. | Aikawa, K.; Nakamura, Y.; Yokota, Y.; Toya, W.; Mikami, K. Chem. – Eur. J. 2015, 21, 96–100. doi:10.1002/chem.201405677 |
| 32. | Popov, I.; Lindeman, S.; Daugulis, O. J. Am. Chem. Soc. 2011, 133, 9286–9289. doi:10.1021/ja2041942 |
| 33. | Burton, D. J.; Jairaj, V. J. Fluorine Chem. 2004, 125, 673–680. doi:10.1016/j.jfluchem.2003.11.010 |
| 38. | We also carried out the reaction of 2-Zn with 3a under the influence of 100 mol % of Cu2O in DMF at 100 °C for 1 h. As a result, it was found that 4a could be obtained in 58% yield. This result indicates that this catalytic cycle proceeds quickly. |
| 36. | Jiang, H.; Lu, W.; Yang, K.; Ma, G.; Xu, M.; Li, J.; Yao, J.; Wan, W.; Deng, H.; Wu, S.; Zhu, S.; Hao, J. Chem. – Eur. J. 2014, 20, 10084–10092. doi:10.1002/chem.201402205 |
| 36. | Jiang, H.; Lu, W.; Yang, K.; Ma, G.; Xu, M.; Li, J.; Yao, J.; Wan, W.; Deng, H.; Wu, S.; Zhu, S.; Hao, J. Chem. – Eur. J. 2014, 20, 10084–10092. doi:10.1002/chem.201402205 |
| 37. | Li, J.-H.; Tang, B.-X.; Tao, L.-M.; Xie, Y.-X.; Liang, Y.; Zhang, M.-B. J. Org. Chem. 2006, 71, 7488–7490. doi:10.1021/jo061220j |
| 12. | Konno, T.; Hoshino, T.; Kida, T.; Takano, S.; Ishihara, T. J. Fluorine Chem. 2013, 152, 106–113. doi:10.1016/j.jfluchem.2013.02.013 |
| 25. | Konno, T.; Takano, S.; Takahashi, Y.; Konishi, H.; Tanaka, Y.; Ishihara, T. Synthesis 2011, 33–44. doi:10.1055/s-0030-1258330 |
| 22. | Voltrová, S.; Muselli, M.; Filgas, J.; Matoušek, V.; Klepetářová, B.; Beier, P. Org. Biomol. Chem. 2017, 15, 4962–4965. doi:10.1039/c7ob01151b |
| 23. | Budinská, A.; Václavík, J.; Matoušek, V.; Beier, P. Org. Lett. 2016, 18, 5844–5847. doi:10.1021/acs.orglett.6b02890 |
| 35. | After storage in the refrigerator for 1.5 years, the 2-Zn solution was found to have the same concentration as the freshly prepared one, as measured by quantitative titration. Furthermore, the one and a half-year-old reagent was also quantitatively coupled with ethyl 2-iodobenzoate (3j) to give the corresponding 4j. The details are described in Scheme S1 in Supporting Information File 1. |
| 22. | Voltrová, S.; Muselli, M.; Filgas, J.; Matoušek, V.; Klepetářová, B.; Beier, P. Org. Biomol. Chem. 2017, 15, 4962–4965. doi:10.1039/c7ob01151b |
| 23. | Budinská, A.; Václavík, J.; Matoušek, V.; Beier, P. Org. Lett. 2016, 18, 5844–5847. doi:10.1021/acs.orglett.6b02890 |
© 2018 Tamamoto et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)