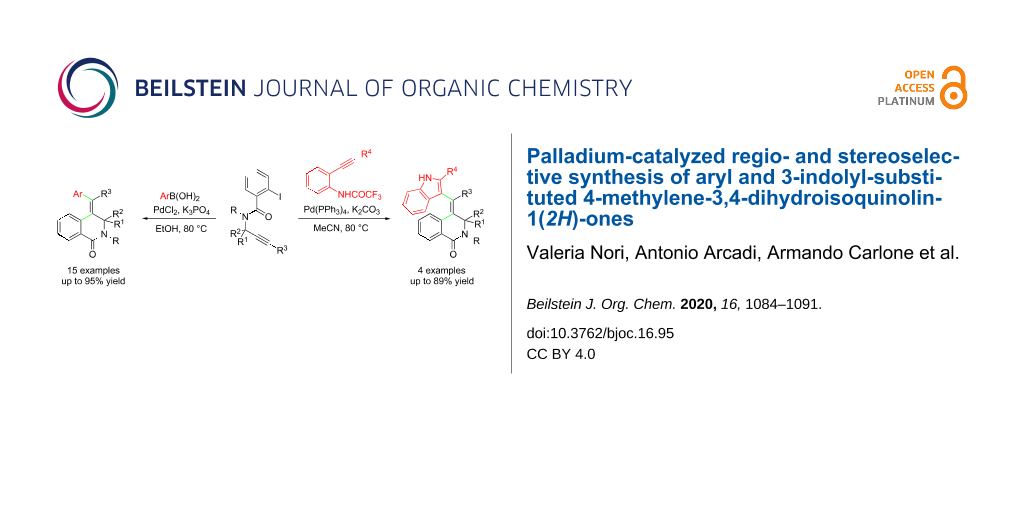
Abstract
Cascade cyclocarbopalladation of the readily available aryl/alkyl-substituted propargylic amides containing an aryl iodide moiety, followed by Suzuki–Miyaura coupling with arylboronic acids, allowed an efficient regio- and stereoselective synthesis of tetrasubstituted 4-methylene-3,4-dihydroisoquinolin-1(2H)-ones. Moreover, cascade cyclocarbopalladation, followed by the reaction with 2-alkynyltrifluoroacetanilides, accomplished a double cyclization to afford challenging 4-methylene-3,4-dihydroisoquinolin-1(2H)-ones bearing a 3-indolyl substituent through aminopalladation/reductive elimination.

Graphical Abstract
Introduction
The isoquinolinone nucleus is a key constituent of many natural products [1-3] and pharmaceuticals [4-6]. Substituted isoquinolinones have been found in biologically active small molecules that exhibit antihypertensive activity [7,8]. Moreover, these heterocycles can be used as 5-HT3 antagonists [9], rho kinase inhibitors [10], thymidylate synthetase inhibitors [11], PARP-1 inhibitors [12], melatonin MT1 and MT2 receptor agonist [13], and fascin-targeted antimetastatic agents [14]. Fittingly, the development of efficient strategies for their construction and peripheral functionalization represents still an active research area aimed to achieve structural diversity [15-18].
Carbometalations of alkynes constitute a powerful tool for the regio- and stereoselective formation of carbon–carbon bonds [19]. Intramolecular palladium-catalyzed versions are particularly attractive, since they afford polycarbo- and heterocyclic systems via sequential reactions of the vinylpalladium intermediate [20-25]. In this field, a variety of regio- and stereoselective Pd-catalyzed cascade reactions, consisting of the addition of in situ-generated arylpalladium complexes over a proximate carbon–carbon triple bond, followed by cross-coupling reactions, have been reported [26-31].
Our continuing interest in the palladium-catalyzed reactions of functionalized alkynes with boronic acids [32,33] prompted us to explore the palladium-catalyzed reaction of the readily available alkynyliodobenzamides 2 with boronic acids 3 as a viable route to the regio- and stereoselective synthesis of 4-alkylidene-3,4-dihydroisoquinolin-1(2H)-ones 3 (Scheme 1a).
![[1860-5397-16-95-i1]](/bjoc/content/inline/1860-5397-16-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Planned approach to tetrasubstituted-4-methylene-3,4-dihydroisoquinolin-1(2H)-ones 4 and 6.
Scheme 1: Planned approach to tetrasubstituted-4-methylene-3,4-dihydroisoquinolin-1(2H)-ones 4 and 6.
We are pleased to report here that this cascade reaction takes place efficiently, resulting in the regio- and stereoselective formation of the poly-substituted isoquinolinones 4 in good to high yield. Applications of this reaction can be relevant for improvements of structure diversity and fine tuning of the chemical and physical properties of the products.
Furthermore, over the years, we have reported a general methodology for the Pd-catalyzed synthesis of 3-substituted indoles, now referred to as the “Cacchi reaction” [34], through an aminopalladation/reductive elimination sequence starting from 2-alkynyltrifluoroacetanilides. In all these procedures, the activation of the triple bond was achieved by means of a σ-organyl palladium complex, in turn generated in situ by oxidative addition of a Pd(0) species to suitable organic electrophiles (aryl and vinyl halides or triflates [35,36], alkyl halides [37], alkynyl halides [38], α-iodoenones [39], or by transmetalation of a Pd(II) species with boronic acids [33]. In this context, we decided to explore the use of substrates 2 in the reaction with 2-alkynyltrifluoroacetanilides 5 through a sequential cyclocarbopalladation/aminopalladation/reductive elimination process, widening in such a way the scope of the methodology and allowing challenging synthesis of indoles 6 bearing a 4-alkylidene-3,4-dihydroisoquinolin-1(2H)-one substituent (Scheme 1b). It is worth noting that an aerobic Pd/Cu-catalyzed cyclizative cross-coupling between 2-alkynylanilines and 2-alkynylbenzamides, affording indoles bearing an alkylidene-iminoisobenzofurane moiety, has been reported [40].
Results and Discussion
The starting N-propargyl-2-iodobenzamides 2 were easily obtained by the reaction of the readily available [41] propargylamines 1 with 2-iodobenzoyl chloride in CH2Cl2 at room temperature (Scheme 2).
![[1860-5397-16-95-i2]](/bjoc/content/inline/1860-5397-16-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of the starting N-propargyl-2-iodobenzamides 2.
Scheme 2: Preparation of the starting N-propargyl-2-iodobenzamides 2.
Initially, we explored the reaction of the N-(4-(4-acetylphenyl)-2-methylbut-3-yn-2-yl)-N-benzyl-2-iodobenzamide (2a) with a variable excess of the phenylboronic acid (3a) in the presence of K3PO4 as the base (K3PO4: 3 equiv) by using 5 mol % of different palladium catalysts/solvent/temperature combinations. The results are reported in Table 1.
Table 1: Optimization of the reaction of propargyl 2-iodobenzamide 2a with phenylboronic acid (3a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-95-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent/temp. (°C) | 3a:2a ratio | catalyst | time (h) |
4aa
yield (%)b |
| 1 | dioxane/100 | 1.5 | PdCl2(PPh3)2 | 7 | 51 |
| 2 | dioxane/100 | 2.0 | PdCl2(PPh3)2 | 2 | 94 |
| 3 | dioxane/100 | 3.0 | PdCl2(PPh3)2 | 2 | 97 |
| 4 | dioxane/100 | 1.5 | PdCl2(PPh3)2 | 4 | 67c |
| 5 | dioxane/water (9:1)/100 | 1.5 | PdCl2(PPh3)2 | 2 | 80c |
| 6 | MeCN/80 | 1.5 | PdCl2(PPh3)2 | 7 | 41c |
| 7 | THF/60 | 1.5 | PdCl2(PPh3)2 | 7 | 27c |
| 8 | DMF/110 | 1.5 | PdCl2(PPh3)2 | 2.5 | 58c |
| 9 | DMSO/110 | 1.5 | PdCl2(PPh3)2 | 2.5 | 41c |
| 10 | EtOH/80 | 1.5 | PdCl2(PPh3)2 | 3 | 85c |
| 11 | EtOH/80 | 1.5 | Pd(PPh3)4 | 3 | 74c |
| 12 | EtOH/80 | 1.5 | Pd/C | 3 | 66c |
| 13 | EtOH/80 | 1.5 | Pd(OAc)2 | 2.5 | 77c |
| 14 | EtOH/80 | 1.5 | PdCl2 | 2.5 | 91c |
aReactions were carried out on a 0.19 mmol scale, using 3 equiv of base, 0.10 equiv of ligand and 0.05 equiv of the palladium catalyst in 2.0 mL of solvent under nitrogen atmosphere. bYields are given for isolated products. c1.0 mL of solvent.
When 1,4-dioxane was used as the solvent in the presence of commercially available PdCl2(PPh3)2 as the catalyst at 100 °C, the reaction of 2a with 1.5 equiv of the phenylboronic acid (3a) delivered the target (Z)-dihydroisoquinolin-1(2H)-one 4aa in 51% yield. Better yields were observed by increasing the excess of the phenylboronic acid (Table 1, entries 1–3) or by halving the amount of the solvent in the presence 1.5 equiv of 3a (Table 1, entry 4). Under these latter conditions a beneficial effect was obtained by using a 9:1 mixture of 1,4-dioxane/H2O as the reaction medium (Table 1, entry 5). While MeCN, DMF, THF and DMSO as solvents gave worse results (Table 1, entries 6–9), the environmentally friendly EtOH proved to be the most efficient reaction medium (Table 1, entry 10). Further attempts to increase the yield of 4aa by tuning the catalytic system showed that the ligand-free PdCl2 was the most effective catalyst (Table 1, entry 14). Other catalysts such as Pd(PPh3)4, Pd/C or Pd(OAc)2 provided inferior results (Table 1, entries 11–13).
We then examined the reaction of 2a with 1.5 equiv of a variety of arylboronic acids. Using the optimized reaction conditions of Table 1, entry 14, 2a reacted smoothly with diversely substituted arylboronic acids 3a–i to regio- and stereoselectively afford the corresponding tetrasubstituted 4-methylene-3,4-dihydroisoquinolin-1(2H)-ones 4ab–ai in moderate to good yields (Scheme 3).
![[1860-5397-16-95-i3]](/bjoc/content/inline/1860-5397-16-95-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of the reaction of N-propargyl-2-iodobenzamide 2a with arylboronic acids 3b–i.
Scheme 3: Substrate scope of the reaction of N-propargyl-2-iodobenzamide 2a with arylboronic acids 3b–i.
Gratifyingly, various functional groups such as amino, acetamido, F, Cl, OMe, CF3, and CN moieties were found to be compatible with these reaction conditions, and only the homocoupling of the arylboronic acids was observed as side reaction to some extent [42]. The best results were obtained with arylboronic acids containing electron-donating substituents; electron-poor arylboronic acids proved to be slightly less effective, probably because of their lower nucleophilicity that could have affected the transmetalation step.
Moreover, we screened the reaction of a number of aromatic boronic acids 3a–j with a set of N-propargyl-2-iodobenzamides 2c–f (Scheme 4).
![[1860-5397-16-95-i4]](/bjoc/content/inline/1860-5397-16-95-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Substrate scope of the reaction of N-propargyl-2-iodobenzamides 2c–f with arylboronic acids 3a–c/j.
Scheme 4: Substrate scope of the reaction of N-propargyl-2-iodobenzamides 2c–f with arylboronic acids 3a–c/j.
All reactions successfully delivered the desired products 4 in moderate to good yields. Remarkably, an alkyl substituent was tolerated at the terminal sp carbon atom of the starting N-propargyl-2-iodobenzamides 2. Furthermore, 2-iodobenzamides 2e and 2f (mono-substituted and unsubstituted at the propargylic position) were successfully used as starting materials, affording the corresponding products 4eb, 4fc and 4fj in good yields. According to the literature, the highly stereoselective formation of products 4 resulted from the intramolecular syn-addition of the in situ-generated arylpalladium iodide complex to the triple bond to give an (E)-vinylpalladium intermediate, which underwent cross-coupling with an arylboronic acid leading to the final product by reductive elimination, with the regeneration of the Pd(0) catalyst.
Finally, we envisaged that the above mentioned (E)-vinylpalladium intermediate A (generated in situ from the insertion of a carbon–carbon triple bond in the initially formed arylpalladium complex) could also be involved in the aminopalladations with the alkynyltrifluoroacetanilides 5 through the formation of the π-complex B, followed by base-assisted cyclization and reductive elimination from the resulting σ-indolylpalladium complex C (Scheme 5).
![[1860-5397-16-95-i5]](/bjoc/content/inline/1860-5397-16-95-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of N-propargyl-2-iodobenzamides 2b,f with the 2-alkynyltrifluoroacetanilides 5a–c.
Scheme 5: Reaction of N-propargyl-2-iodobenzamides 2b,f with the 2-alkynyltrifluoroacetanilides 5a–c.
The reaction led to the stereoselective formation of indole derivatives 6ba–fc (aryl, heteroaryl and vinyl groups were allowed in substrates 5) in good to high yield.
The stereochemistry of compounds 4 and 6 was unambiguously confirmed by NMR spectroscopy [43].
Conclusion
In conclusion, we have demonstrated that cascade cyclocarbopalladations of the readily available aryl/alkyl-substituted N-propargyl-2-iodobenzamides 2 followed by Suzuki–Miyaura coupling reactions with arylboronic acids, in the presence of a catalytic amount of the ligand-free PdCl2 in environmentally friendly ethanol, achieve an efficient regio- and stereoselective synthesis of 4-methylene-3,4-dihydroisoquinolin-1(2H)-ones 4. It is worth noting that, during the preparation of this paper, a related article focused on the palladium-catalyzed regioselective cascade cyclization of propargylamides/coupling with ArB(OH)2 in dioxane/water, to give trisubstituted arylidene-isoquinolinones [44] was published. However, the Ugi four-component reaction used to construct the starting building blocks was limited to the preparation of propargylic 2-halobenzamides unsubstituted at the propargyl carbon, allowing the synthesis of isoquinolinones without substituents at C-3; but the present methodology overcame these limitations.
Moreover, the previously developed strategy of indole synthesis through an aminopalladation/reductive elimination process has been significantly extended to include σ-vinyl Pd(II) intermediates B obtained through oxidative addition/insertion of substrates 2 with Pd(0). This reaction efficiently led to challenging indoloquinolinones 6 through a sequential double cyclization. It is worth noting that, in both cases, the intramolecular alkyne insertion in the initially formed arylpalladium iodide A (leading to B) occured faster than the direct reaction of A with arylboronic acids or with 2-alkynyltrifluoroacetanilides.
Experimental
General methods
Melting points are uncorrected. IR spectra were recorded with a Perkin Elmer Spectrum Two FT/IR spectrometer. 1H NMR spectra were recorded in CDCl3 at 400 MHz on Bruker Avance 400 instrument. Chemical shifts (in ppm) were referenced to tetramethylsilane (δ = 0 ppm) as an internal standard. 13C NMR spectra were recorded in CDCl3 at 100.6 MHz and were calibrated with CDCl3 (δ = 77.00 ppm) or tetramethylsilane (δ = 0 ppm). Mass spectrometry was performed using a MALDI–TOF spectrometer AB SCIEX TOF/TOF 5800 system using 3-hydroxycoumarin or α-cyano-4-hydroxycinnamic acid as a matrix in combination with KI for the ionization. Unless otherwise stated, all starting materials, catalysts, and solvents were commercially available and were used as purchased. The reaction products were purified by flash chromatography on silica gel by elution with n-hexane/EtOAc mixtures. Compounds 1a [44], 1b,c [41], 1d [45], 1e [46] and 5a–c [47] are known products and were identified by comparison of their physical and spectral data obtained with those reported in the cited references.
Procedures
Procedure for the preparation of 1-(4-(3-(benzylamino)prop-1-yn-1-yl)phenyl)ethanone (1f): To a solution of N-benzylprop-2-yn-1-amine (0.25 g, 1,72 mmol) in 3 mL of anhydrous THF were added diisopropylamine (1.2 mL, 8.6 mmol), 4-iodoacetophenone (0.505 g, 2.06 mmol), PdCl2(PPh3)2 (18,1 mg, 0.026 mmol) and CuI (9.8 mg, 0.051 mmol). The mixture was stirred at room temperature under an N2 atmosphere for 2 hours. Then the reaction mixture was diluted with ethyl acetate, washed with a solution of NH4Cl 0.5 M, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/EtOAc, 65:35 v/v) to afford 1-(4-(3-(benzylamino)prop-1-yn-1-yl)phenyl)ethanone (1f, 385.0 mg, 85%).
Typical procedure for the preparation of N-benzyl-2-iodobenzamides (2a–f): To a solution (0.25 M) of the propargylamine 1 (1 equiv) [48] in anhydrous dichloromethane (5 mL) were added at room temperature, under N2 atmosphere, 2-iodobenzoyl chloride (1.5 equiv) and anhydrous triethylamine (2 equiv). The reaction mixture was stirred under N2 until complete consumption of the starting propargylamine (monitored by TLC). Then the reaction mixture was diluted with ethyl acetate, washed with a solution of NH4Cl (0.5 M), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/EtOAc) to afford the N-benzyl-2-iodobenzamide 2.
Typical procedure for the preparation of-2-benzyl-3,4-dihydroisoquinolin-1(2H)-ones (4): preparation of (Z)-4-((4-acetylphenyl)(phenyl)methylene)-2-benzyl-3,3-dimethyl-3,4-dihydroisoquinolin-1(2H)-one (4aa): To a stirred solution (0.2 M) of N-(4-(4-acetylphenyl)-2-methylbut-3-yn-2-yl)-N-benzyl-2-iodobenzamide (2a, 100 mg, 0.19 mmol) in EtOH (1 mL), were added phenylboronic acid (3a, 34.7 mg, 0.285 mmol) and K3PO4 (120.9 mg, 0.57 mmol); after 5 min stirring at room temperature, PdCl2 (2 mg, 0.0095 mmol) was added. The mixture was stirred at 79 °C under an N2 atmosphere and stirring was continued at that temperature until complete consumption of the starting propargylamide 2a (monitored by TLC). Then the reaction mixture was cooled to room temperature, diluted with ethyl acetate (EtOAc), washed with a solution of NH4Cl (0.5 M), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/EtOAc, 70:30 v/v) to afford the dihydroisoquinolin-1(2H)-one 4aa.
General procedure for the preparation of indole-substituted dihydroisoquinolin-1(2H)-ones 6: To a stirred solution of propargylamide 2 (0.1 mmol) in MeCN (2 mL) were added 2-alkynyltrifluoroacetylanilide 5 (0.12 mmol) and K2CO3 (0.3 mmol); after 5 min stirring at room temperature Pd(PPh3)4 (0.005 mmol) was added. The mixture was stirred at 80 °C under N2 atmosphere until complete consumption of the starting propargylamide (monitored by TLC). Then the reaction mixture was cooled to room temperature, diluted with ethyl acetate, washed with a 0.5 M solution of NH4Cl, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/EtOAc, 80:20–50:50 v/v) to afford the desired dihydroisoquinolin-1(2H)-one 6.
Supporting Information
| Supporting Information File 1: Characterization of all new compounds, copies of 1H and 13C NMR spectra, 2D NOESY experiments. | ||
| Format: PDF | Size: 4.4 MB | Download |
References
-
Pettit, G. R.; Meng, Y.; Herald, D. L.; Graham, K. A. N.; Pettit, R. K.; Doubek, D. L. J. Nat. Prod. 2003, 66, 1065–1069. doi:10.1021/np0300986
Return to citation in text: [1] -
Glushkov, V. A.; Shklyaev, Y. V. Chem. Heterocycl. Compd. 2001, 37, 663–687. doi:10.1023/a:1011958810129
Return to citation in text: [1] -
Jin, Z. Nat. Prod. Rep. 2013, 30, 849–868. doi:10.1039/c3np70005d
Return to citation in text: [1] -
Bhadra, K.; Suresh Kumar, G. Mini-Rev. Med. Chem. 2010, 10, 1235–1247. doi:10.2174/13895575110091235
Return to citation in text: [1] -
Giri, P.; Suresh Kumar, G. Mini-Rev. Med. Chem. 2010, 10, 568–577. doi:10.2174/138955710791384009
Return to citation in text: [1] -
Ruchelman, A. L.; Houghton, P. J.; Zhou, N.; Liu, A.; Liu, L. F.; LaVoie, E. J. J. Med. Chem. 2005, 48, 792–804. doi:10.1021/jm049447z
Return to citation in text: [1] -
Saeed, A.; Ashraf, Z. Pharm. Chem. J. 2008, 42, 277–280. doi:10.1007/s11094-008-0105-y
Return to citation in text: [1] -
Guastavino, J. F.; Barolo, S. M.; Rossi, R. A. Eur. J. Org. Chem. 2006, 3898–3902. doi:10.1002/ejoc.200600244
Return to citation in text: [1] -
Matsui, T.; Sugiura, T.; Nakai, H.; Iguchi, S.; Shigeoka, S.; Takada, H.; Odagaki, Y.; Nagao, Y.; Ushio, Y.; Ohmoto, K.; Iwamura, H.; Yamazaki, S.; Arai, Y.; Kawamura, M. J. Med. Chem. 1992, 35, 3307–3319. doi:10.1021/jm00096a001
Return to citation in text: [1] -
Plettenburg, O.; Lorenz, K.; Goerlitzer, J.; Löhn, M. Substituted isoquinolines and their use as rho-kinase inhibitors. WO Patent WO2008077555A2, July 3, 2008.
Return to citation in text: [1] -
Li, S. W.; Nair, M. G.; Edwards, D. M.; Kisliuk, R. L.; Gaumont, Y.; Dev, I. K.; Duch, D. S.; Humphreys, J.; Smith, G. K.; Ferone, R. J. Med. Chem. 1991, 34, 2746–2754. doi:10.1021/jm00113a011
Return to citation in text: [1] -
Vinod, K. R.; Chandra, S.; Sharma, S. K. Toxicol. Mech. Methods 2010, 20, 90–95. doi:10.3109/15376510903572870
Return to citation in text: [1] -
Hudlicky, T.; Rinner, U.; Gonzalez, D.; Akgun, H.; Schilling, S.; Siengalewicz, P.; Martinot, T. A.; Pettit, G. R. J. Org. Chem. 2002, 67, 8726–8743. doi:10.1021/jo020129m
Return to citation in text: [1] -
Francis, S.; Croft, D.; Schüttelkopf, A. W.; Parry, C.; Pugliese, A.; Cameron, K.; Claydon, S.; Drysdale, M.; Gardner, C.; Gohlke, A.; Goodwin, G.; Gray, C. H.; Konczal, J.; McDonald, L.; Mezna, M.; Pannifer, A.; Paul, N. R.; Machesky, L.; McKinnon, H.; Bower, J. Bioorg. Med. Chem. Lett. 2019, 29, 1023–1029. doi:10.1016/j.bmcl.2019.01.035
Return to citation in text: [1] -
Takwale, A. D.; Jeon, Y. U.; Lee, D. H.; Kim, H. J.; Hwang, J. Y. Tetrahedron Lett. 2019, 60, 1259–1261. doi:10.1016/j.tetlet.2019.03.073
Return to citation in text: [1] -
Beng, T. K.; Langevin, S.; Farah, A. O.; Goodsell, J.; Wyatt, K. New J. Chem. 2019, 43, 5282–5286. doi:10.1039/c8nj06539j
Return to citation in text: [1] -
Xie, H.; Xing, Q.; Shan, Z.; Xiao, F.; Deng, G.-J. Adv. Synth. Catal. 2019, 361, 1896–1901. doi:10.1002/adsc.201801635
Return to citation in text: [1] -
Yang, Z.; Jie, L.; Yao, Z.; Yang, Z.; Cui, X. Adv. Synth. Catal. 2019, 361, 214–218. doi:10.1002/adsc.201801217
Return to citation in text: [1] -
Ma, S. Handbook of Cyclization Reactions; Wiley-VCH: Weinheim, Germany, 2010; Vol. 1.
Return to citation in text: [1] -
Sperger, T.; Le, C. M.; Lautens, M.; Schoenebeck, F. Chem. Sci. 2017, 8, 2914–2922. doi:10.1039/c6sc05001h
Return to citation in text: [1] -
Blouin, S.; Gandon, V.; Blond, G.; Suffert, J. Angew. Chem., Int. Ed. 2016, 55, 7208–7211. doi:10.1002/anie.201602586
Return to citation in text: [1] -
Le, C. M.; Hou, X.; Sperger, T.; Schoenebeck, F.; Lautens, M. Angew. Chem., Int. Ed. 2015, 54, 15897–15900. doi:10.1002/anie.201507883
Return to citation in text: [1] -
Milde, B.; Leibeling, M.; Pawliczek, M.; Grunenberg, J.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2015, 54, 1331–1335. doi:10.1002/anie.201408637
Return to citation in text: [1] -
Kan, S. B. J.; Anderson, E. A. Org. Lett. 2008, 10, 2323–2326. doi:10.1021/ol8007952
Return to citation in text: [1] -
Bour, C.; Blond, G.; Salem, B.; Suffert, J. Tetrahedron 2006, 62, 10567–10581. doi:10.1016/j.tet.2006.06.116
Return to citation in text: [1] -
Düfert, A.; Werz, D. B. Chem. – Eur. J. 2016, 22, 16718–16732. doi:10.1002/chem.201603044
Return to citation in text: [1] -
Peshkov, A. A.; Peshkov, V. A.; Pereshivko, O. P.; Van Hecke, K.; Kumar, R.; Van der Eycken, E. V. J. Org. Chem. 2015, 80, 6598–6608. doi:10.1021/acs.joc.5b00670
Return to citation in text: [1] -
Nandakumar, A.; Perumal, P. T. Org. Lett. 2013, 15, 382–385. doi:10.1021/ol303326g
Return to citation in text: [1] -
Charpenay, M.; Boudhar, A.; Siby, A.; Schigand, S.; Blond, G.; Suffert, J. Adv. Synth. Catal. 2011, 353, 3151–3156. doi:10.1002/adsc.201100465
Return to citation in text: [1] -
Richey, R. N.; Yu, H. Org. Process Res. Dev. 2009, 13, 315–320. doi:10.1021/op800231b
Return to citation in text: [1] -
Mondal, A.; Kundu, P.; Jash, M.; Chowdhury, C. Org. Biomol. Chem. 2018, 16, 963–980. doi:10.1039/c7ob02788e
Return to citation in text: [1] -
Arcadi, A.; Blesi, F.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Marinelli, F. J. Org. Chem. 2013, 78, 4490–4498. doi:10.1021/jo400503f
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/c2ob27125g
Return to citation in text: [1] [2] -
He, Y.-P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2020, 59, 2105–2109. doi:10.1002/anie.201914049
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Marinelli, F. Tetrahedron Lett. 1992, 33, 3915–3918. doi:10.1016/s0040-4039(00)74818-0
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G.; Lamba, D.; Marinelli, F.; Parisi, L. M. Synthesis 2003, 728–734. doi:10.1055/s-2003-38079
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F. Synlett 2000, 394–396. doi:10.1055/s-2000-6529
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Parisi, L. M. J. Org. Chem. 2005, 70, 6213–6217. doi:10.1021/jo050517z
Return to citation in text: [1] -
Arcadi, A.; Cianci, R.; Ferrara, G.; Marinelli, F. Tetrahedron 2010, 66, 2378–2383. doi:10.1016/j.tet.2010.01.096
Return to citation in text: [1] -
Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2013, 52, 12992–12996. doi:10.1002/anie.201307738
Return to citation in text: [1] -
Arcadi, A.; Aschi, M.; Chiarini, M.; Ferrara, G.; Marinelli, F. Adv. Synth. Catal. 2010, 352, 493–498. doi:10.1002/adsc.200900773
Return to citation in text: [1] [2] -
Ostrowska, S.; Rogalski, S.; Lorkowski, J.; Walkowiak, J.; Pietraszuk, C. Synlett 2018, 29, 1735–1740. doi:10.1055/s-0036-1591602
Return to citation in text: [1] -
The double bond configurations of 4fj and 6fa, as model of compounds, were unambiguously assigned as E-isomer by 1H NOESY spectroscopy. NOESY was recorded using standard Bruker pulse sequences and mixing time of 800 ms. See the section for 2D spectra in Supporting Information File 1.
Return to citation in text: [1] -
Asgari, M. S.; Mirzazadeh, R.; Larijani, B.; Rashidi Ranjbar, P.; Rahimi, R.; Mahdavi, M. Synlett 2019, 30, 1073–1076. doi:10.1055/s-0037-1611804
Return to citation in text: [1] [2] -
Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e
Return to citation in text: [1] -
Ranjan, A.; Deore, A. S.; Yerande, S. G.; Dethe, D. H. Eur. J. Org. Chem. 2017, 4130–4139. doi:10.1002/ejoc.201700603
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Carnicelli, V.; Marinelli, F. Tetrahedron 1994, 50, 437–452. doi:10.1016/s0040-4020(01)80766-3
Return to citation in text: [1] -
Montgomery, S. L.; Mangas-Sanchez, J.; Thompson, M. P.; Aleku, G. A.; Dominguez, B.; Turner, N. J. Angew. Chem., Int. Ed. 2017, 56, 10491–10494. doi:10.1002/anie.201705848
Return to citation in text: [1]
| 45. | Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e |
| 46. | Ranjan, A.; Deore, A. S.; Yerande, S. G.; Dethe, D. H. Eur. J. Org. Chem. 2017, 4130–4139. doi:10.1002/ejoc.201700603 |
| 47. | Arcadi, A.; Cacchi, S.; Carnicelli, V.; Marinelli, F. Tetrahedron 1994, 50, 437–452. doi:10.1016/s0040-4020(01)80766-3 |
| 1. | Pettit, G. R.; Meng, Y.; Herald, D. L.; Graham, K. A. N.; Pettit, R. K.; Doubek, D. L. J. Nat. Prod. 2003, 66, 1065–1069. doi:10.1021/np0300986 |
| 2. | Glushkov, V. A.; Shklyaev, Y. V. Chem. Heterocycl. Compd. 2001, 37, 663–687. doi:10.1023/a:1011958810129 |
| 3. | Jin, Z. Nat. Prod. Rep. 2013, 30, 849–868. doi:10.1039/c3np70005d |
| 10. | Plettenburg, O.; Lorenz, K.; Goerlitzer, J.; Löhn, M. Substituted isoquinolines and their use as rho-kinase inhibitors. WO Patent WO2008077555A2, July 3, 2008. |
| 34. | He, Y.-P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2020, 59, 2105–2109. doi:10.1002/anie.201914049 |
| 9. | Matsui, T.; Sugiura, T.; Nakai, H.; Iguchi, S.; Shigeoka, S.; Takada, H.; Odagaki, Y.; Nagao, Y.; Ushio, Y.; Ohmoto, K.; Iwamura, H.; Yamazaki, S.; Arai, Y.; Kawamura, M. J. Med. Chem. 1992, 35, 3307–3319. doi:10.1021/jm00096a001 |
| 35. | Arcadi, A.; Cacchi, S.; Marinelli, F. Tetrahedron Lett. 1992, 33, 3915–3918. doi:10.1016/s0040-4039(00)74818-0 |
| 36. | Cacchi, S.; Fabrizi, G.; Lamba, D.; Marinelli, F.; Parisi, L. M. Synthesis 2003, 728–734. doi:10.1055/s-2003-38079 |
| 7. | Saeed, A.; Ashraf, Z. Pharm. Chem. J. 2008, 42, 277–280. doi:10.1007/s11094-008-0105-y |
| 8. | Guastavino, J. F.; Barolo, S. M.; Rossi, R. A. Eur. J. Org. Chem. 2006, 3898–3902. doi:10.1002/ejoc.200600244 |
| 26. | Düfert, A.; Werz, D. B. Chem. – Eur. J. 2016, 22, 16718–16732. doi:10.1002/chem.201603044 |
| 27. | Peshkov, A. A.; Peshkov, V. A.; Pereshivko, O. P.; Van Hecke, K.; Kumar, R.; Van der Eycken, E. V. J. Org. Chem. 2015, 80, 6598–6608. doi:10.1021/acs.joc.5b00670 |
| 28. | Nandakumar, A.; Perumal, P. T. Org. Lett. 2013, 15, 382–385. doi:10.1021/ol303326g |
| 29. | Charpenay, M.; Boudhar, A.; Siby, A.; Schigand, S.; Blond, G.; Suffert, J. Adv. Synth. Catal. 2011, 353, 3151–3156. doi:10.1002/adsc.201100465 |
| 30. | Richey, R. N.; Yu, H. Org. Process Res. Dev. 2009, 13, 315–320. doi:10.1021/op800231b |
| 31. | Mondal, A.; Kundu, P.; Jash, M.; Chowdhury, C. Org. Biomol. Chem. 2018, 16, 963–980. doi:10.1039/c7ob02788e |
| 4. | Bhadra, K.; Suresh Kumar, G. Mini-Rev. Med. Chem. 2010, 10, 1235–1247. doi:10.2174/13895575110091235 |
| 5. | Giri, P.; Suresh Kumar, G. Mini-Rev. Med. Chem. 2010, 10, 568–577. doi:10.2174/138955710791384009 |
| 6. | Ruchelman, A. L.; Houghton, P. J.; Zhou, N.; Liu, A.; Liu, L. F.; LaVoie, E. J. J. Med. Chem. 2005, 48, 792–804. doi:10.1021/jm049447z |
| 32. | Arcadi, A.; Blesi, F.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Marinelli, F. J. Org. Chem. 2013, 78, 4490–4498. doi:10.1021/jo400503f |
| 33. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/c2ob27125g |
| 14. | Francis, S.; Croft, D.; Schüttelkopf, A. W.; Parry, C.; Pugliese, A.; Cameron, K.; Claydon, S.; Drysdale, M.; Gardner, C.; Gohlke, A.; Goodwin, G.; Gray, C. H.; Konczal, J.; McDonald, L.; Mezna, M.; Pannifer, A.; Paul, N. R.; Machesky, L.; McKinnon, H.; Bower, J. Bioorg. Med. Chem. Lett. 2019, 29, 1023–1029. doi:10.1016/j.bmcl.2019.01.035 |
| 19. | Ma, S. Handbook of Cyclization Reactions; Wiley-VCH: Weinheim, Germany, 2010; Vol. 1. |
| 13. | Hudlicky, T.; Rinner, U.; Gonzalez, D.; Akgun, H.; Schilling, S.; Siengalewicz, P.; Martinot, T. A.; Pettit, G. R. J. Org. Chem. 2002, 67, 8726–8743. doi:10.1021/jo020129m |
| 20. | Sperger, T.; Le, C. M.; Lautens, M.; Schoenebeck, F. Chem. Sci. 2017, 8, 2914–2922. doi:10.1039/c6sc05001h |
| 21. | Blouin, S.; Gandon, V.; Blond, G.; Suffert, J. Angew. Chem., Int. Ed. 2016, 55, 7208–7211. doi:10.1002/anie.201602586 |
| 22. | Le, C. M.; Hou, X.; Sperger, T.; Schoenebeck, F.; Lautens, M. Angew. Chem., Int. Ed. 2015, 54, 15897–15900. doi:10.1002/anie.201507883 |
| 23. | Milde, B.; Leibeling, M.; Pawliczek, M.; Grunenberg, J.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2015, 54, 1331–1335. doi:10.1002/anie.201408637 |
| 24. | Kan, S. B. J.; Anderson, E. A. Org. Lett. 2008, 10, 2323–2326. doi:10.1021/ol8007952 |
| 25. | Bour, C.; Blond, G.; Salem, B.; Suffert, J. Tetrahedron 2006, 62, 10567–10581. doi:10.1016/j.tet.2006.06.116 |
| 12. | Vinod, K. R.; Chandra, S.; Sharma, S. K. Toxicol. Mech. Methods 2010, 20, 90–95. doi:10.3109/15376510903572870 |
| 48. | Montgomery, S. L.; Mangas-Sanchez, J.; Thompson, M. P.; Aleku, G. A.; Dominguez, B.; Turner, N. J. Angew. Chem., Int. Ed. 2017, 56, 10491–10494. doi:10.1002/anie.201705848 |
| 11. | Li, S. W.; Nair, M. G.; Edwards, D. M.; Kisliuk, R. L.; Gaumont, Y.; Dev, I. K.; Duch, D. S.; Humphreys, J.; Smith, G. K.; Ferone, R. J. Med. Chem. 1991, 34, 2746–2754. doi:10.1021/jm00113a011 |
| 15. | Takwale, A. D.; Jeon, Y. U.; Lee, D. H.; Kim, H. J.; Hwang, J. Y. Tetrahedron Lett. 2019, 60, 1259–1261. doi:10.1016/j.tetlet.2019.03.073 |
| 16. | Beng, T. K.; Langevin, S.; Farah, A. O.; Goodsell, J.; Wyatt, K. New J. Chem. 2019, 43, 5282–5286. doi:10.1039/c8nj06539j |
| 17. | Xie, H.; Xing, Q.; Shan, Z.; Xiao, F.; Deng, G.-J. Adv. Synth. Catal. 2019, 361, 1896–1901. doi:10.1002/adsc.201801635 |
| 18. | Yang, Z.; Jie, L.; Yao, Z.; Yang, Z.; Cui, X. Adv. Synth. Catal. 2019, 361, 214–218. doi:10.1002/adsc.201801217 |
| 39. | Arcadi, A.; Cianci, R.; Ferrara, G.; Marinelli, F. Tetrahedron 2010, 66, 2378–2383. doi:10.1016/j.tet.2010.01.096 |
| 37. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F. Synlett 2000, 394–396. doi:10.1055/s-2000-6529 |
| 38. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Parisi, L. M. J. Org. Chem. 2005, 70, 6213–6217. doi:10.1021/jo050517z |
| 44. | Asgari, M. S.; Mirzazadeh, R.; Larijani, B.; Rashidi Ranjbar, P.; Rahimi, R.; Mahdavi, M. Synlett 2019, 30, 1073–1076. doi:10.1055/s-0037-1611804 |
| 41. | Arcadi, A.; Aschi, M.; Chiarini, M.; Ferrara, G.; Marinelli, F. Adv. Synth. Catal. 2010, 352, 493–498. doi:10.1002/adsc.200900773 |
| 43. | The double bond configurations of 4fj and 6fa, as model of compounds, were unambiguously assigned as E-isomer by 1H NOESY spectroscopy. NOESY was recorded using standard Bruker pulse sequences and mixing time of 800 ms. See the section for 2D spectra in Supporting Information File 1. |
| 44. | Asgari, M. S.; Mirzazadeh, R.; Larijani, B.; Rashidi Ranjbar, P.; Rahimi, R.; Mahdavi, M. Synlett 2019, 30, 1073–1076. doi:10.1055/s-0037-1611804 |
| 41. | Arcadi, A.; Aschi, M.; Chiarini, M.; Ferrara, G.; Marinelli, F. Adv. Synth. Catal. 2010, 352, 493–498. doi:10.1002/adsc.200900773 |
| 42. | Ostrowska, S.; Rogalski, S.; Lorkowski, J.; Walkowiak, J.; Pietraszuk, C. Synlett 2018, 29, 1735–1740. doi:10.1055/s-0036-1591602 |
| 33. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/c2ob27125g |
| 40. | Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2013, 52, 12992–12996. doi:10.1002/anie.201307738 |
© 2020 Nori et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)