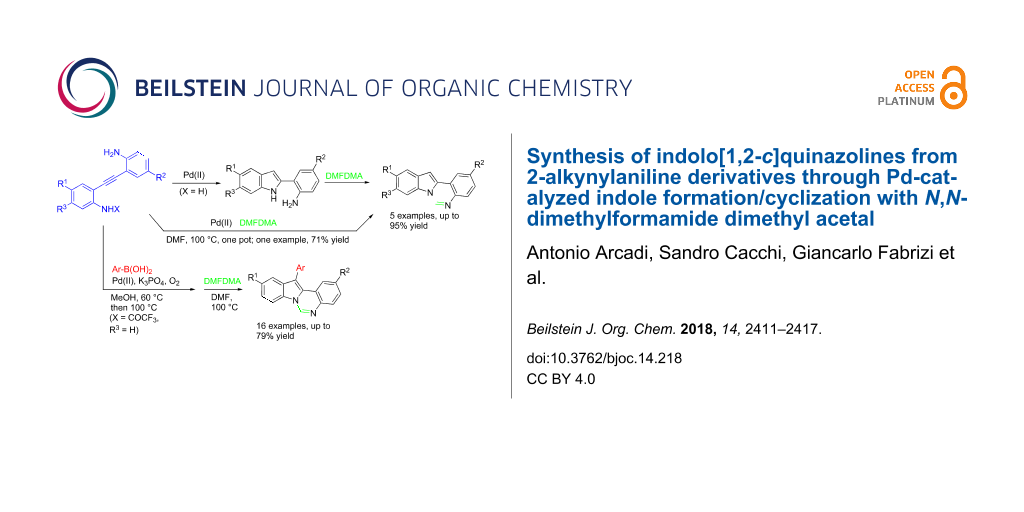
Abstract
An efficient strategy for the synthesis of 6-unsubstituted indolo[1,2-c]quinazolines is described. The Pd-catalyzed reaction of o-(o-aminophenylethynyl) trifluoroacetanilides with Ar–B(OH)2 afforded 2-(o-aminophenyl)-3-arylindoles, that were converted to 12-arylindolo[1,2-c]quinazolines by adding dimethylformamide dimethyl acetal (DMFDMA) to the reaction mixture after extractive work-up. This reaction outcome is different from the previously reported Pd-catalyzed sequential reaction of the same substrates with Ar–I, Ar–Br and ArN2+BF4−, that afforded 12-arylindolo[1,2-c]quinazolin-6(5H)-ones. Moreover, 12-unsubstituted indolo[1,2-c]quinazolines can be obtained both by reacting 2-(o-aminophenyl)indoles with DMFDMA or by sequential Pd-catalyzed reaction of o-(o-aminophenylethynyl)aniline with DMFDMA.

Graphical Abstract
Introduction
Indoloquinazoline derivatives constitute an important class of compounds which exhibit a wide range of biological activities [1-4]. Then, extensive searches aimed at discovering pharmacologically active compounds encouraged the synthesis of some new products containing the indolequinazoline nucleus with the aim to discover novel drug candidates [5-8]. In particular, libraries of 6-substituted indolo[1,2-c]quinazolines were synthesized and exhibited good antimicrobial as well as notable antifungal activities [9-11]. Since the isolation of hinckdentine A, an unusual marine alkaloid from the bryozoan Hincksinoflustra denticulata collected off the eastern coast of Tasmania [12,13], indolo[1,2-c]quinazolines related to hinckdentine A received increasing attention as a source of new and useful pharmaceuticals. One well-established approach is based on the elaboration of a preformed indole ring, for example through cyclocondensation of 2-(o-aminophenyl)indoles with 2-cyanobenzothiazoles [14], aldehydes [9] and formic acid [15,16], or reactions of 2-(2-bromoaryl)-1H-indoles with aldehydes and aqueous ammonia [17] or with amino acids [18]. An alternative approach to indoloquinazolines is represented by sequential procedures that use 2-alkynylaniline derivatives as starting materials, via their conversion to 2-(o-aminophenyl)indole derivatives followed by further cyclization [19-22]. Our interest in this field led to the development of an original approach to 6-trifluoromethyl-12-aryl(vinyl)indolo[1,2-c]quinazolines 4 through sequential Pd-catalyzed reactions of bis(o-trifluoroacetamidophenyl)acetylene (1) with aryl or vinyl halides and triflates followed by cyclization reactions (Scheme 1) [19].
![[1860-5397-14-218-i1]](/bjoc/content/inline/1860-5397-14-218-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 6-trifluoromethyl-12-aryl(vinyl)indolo[1,2-c]quinazolines 4.
Scheme 1: Synthesis of 6-trifluoromethyl-12-aryl(vinyl)indolo[1,2-c]quinazolines 4.
The reaction, which tolerates a variety of important functional groups, likely involves the formation of the indole intermediates 2 (through aminopalladation/reductive elimination) [20,21] followed by cyclization to 3 and elimination of trifluoroacetic acid (Scheme 1). Later, a procedure that allows the introduction of a variety of substituents other than CF3 in the 6-position (without the substituent in the 12-position) was reported by Wang and co-workers [22]. More recently, we showed that the Pd-catalyzed reaction of o-(o-aminophenylethynyl) trifluoroacetanilides 5 with aryl halides and aryldiazonium salts led to the formation of 12-arylindolo[1,2-c]quinazolin-6(5H)-ones 8 in moderate to excellent yields (Scheme 2a) [23]. Very likely, under these reaction conditions, the palladium-catalyzed reaction of 5 gave intermediates 6; then, cyclization to 7 followed by elimination of trifluoromethane afforded products 8.
![[1860-5397-14-218-i2]](/bjoc/content/inline/1860-5397-14-218-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Present and previously reported reactions starting from 5. DMFDMA: dimethylformamide dimethyl acetal.
Scheme 2: Present and previously reported reactions starting from 5. DMFDMA: dimethylformamide dimethyl aceta...
Since the unique molecular skeleton of hinckdentine A is constituted of a 6-unsubstituted indolo[1,2-c]quinazoline nucleus [12,13], we planned to modify our previous procedures to address the 6-unsubstituted-12-arylindolo[1,2-c]quinazolines 10. We wish to report here that, under suitable reaction conditions, substrates 5 can be easily converted into target products 10 (Scheme 2b). Moreover, 12-unsubstituted analogues 13 are also available from 5 or from o-(o-aminophenylethynyl)anilines 15 (see below).
Results and Discussion
We have previously reported that arylboronic acids 12 can be used in place of aryl halides in the Pd-catalyzed synthesis of indoles through aminopalladation/reductive elimination reaction from 2-alkynyltrifluoroacetanilides [24]. This reaction is carried out in MeOH at 60 °C in the presence of K3PO4 under an oxygen atmosphere. We then decided to react substrate 5 under these reaction conditions, in order to ascertain if the presence of methanol and base would result in the cleavage of the trifluoroacetylamide group after the formation of the indole ring, affording 2-(1H-indolo-2-yl)benzenamine derivatives 9 instead of products 8. We were pleased to observe the formation of 9a in good yields when 5 was reacted with 4-methoxyphenylboronic acid. A minor amount of product 11a (likely deriving from a transamidation reaction [25] of initially formed intermediate 6a) was also isolated. Then, 9a was treated with dimethylformamide dimethyl acetal (DMFDMA) as a source of an electrophilic one-carbon unit at the formate oxidation level [26,27], affording 12-arylindolo[1,2-c]quinazoline 10a in good yield (Scheme 3).
![[1860-5397-14-218-i3]](/bjoc/content/inline/1860-5397-14-218-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Two-step synthesis of 12-arylindolo[1,2-c]quinazoline 10a.
Scheme 3: Two-step synthesis of 12-arylindolo[1,2-c]quinazoline 10a.
Based on this result, we tested an approach to target products 10 avoiding the isolation of intermediates 9. After some experimentation, we found that the best results were obtained from the following procedure: 1) Pd-catalyzed reaction of 5 with ArB(OH)2/K3PO4 in MeOH at 60 °C; 2) heating of the reaction mixture at 100 °C to allow complete hydrolysis of byproducts 11; 3) after extractive work-up, treatment of the crude reaction mixture with an excess of DMFDMA (5 equiv) [28] in DMF at 100 °C to obtain indoloquinazolines 10. The results of this procedure, starting from a variety of o-(o-aminophenylethynyl)trifluoroacetanilides 5 and arylboronic acids 12, are summarized in Table 1.
Table 1: Synthesis of 12-arylindolo[1,2-c]quinazolines 10.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-218-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | R1 | R2 | 5 | Ar | Time (h)b | 10 (% yield)c | 13 (% yield)c |
| 1 | H | H | 5a | 4-MeO-C6H4- | 3, 16, 16 | 10a (76) | – |
| 2 | H | H | 5a | 4-MeO-C6H4- | 24, 18, 16 | 10a (49)d | – |
| 3 | H | H | 5a | 4-Me-C6H4- | 5, 16, 16 | 10b (61) | – |
| 4 | H | H | 5a | 2-Me-C6H4- | 5, 16, 16 | 10j (56) | 13j (11) |
| 5 | H | H | 5a | phenyl | 24, 8, 12 | 10c (75) | – |
| 6 | H | H | 5a | 1-naphthyl | 2, 16, 2 | 10d (60) | – |
| 7 | H | H | 5a | 4-CF3-C6H4- | 4, 7, 16 | 10e (69) | – |
| 8 | H | H | 5a | 4-Br-C6H4- | 2, 16, 6 | 10f (60) | – |
| 9 | H | H | 5a | 4-MeO2C-C6H4- | 3, 28, 2 | 10g (59) | – |
| 10 | H | H | 5a | 4-Cl-C6H4- | 6, 16, 3 | 10h (52) | 13h (12) |
| 11 | H | H | 5a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-218-i7.svg?max-width=637&scale=1.0)
|
18, 16, 2 | 10i (65) | 13i (12) |
| 12 | Me | H | 5b | 4-MeO-C6H4- | 18, 6, 1 | 10k (63) | 13k (5) |
| 13 | Me | H | 5b | phenyl | 24, 5, 4 | 10l (72) | – |
| 14 | Me | H | 5b |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-218-i8.svg?max-width=637&scale=1.0)
|
1, 16, 8 | 10m (76) | – |
| 15 | Me | COOMe | 5c | 4-MeO-C6H4- | 5, 16, 3 | 10n (60) | – |
| 16 | H | Me | 5d | 4-Br-C6H4- | 6, 16, 1.5 | 10o (58) | – |
| 17 | H | H | 5a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-218-i9.svg?max-width=637&scale=1.0)
|
24, 24, 8 | 10p (12) | 13p (38) |
aReactions were carried out at 60 °C on 0.345 mmol scale using 1.0 equiv of 5, 2.0 equiv of 13, 0.05 equiv of Pd(OAc)2, 0.05 equiv of dppp and 2 equiv of K3PO4 in MeOH (2 mL) under an atmosphere of O2; then 100 °C; work-up; then 5 equiv of DMFDMA in DMF (2 mL), 100 °C. bNumbers refer to the 1st, 2nd and 3rd step, respectively. cIsolated yields. dCarried out under air atmosphere.
In some cases, 12-unsubstituted indolo[1,2-c]quinazoline derivatives 13 were also obtained as byproducts. Electron-rich arylboronic acids worked quite well (Table 1, entries 1, 3, and 4). However, an attempt to carry out the reaction under air, instead of under O2 atmosphere, resulted in a loss of efficiency (Table 1, entry 2). Good results were also observed with phenyl- and 1-naphthylboronic acids (Table 1, entries 5 and 6). Electron-poor arylboronic acids proved to be effective (Table 1, entries 7–10), and only (4-chlorophenyl)boronic acid gave a moderate yield of product (Table 1, entry 10).
Products substituted in the indole and quinazoline frameworks were also obtained (Table 1, entries 12–16). It is worth noting that the reaction tolerates a -Br substituent (Table 1, entries 8 and 16) which may serve as a useful handle for increasing molecular complexity through cross-coupling reactions. Only in the case of two ortho-substituents on the aryl residue of the boronic acid the reaction afforded the target product 10p in low yield (Table 1, entry 17), and 12-unsubstituted derivative 13p as the major product.
Then, with the aim of obtaining 12-unsubstituted indoloquinazolines 13 selectively, we tested the use of DMFDMA for the cyclization of 2-(o-aminophenyl)indoles 14. This reaction required only a slight excess of DMFDMA (1.2 equiv), and afforded derivatives 13 as sole products in high yields (Table 2).
Table 2: Synthesis of 12-unsubstituted indolo[1,2-c]-quinazolines 13.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-218-i10.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | R3 | 14 | Time (h) | 13 (% yield)b |
| 1 | H | H | H | 14a | 5 | 13a (86) |
| 2 | Me | H | Me | 14b | 24 | 13b (87) |
| 3 | COOMe | H | COOMe | 14c | 6 | 13c (81) |
| 4 | H | H | Me | 14d | 25 | 13d (95) |
| 5 | Me | Me | H | 14e | 24 | 13e (73) |
aReactions were carried out at 100 °C on 0.481 mmol scale using 1.2 equiv of DMFDMA in DMF (2 mL). bIsolated yields.
As described in Supporting Information File 1, indoles 14a–c (R1 = R3, R2 = H) were obtained by cyclization of the corresponding o-(o-aminophenylethynyl)anilines 15a–c with PdCl2(MeCN)2 [29,30]. Indoles 14d,e (R1 ≠ R3) could not be obtained in such way (due to the lack of selectivity between the two different NH2 groups in the hydroamination reaction), and were obtained by cyclization of the corresponding o-(o-aminophenylethynyl) trifluoroacetanilides 5 with PdCl2(MeCN)2 (it was previously demonstrated that the selective formation of 16 occurs under these conditions [31]) followed by the hydrolysis of the COCF3 group of the crude product (Scheme 4).
![[1860-5397-14-218-i4]](/bjoc/content/inline/1860-5397-14-218-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Moreover, the palladium-catalyzed reaction of 15a in the presence of DMFDMA led directly to the formation of quinazoline 13a in 71% yield through a sequential process (Scheme 5a).
![[1860-5397-14-218-i5]](/bjoc/content/inline/1860-5397-14-218-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Sequential preparation of 13a from 15a.
Scheme 5: Sequential preparation of 13a from 15a.
The selective formation of products 13 from 15 (and also from 3-unsubstituted indoles 14) is not trivial, since the previously reported gold-catalyzed reaction of 15a with aldehydes as electrophiles resulted in the divergent formation of 11H-indolo[3,2-c]quinolines 17 (Scheme 5b) [32] through functionalization of C-3 position of the indole ring instead of N-1.
The sequential reaction shown in Scheme 5, path a, probably occurs through cyclization of 15a to indole 14a, followed by the reaction with DMFDMA; however, an alternative path in which one amino group interact with DMFDMA to give a formamidine intermediate [27] before cyclization cannot be ruled out.
Conclusion
We have reported here an efficient procedure for the preparation of 12-arylindolo[1,2-c]quinazolines 10 from o-(o-aminophenylethynyl) trifluoroacetanilides 5 and arylboronic acids, by avoiding the isolation of intermediate indoles 9. Starting from indoles 14, or from o-(o-aminophenylethynyl)aniline 15a, selective formation of 12-unsubstituted[1,2-c]quinazolines 13 was accomplished, without competitive formation of products derived from C-3 functionalization at the indole moiety.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Demeunynck, M.; Baussanne, I. Curr. Med. Chem. 2013, 20, 794–814. doi:10.2174/0929867311320060006
Return to citation in text: [1] -
Tucker, A. M.; Grundt, P. ARKIVOC 2012, No. i, 546–569.
Return to citation in text: [1] -
Adams, M.; Mahringer, A.; Kunert, O.; Fricker, G.; Efferth, T.; Bauer, R. Planta Med. 2007, 73, 1554–1557. doi:10.1055/s-2007-993743
Return to citation in text: [1] -
Hochfellner, C.; Evangelopoulos, D.; Zloh, M.; Wube, A.; Guzman, J. D.; McHugh, T. D.; Kunert, O.; Bhakta, S.; Bucar, F. J. Appl. Microbiol. 2015, 118, 864–872. doi:10.1111/jam.12753
Return to citation in text: [1] -
Vaidya, S. D.; Argade, N. P. Org. Lett. 2013, 15, 4006–4009. doi:10.1021/ol4018062
Return to citation in text: [1] -
Wang, C.; Zhang, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2013, 15, 2982–2985. doi:10.1021/ol401144m
Return to citation in text: [1] -
Nelson, A. C.; Kalinowski, E. S.; Jacobson, T. L.; Grundt, P. Tetrahedron Lett. 2013, 54, 6804–6806. doi:10.1016/j.tetlet.2013.09.124
Return to citation in text: [1] -
Sharma, V. M.; Prasanna, P.; Seshu, K. V. A.; Renuka, B.; Rao, C. V. L.; Kumar, G. S.; Narasimhulu, C. P.; Babu, P. A.; Puranik, R. C.; Subramanyam, D.; Venkateswarlu, A.; Rajagopal, S.; Kumar, K. B. S.; Rao, C. S.; Mamidi, N. V. S. R.; Deevi, D. S.; Ajaykumar, R.; Rajagopalan, R. Bioorg. Med. Chem. Lett. 2002, 12, 2303–2307. doi:10.1016/S0960-894X(02)00431-6
Return to citation in text: [1] -
Rohini, R.; Reddy, P. M.; Shanker, K.; Hu, A.; Ravinder, V. Eur. J. Med. Chem. 2010, 45, 1200–1205. doi:10.1016/j.ejmech.2009.11.038
Return to citation in text: [1] [2] -
Rohini, R.; Reddy, P. M.; Shanker, K.; Hu, A.; Ravinder, V. J. Braz. Chem. Soc. 2010, 21, 897–904. doi:10.1590/S0103-50532010000500018
Return to citation in text: [1] -
Rohini, R.; Shanker, K.; Reddy, P. M.; Sekhar, V. C.; Ravinder, V. Arch. Pharm. 2009, 342, 533–540. doi:10.1002/ardp.200900068
Return to citation in text: [1] -
Blackman, A. J.; Hambley, T. W.; Picker, K.; Taylor, W. C.; Thirasasana, N. Tetrahedron Lett. 1987, 28, 5561–5562. doi:10.1016/S0040-4039(00)96781-9
Return to citation in text: [1] [2] -
Douki, K.; Ono, H.; Taniguchi, T.; Shimokawa, J.; Kitamura, M.; Fukuyama, T. J. Am. Chem. Soc. 2016, 138, 14578–14581. doi:10.1021/jacs.6b10237
Return to citation in text: [1] [2] -
Frère, S.; Thiéry, V.; Bailly, C.; Besson, T. Tetrahedron 2003, 59, 773–779. doi:10.1016/S0040-4020(02)01593-4
Return to citation in text: [1] -
Billimoria, A. D.; Cava, M. P. J. Org. Chem. 1994, 59, 6777–6782. doi:10.1021/jo00101a043
Return to citation in text: [1] -
Dai, X.; Liu, H.; Palani, A.; He, S.; Lai, Z.; Nargund, R.; Marcantonio, K.; Xiao, D.; Brockunier, L. L.; Zorn, N.; Dang, Q.; Peng, X.; Li, P. Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c. WO Patent WO2014/123794A1, Aug 14, 2014.
Return to citation in text: [1] -
Guo, S.; Tao, L.; Zhang, W.; Zhang, X.; Fan, X. J. Org. Chem. 2015, 80, 10955–10964. doi:10.1021/acs.joc.5b02076
Return to citation in text: [1] -
Liu, Q.; Yang, H.; Jiang, Y.; Zhao, Y.; Fu, H. RSC Adv. 2013, 3, 15636–15644. doi:10.1039/c3ra41644e
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Cassetta, A.; Fabrizi, G.; Parisi, L. M. Synlett 2001, 1605–1607. doi:10.1055/s-2001-17458
Return to citation in text: [1] [2] -
Cacchi, S.; Marinelli, F. In Organopalladium chemistry for Organic Synthesis; Neghishi, E.; de Meijere, A., Eds.; John Wiley & Sons: New York, 2002; Vol. 2, pp 2227–2244.
Return to citation in text: [1] [2] -
Battistuzzi, G.; Cacchi, S.; Fabrizi, G. Eur. J. Org. Chem. 2002, 2671–2681. doi:10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X
See for a review.
Return to citation in text: [1] [2] -
Xu, M.; Xu, K.; Wang, S.; Yao, Z.-J. Tetrahedron Lett. 2013, 54, 4675–4678. doi:10.1016/j.tetlet.2013.06.079
Return to citation in text: [1] [2] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Ghirga, F.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Synthesis 2018, 1133–1140. doi:10.1055/s-0036-1589158
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/C2OB27125G
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G.; Pace, P.; Marinelli, F. Synlett 1999, 620–622. doi:10.1055/s-1999-2659
Return to citation in text: [1] -
Kidjemet, D. Synlett 2002, 1741–1742. doi:10.1055/s-2002-34251
Return to citation in text: [1] -
Nathubhai, A.; Patterson, R.; Woodman, T. J.; Sharp, H. E. C.; Chui, M. T. Y.; Chung, H. H. K.; Lau, S. W. S.; Zheng, J.; Lloyd, M. D.; Thompson, A. S.; Threadgill, M. D. Org. Biomol. Chem. 2011, 9, 6089–6099. doi:10.1039/c1ob05430a
Return to citation in text: [1] [2] -
The use of a lower excess of DMFDMA decreased the yields of products.
Return to citation in text: [1] -
Utimoto, K.; Miwa, H.; Nozaki, H. Tetrahedron Lett. 1981, 22, 4277–4278. doi:10.1016/S0040-4039(01)82932-4
Return to citation in text: [1] -
Iritani, K.; Matsubara, S.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1799–1802. doi:10.1016/S0040-4039(00)82047-X
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Parisi, L. M. Heterocycles 2004, 64, 475–482. doi:10.3987/COM-04-S(P)23
Return to citation in text: [1] -
Abbiati, G.; Arcadi, A.; Chiarini, M.; Marinelli, F.; Pietropaolo, E.; Rossi, E. Org. Biomol. Chem. 2012, 10, 7801–7808. doi:10.1039/c2ob26380g
Return to citation in text: [1]
| 25. | Cacchi, S.; Fabrizi, G.; Pace, P.; Marinelli, F. Synlett 1999, 620–622. doi:10.1055/s-1999-2659 |
| 26. | Kidjemet, D. Synlett 2002, 1741–1742. doi:10.1055/s-2002-34251 |
| 27. | Nathubhai, A.; Patterson, R.; Woodman, T. J.; Sharp, H. E. C.; Chui, M. T. Y.; Chung, H. H. K.; Lau, S. W. S.; Zheng, J.; Lloyd, M. D.; Thompson, A. S.; Threadgill, M. D. Org. Biomol. Chem. 2011, 9, 6089–6099. doi:10.1039/c1ob05430a |
| 1. | Demeunynck, M.; Baussanne, I. Curr. Med. Chem. 2013, 20, 794–814. doi:10.2174/0929867311320060006 |
| 2. | Tucker, A. M.; Grundt, P. ARKIVOC 2012, No. i, 546–569. |
| 3. | Adams, M.; Mahringer, A.; Kunert, O.; Fricker, G.; Efferth, T.; Bauer, R. Planta Med. 2007, 73, 1554–1557. doi:10.1055/s-2007-993743 |
| 4. | Hochfellner, C.; Evangelopoulos, D.; Zloh, M.; Wube, A.; Guzman, J. D.; McHugh, T. D.; Kunert, O.; Bhakta, S.; Bucar, F. J. Appl. Microbiol. 2015, 118, 864–872. doi:10.1111/jam.12753 |
| 14. | Frère, S.; Thiéry, V.; Bailly, C.; Besson, T. Tetrahedron 2003, 59, 773–779. doi:10.1016/S0040-4020(02)01593-4 |
| 12. | Blackman, A. J.; Hambley, T. W.; Picker, K.; Taylor, W. C.; Thirasasana, N. Tetrahedron Lett. 1987, 28, 5561–5562. doi:10.1016/S0040-4039(00)96781-9 |
| 13. | Douki, K.; Ono, H.; Taniguchi, T.; Shimokawa, J.; Kitamura, M.; Fukuyama, T. J. Am. Chem. Soc. 2016, 138, 14578–14581. doi:10.1021/jacs.6b10237 |
| 12. | Blackman, A. J.; Hambley, T. W.; Picker, K.; Taylor, W. C.; Thirasasana, N. Tetrahedron Lett. 1987, 28, 5561–5562. doi:10.1016/S0040-4039(00)96781-9 |
| 13. | Douki, K.; Ono, H.; Taniguchi, T.; Shimokawa, J.; Kitamura, M.; Fukuyama, T. J. Am. Chem. Soc. 2016, 138, 14578–14581. doi:10.1021/jacs.6b10237 |
| 24. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/C2OB27125G |
| 9. | Rohini, R.; Reddy, P. M.; Shanker, K.; Hu, A.; Ravinder, V. Eur. J. Med. Chem. 2010, 45, 1200–1205. doi:10.1016/j.ejmech.2009.11.038 |
| 10. | Rohini, R.; Reddy, P. M.; Shanker, K.; Hu, A.; Ravinder, V. J. Braz. Chem. Soc. 2010, 21, 897–904. doi:10.1590/S0103-50532010000500018 |
| 11. | Rohini, R.; Shanker, K.; Reddy, P. M.; Sekhar, V. C.; Ravinder, V. Arch. Pharm. 2009, 342, 533–540. doi:10.1002/ardp.200900068 |
| 22. | Xu, M.; Xu, K.; Wang, S.; Yao, Z.-J. Tetrahedron Lett. 2013, 54, 4675–4678. doi:10.1016/j.tetlet.2013.06.079 |
| 5. | Vaidya, S. D.; Argade, N. P. Org. Lett. 2013, 15, 4006–4009. doi:10.1021/ol4018062 |
| 6. | Wang, C.; Zhang, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2013, 15, 2982–2985. doi:10.1021/ol401144m |
| 7. | Nelson, A. C.; Kalinowski, E. S.; Jacobson, T. L.; Grundt, P. Tetrahedron Lett. 2013, 54, 6804–6806. doi:10.1016/j.tetlet.2013.09.124 |
| 8. | Sharma, V. M.; Prasanna, P.; Seshu, K. V. A.; Renuka, B.; Rao, C. V. L.; Kumar, G. S.; Narasimhulu, C. P.; Babu, P. A.; Puranik, R. C.; Subramanyam, D.; Venkateswarlu, A.; Rajagopal, S.; Kumar, K. B. S.; Rao, C. S.; Mamidi, N. V. S. R.; Deevi, D. S.; Ajaykumar, R.; Rajagopalan, R. Bioorg. Med. Chem. Lett. 2002, 12, 2303–2307. doi:10.1016/S0960-894X(02)00431-6 |
| 23. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Ghirga, F.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Synthesis 2018, 1133–1140. doi:10.1055/s-0036-1589158 |
| 18. | Liu, Q.; Yang, H.; Jiang, Y.; Zhao, Y.; Fu, H. RSC Adv. 2013, 3, 15636–15644. doi:10.1039/c3ra41644e |
| 19. | Arcadi, A.; Cacchi, S.; Cassetta, A.; Fabrizi, G.; Parisi, L. M. Synlett 2001, 1605–1607. doi:10.1055/s-2001-17458 |
| 32. | Abbiati, G.; Arcadi, A.; Chiarini, M.; Marinelli, F.; Pietropaolo, E.; Rossi, E. Org. Biomol. Chem. 2012, 10, 7801–7808. doi:10.1039/c2ob26380g |
| 17. | Guo, S.; Tao, L.; Zhang, W.; Zhang, X.; Fan, X. J. Org. Chem. 2015, 80, 10955–10964. doi:10.1021/acs.joc.5b02076 |
| 20. | Cacchi, S.; Marinelli, F. In Organopalladium chemistry for Organic Synthesis; Neghishi, E.; de Meijere, A., Eds.; John Wiley & Sons: New York, 2002; Vol. 2, pp 2227–2244. |
| 21. |
Battistuzzi, G.; Cacchi, S.; Fabrizi, G. Eur. J. Org. Chem. 2002, 2671–2681. doi:10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X
See for a review. |
| 27. | Nathubhai, A.; Patterson, R.; Woodman, T. J.; Sharp, H. E. C.; Chui, M. T. Y.; Chung, H. H. K.; Lau, S. W. S.; Zheng, J.; Lloyd, M. D.; Thompson, A. S.; Threadgill, M. D. Org. Biomol. Chem. 2011, 9, 6089–6099. doi:10.1039/c1ob05430a |
| 15. | Billimoria, A. D.; Cava, M. P. J. Org. Chem. 1994, 59, 6777–6782. doi:10.1021/jo00101a043 |
| 16. | Dai, X.; Liu, H.; Palani, A.; He, S.; Lai, Z.; Nargund, R.; Marcantonio, K.; Xiao, D.; Brockunier, L. L.; Zorn, N.; Dang, Q.; Peng, X.; Li, P. Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c. WO Patent WO2014/123794A1, Aug 14, 2014. |
| 29. | Utimoto, K.; Miwa, H.; Nozaki, H. Tetrahedron Lett. 1981, 22, 4277–4278. doi:10.1016/S0040-4039(01)82932-4 |
| 30. | Iritani, K.; Matsubara, S.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1799–1802. doi:10.1016/S0040-4039(00)82047-X |
| 9. | Rohini, R.; Reddy, P. M.; Shanker, K.; Hu, A.; Ravinder, V. Eur. J. Med. Chem. 2010, 45, 1200–1205. doi:10.1016/j.ejmech.2009.11.038 |
| 19. | Arcadi, A.; Cacchi, S.; Cassetta, A.; Fabrizi, G.; Parisi, L. M. Synlett 2001, 1605–1607. doi:10.1055/s-2001-17458 |
| 20. | Cacchi, S.; Marinelli, F. In Organopalladium chemistry for Organic Synthesis; Neghishi, E.; de Meijere, A., Eds.; John Wiley & Sons: New York, 2002; Vol. 2, pp 2227–2244. |
| 21. |
Battistuzzi, G.; Cacchi, S.; Fabrizi, G. Eur. J. Org. Chem. 2002, 2671–2681. doi:10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X
See for a review. |
| 22. | Xu, M.; Xu, K.; Wang, S.; Yao, Z.-J. Tetrahedron Lett. 2013, 54, 4675–4678. doi:10.1016/j.tetlet.2013.06.079 |
| 31. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Parisi, L. M. Heterocycles 2004, 64, 475–482. doi:10.3987/COM-04-S(P)23 |
© 2018 Arcadi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)