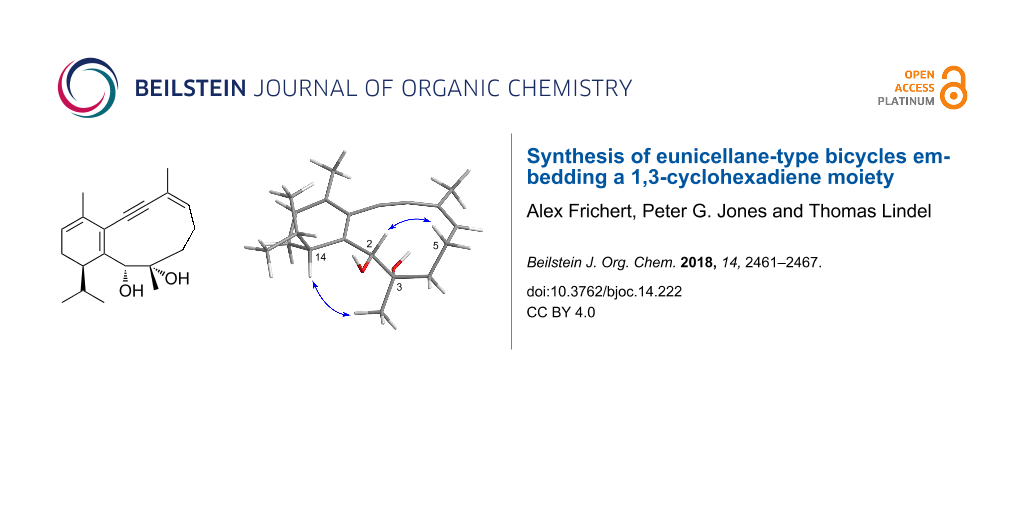
Abstract
The first synthesis of diterpenoid eunicellane skeletons incorporating a 1,3-cyclohexadiene moiety is presented. Key step is a low-valent titanium-induced pinacol cyclization that proved to be perfectly diastereoselective. Determination of the relative configuration of the diol was aided by the conversion to the diastereomer by oxidation and reduction. Conformational analysis of some of the resulting diols obtained under McMurry conditions was complicated by the presence of several conformers of similar energy. The pinacol coupling appears to start at the ketone, as indicated by the selective reduction of non-cyclizing cyclohexane systems that were synthesized from limonene oxide. The title compounds and their synthetic precursors are prone to aromatization on contact with air oxygen. Attempted synthesis of cyclohexene-containing eunicellane bicycles by elimination of water from tertiary alkynyl carbinols afforded novel allene systems. Our study may be of help towards the total synthesis of solenopodin or klysimplexin derivatives.

Graphical Abstract
Introduction
Eunicellane-type diterpenoids share an [8.4.0] bicyclic skeleton (1, Figure 1). In many cases, an additional oxygen bridge is present between positions 4 and 7, or 2 and 9, but there are also interesting eunicellanes without oxygen bridge, the majority of which has been isolated from marine corals. These comprise the solenopodins A–D (2, solenopodin D) from Solenopodium stechei [1], an unnamed eunicellane [2] and the klysimplexins Q and R (3, klysimplexin R) [3] from Eunicella labiata, cladieunicelline F from Cladiella sp. [4] and eunicellol A (4) from the soft coral Gersemia fruticose [5]. Magdalenic acid (5) was isolated from the plant Vellozia magdalenae [6]. Recently, prehydropyrene (6) was discovered as biosynthetic intermediate towards the diterpene hydropyrene from the Gram-positive bacterium Streptomyces clavuligerus [7]. The six- and ten-membered rings of eunicellane diterpenoids can be either cis or trans fused, and the ten-membered ring may contain (Z)- or (E)-double bonds. None of them has been synthesized.
![[1860-5397-14-222-1]](/bjoc/content/figures/1860-5397-14-222-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bicyclic eunicellane-type diterpenes.
Figure 1: Bicyclic eunicellane-type diterpenes.
A series of eunicellane-type bicycles containing a cyclohexene partial structure served as intermediates of the total syntheses of the sarcodictyins, the eleuthesides, and of eleutherobin [8,9]. The benzene-containing eunicellane derivative 7 (Figure 2) was obtained synthetically starting from the cembranoid sarcophytoxide from the soft coral Sarcophytum glaucum [10].
![[1860-5397-14-222-2]](/bjoc/content/figures/1860-5397-14-222-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthetic eunicellane-type compounds with benzene partial structure.
Figure 2: Synthetic eunicellane-type compounds with benzene partial structure.
We have shown that eunicellane 8 containing a benzene partial structure can be accessed efficiently via pinacol cyclization of a ketoaldehyde precursor [11]. However, compound 8 proved to be very resistant towards any attempt of partial hydrogenation of the benzene moiety. Systems embedding a cyclohexadiene ring should be more versatile, e.g., by allowing regio- and stereoselective hydrogenation and oxygenation towards partial structures present in compounds 2–6. In addition, we expected that the higher hydrogen count and the non-planarity of the envisaged 1,3-cyclohexadiene ring would allow the determination of the diastereoselectivity of the coupling step more precisely than in the case of aromatic 8 that exists as mixture of two conformers in CDCl3.
Thus, it was to be explored how open-ring cyclohexadiene precursors would be synthesized and behave under McMurry conditions, and how stable the resulting [8.4.0]bicycles would be. Normally, McMurry conditions lead to the formation of alkenes, but medium-sized ring 1,2-diols have also been obtained (TiCl4/Zn [12-17] or TiCl3/Zn-Cu [18-20]), often as mixture of diastereomers. The use of samarium diiodide to achieve the pinacol coupling was not advised, since we had observed that intially formed ketyl radicals would add to the alkyne moiety [11], even if there are examples, where this was not the case [21,22]. Access to partially unsaturated eunicellane systems could also be of interest for studies on biosynthesis and chemical interconversion [7,10].
Results and Discussion
Dihydrocarvone 9 was converted to the enolate and quenched with ethyl cyanoformate in the presence of DMPU affording an inconsequential 5.6:1 mixture of diastereomers favoring 10 (3J3H-4H 12.2 Hz vs 4.8 Hz, Scheme 1). The cyclohexadiene system of 11 was formed after deprotonation of 10 (LiHMDS) and quenching with triflic anhydride. All compounds carrying the 1,3-cyclohexadiene motif were prone to aromatization and had to be protected from contact with air and higher temperatures. Removal of solvents was performed below 21 °C and all compounds were stored under argon at −18 °C. Sonogashira coupling (PdCl2(PPh3)2) of dienol triflate 11 with alkyne 12 [11] provided C20 ester 13 in a good yield of 86%. However, after hydrolysis of the 1,3-dioxolane moiety to ketoester 14 it proved to be impossible to induce any McMurry cyclization involving the two carbonyl groups of the system. Only starting material was isolated. We had hoped that the ester might participate in the cyclization, since there is precedence of accessing medium-sized ring ethyl vinyl ethers when employing TiCl3/LiAlH4/NEt3 in DME instead of TiCl4/Zn/pyridine [23].
![[1860-5397-14-222-i1]](/bjoc/content/inline/1860-5397-14-222-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Access to ketoester 14 that did not cyclize to the ethyl vinyl ether under McMurry conditions.
Scheme 1: Access to ketoester 14 that did not cyclize to the ethyl vinyl ether under McMurry conditions.
Direct reduction of ester 14 to the aldehyde proved to be surprisingly difficult with either DIBAL-H or LiAlH4, when the alkynyl side chain was in place. Thus, we reduced the ester function of dienol triflate 11 to the alcohol (DIBAL-H, DCM, −78 °C), followed by oxidation to aldehyde 15 (IBX, Scheme 2). Fortunately, the cyclohexadiene moiety survived the oxidation conditions, which was not the case when using PCC or MnO2. Subsequent Sonogashira coupling to 16 and deprotection to 17 worked satisfyingly. As in the case of a benzene partial structure (8), it was a diol that was formed from 17 under McMurry conditions (20 equiv TiCl4, 40 equiv Zn, THF, rt). No alkene was detected.
![[1860-5397-14-222-i2]](/bjoc/content/inline/1860-5397-14-222-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the 1,3-cyclohexadiene-containing eunicellane-type [8.4.0]bicycle 18 by McMurry coupling to the diol, followed by two-step epimerization at C2.
Scheme 2: Synthesis of the 1,3-cyclohexadiene-containing eunicellane-type [8.4.0]bicycle 18 by McMurry coupli...
McMurry-type pinacol cyclizations have shown varying degrees of diastereoselectivity, which means that every single example has to be explored independently. The assignment of the configuration and preferred conformation of product 18 was conducted on the basis of NOESY NMR experiments. Our analysis was aided by the observation that the 2-epimer 19 was accessible by oxidation of 18 to the acyloin (IBX, MeCN) and subsequent reduction (LiAlH4, THF, Scheme 2). Diastereomers 18 and 19 show distinct sets of NMR signals. The largest chemical shift differences are observed for the secondary carbinol group (δ2-H 5.18, δC2 73.1 for 18 vs δ2-H 4.14, δC2 87.2 for 19). For diol 18, we observed key NOESY correlations between carbinol 2-H (δ 5.18) and one of the C5 methylene hydrogens (δ 2.37) and between 14-H (δ 2.43) and 3-CH3 (δ 1.20, Figure 3). For each of the four diastereomers with 14R configuration we found one conformation (MM2) placing 2-H and one 5-H in proximity. Moreover, those conformations are within 10 kJ/mol range of each other. However, only one of those conformations shows the required short distance between 14-H and 3-CH3, making the configuration (2R,3S,14R) probable for diastereomer 18. In the preferred conformation of 18, all non-sp3-hybridized carbon atoms of the ring system and the adjacent atoms are located almost in plane, whereas the isopropyl group and ring carbons C3 and C4 are located on opposite sides of that plane. Since the oxidation/reduction sequence has affected the configuration at C2, diastereomer 19 is assigned the configuration (2S,3S,14R). For diastereomer 19, 2-H did not show a NOESY correlation to 5-Hβ, but instead a correlation to 14-H, to the isopropyl methine hydrogen, and to 3-CH3. In addition, 3-CH3 correlates with both methylene hydrogens at C4. We found only two conformers of the (2S,3S,14R) diastereomer that meet those constraints. One of them is shown in Figure 3.
![[1860-5397-14-222-3]](/bjoc/content/figures/1860-5397-14-222-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Preferred conformations of diastereomeric diols 18 and 19 including decisive NOESY correlations.
Figure 3: Preferred conformations of diastereomeric diols 18 and 19 including decisive NOESY correlations.
The transfer of the pinacol McMurry route to the formation of eunicellane-type bicycles with two sp3 centers as bridgeheads will not be straight-forward. This became clear on our attempts to cyclize model compound 25 that was synthesized within nine steps (Scheme 3). For the synthesis of 25, we started from the known limonene oxide-derived diol 20 [24] that was hydrogenated, oxidized, and silylated at the tertiary alcohol moiety (81%). Reaction of deprotonated 21 with ethyl cyanoformate afforded cyanohydrin 22 by attack of liberated cyanide at the carbonyl carbon following the ethoxycarbonylation. The 3JHH coupling constant proved that the isopropyl and ethoxycarbonyl groups both assume an equatorial position in a chair conformation.
![[1860-5397-14-222-i3]](/bjoc/content/inline/1860-5397-14-222-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Assembly of the envisaged cyclization precursor 27.
Scheme 3: Assembly of the envisaged cyclization precursor 27.
We were not able to obtain an X-ray analysis of cyanohydrin 22, but of one diastereomer (30) of an analog where the OTBS was replaced by a methoxy group (Scheme 4, obtained by ethoxycarbonylation of 28) [25]. In agreement with the NMR data, cyanohydrin 30 adopts a chair conformation in the crystal. The 1H NMR spectra of diastereomers 29 and 30 differ characteristically regarding the chemical shift of the hydroxy proton which appeared as a sharp signal at 5.25 ppm (CDCl3) for 29 and as a broad peak at 3.00 ppm for 30. This can be explained by the presence of an intramolecular hydrogen bond that is possible only in the case of 29. Since the 1H NMR spectrum of cyanohydrin 22 exhibits a sharp hydroxy peak at 5.29 ppm, we derive the relative configuration shown in Scheme 4.
![[1860-5397-14-222-i4]](/bjoc/content/inline/1860-5397-14-222-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Structure analysis of diastereomeric cyanohydrins 29 and 30.
Scheme 4: Structure analysis of diastereomeric cyanohydrins 29 and 30.
From cyanohydrin 22 HCN was eliminated by treatment with diluted NaOH (100%, Scheme 3). The resulting ketone 23 reacted with lithiated alkyne 12 affording diastereomerically pure tertiary alcohol 24 (63%) that showed a broad hydroxy signal in the 1H NMR spectrum at 2.61 ppm. This indicates that the alkynyl side chain had been introduced from the same side as the ethoxycarbonyl group. By conversion of the ester to an aldehyde group and deprotection we obtained the subject of study, the putative cyclization precursor 25 (71%). In an orienting reaction, treatment of 25 with TiCl4/Zn did not lead to pinacol cyclization and we have evidence that the aldehyde group stayed in place and the keto group had been reduced. Installation of a TMS group at the tertiary alcohol moiety of 25 (TMSOTf, 2,6-lutidine) formed 26, which was simply reduced at the keto function on reaction with TiCl4/Zn/pyridine (27, Scheme 3) without cyclization. As before, the aldehyde had stayed in place. Interestingly, treatment of 25 with samarium diiodide afforded the primary alcohol and left the keto group unchanged. Still, no cyclization took place.
One could think that endocyclic elimination of water from 24 to the α,β-unsaturated ester would afford a cyclohexene system that would regain the ability of undergoing pinacol cyclization, because the bridge of the [8.4.0] system would become a double bond. However, standard protocols (MsCl/Et3N or p-TsOH) failed. We also synthesized the (E)-isomer of 24, compound 31, via Sonogashira coupling with the (E)-isomer [11] of 12. Chlorinated allene 32 was formed from 31 as the only product on treatment with SOCl2/pyridine, presumably after chlorosulfonation, followed by chlorine transfer and loss of SO2 (Scheme 5). There is precedence that elimination towards the cyclohexene can be a competing process [26]. The configuration of the allene moieties could not be elucidated.
![[1860-5397-14-222-i5]](/bjoc/content/inline/1860-5397-14-222-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Formation of allenes 32 and 34 from sterically crowded propargylic alcohol 31.
Scheme 5: Formation of allenes 32 and 34 from sterically crowded propargylic alcohol 31.
Interestingly, treatment of 31 with Tf2O/pyridine afforded allenyl triflone 34, with 13C NMR signals of the allene center and the triflyl-substituted carbon at δ 203.7 and 113.5 ppm, respectively. The sequence probably commences with pyridine-assisted conversion of propargylic alcohol 31 to the propargyl triflate, which looses triflate, forming propargyl cation 33. Since a triflone is formed rather than a triflate, reduction of the incoming nucleophile must have taken place, probably before incorporation. Corey and Tian reported the formation of 4-substituted N-triflyldihydropyridine derivatives on reaction of pyridine (4 equiv) with Tf2O (1.5 equiv) in the presence of aryl nucleophiles that occurred already at −30 °C within 30 min. Conversion to the corresponding pyridine derivatives was possible on treatment with KOt-Bu at −23 °C, presumably with formation of potassium triflinate [27]. In our case, pyridine was used as solvent and the reaction mixture was heated up to 70 °C. This could allow pyridine itself taking the double role of nucleophile and base, leading to the formation of pyridinium triflinate. It has also been reported that the triflinate anion can be generated from 2,6-lutidine/Tf2O [28] or Et3N/Tf2O [29]. Allenyl triflone 34 could be formed by attack of triflinate as S-nucleophile [30] at the chain carbon of propargyl cation 33. An alternative would be the attack of triflinate as O-nucleophile at the cyclohexane carbon, followed by [2,3]-sigmatropic rearrangement [31].
Conclusion
With the synthesis of the [8.4.0]bicycles 18 and 19 that contain a 1,3-cyclohexadiene partial structure, we have made progress towards the synthesis of a small group of bicyclic diterpenoids sharing the eunicellane skeleton. Closure of the ten-membered ring by pinacol cyclization proved to be possible, if the six-membered ring is either aromatic or a 1,3-cyclohexadiene, but failed for systems with two sp3 centers as bridgeheads. The ten-membered ring of benzene-containing 8 adopts two distinct major conformations in CDCl3, whereas diastereomeric diols 18 and 19 prefer only one, which were elucidated by NOESY spectroscopy. In upcoming studies we will address the synthesis of systems that contain a cyclohexene ring keeping the sp2–sp2 bridge of the product from the beginning, and of precursors in which one of the centers will be sp2- and the other sp3-hybridized. Examples of the latter have already been obtained in form of allenes 32 and 34 that will now have to be elaborated further.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectroscopical data, X-ray analysis of 30, and NMR spectra plots. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Bloor, S. J.; Schmitz, F. J.; Hossain, M. B.; Van der Helm, D. J. Org. Chem. 1992, 57, 1205–1216. doi:10.1021/jo00030a031
Return to citation in text: [1] -
Ortega, M. J.; Zubía, E.; Salvá, J. J. Nat. Prod. 1997, 60, 485–487. doi:10.1021/np970026c
Return to citation in text: [1] -
Chen, B.-W.; Chao, C.-H.; Su, J.-H.; Tsai, C.-W.; Wang, W.-H.; Wen, Z.-H.; Huang, C.-Y.; Sung, P.-J.; Wu, Y.-C.; Sheu, J.-H. Org. Biomol. Chem. 2011, 9, 834–844. doi:10.1039/c0ob00351d
Return to citation in text: [1] -
Chen, Y.-H.; Tai, C.-Y.; Su, Y.-D.; Chang, Y.-C.; Lu, M.-C.; Weng, C.-F.; Su, J.-H.; Hwang, T.-L.; Wu, Y.-C.; Sung, P.-J. Mar. Drugs 2011, 9, 934–943. doi:10.3390/md9060934
Return to citation in text: [1] -
Angulo-Preckler, C.; Genta-Jouve, G.; Mahajan, N.; de la Cruz, M.; de Pedro, N.; Reyes, F.; Iken, K.; Avila, C.; Thomas, O. P. J. Nat. Prod. 2016, 79, 1132–1136. doi:10.1021/acs.jnatprod.6b00040
Return to citation in text: [1] -
Pinto, A. C.; Pizzolatti, M. G.; de A. Epifanio, R.; Frankmölle, W.; Fenical, W. Tetrahedron 1997, 53, 2005–2012. doi:10.1016/s0040-4020(96)01183-0
Return to citation in text: [1] -
Rinkel, J.; Rabe, P.; Chen, X.; Köllner, T. G.; Chen, F.; Dickschat, J. S. Chem. – Eur. J. 2017, 23, 10501–10505. doi:10.1002/chem.201702704
Return to citation in text: [1] [2] -
Nicolaou, K. C.; Ohshima, T.; Hosokawa, S.; van Delft, F. L.; Vourloumis, D.; Xu, J. Y.; Pfefferkorn, J.; Kim, S. J. Am. Chem. Soc. 1998, 120, 8674–8680. doi:10.1021/ja9810639
Return to citation in text: [1] -
Castoldi, D.; Caggiano, L.; Panigada, L.; Sharon, O.; Costa, A. M.; Gennari, C. Angew. Chem. 2005, 117, 594–597. doi:10.1002/ange.200461767
Angew. Chem., Int. Ed. 2005, 44, 588–591. doi:10.1002/anie.200461767
Return to citation in text: [1] -
Nii, K.; Tagami, K.; Matsuoka, K.; Munakata, T.; Ooi, T.; Kusumi, T. Org. Lett. 2006, 8, 2957–2960. doi:10.1021/ol0608404
Return to citation in text: [1] [2] -
Al Batal, M.; Jones, P. G.; Lindel, T. Eur. J. Org. Chem. 2013, 2533–2536. doi:10.1002/ejoc.201300178
Return to citation in text: [1] [2] [3] [4] -
Kato, N.; Kataoka, H.; Ohbuchi, S.; Tanaka, S.; Takeshita, H. J. Chem. Soc., Chem. Commun. 1988, 354–356. doi:10.1039/c39880000354
Return to citation in text: [1] -
Swindell, C. S.; Fan, W. J. Org. Chem. 1996, 61, 1109–1118. doi:10.1021/jo9519367
Return to citation in text: [1] -
Yamato, T.; Fujita, K.; Okuyama, K.-i.; Tsuzuki, H. New J. Chem. 2000, 24, 221–228. doi:10.1039/b001145m
Return to citation in text: [1] -
Sergeeva, E. V.; Rozenberg, V. I.; Antonov, D. Y.; Vorontsov, E. V.; Starikova, Z. A.; Fedyanin, I. V.; Hopf, H. Chem. – Eur. J. 2005, 11, 6944–6961. doi:10.1002/chem.200500413
Return to citation in text: [1] -
Saisyo, T.; Shiino, M.; Shimizu, T.; Paudel, A.; Yamato, T. J. Chem. Res. 2008, 479–483. doi:10.3184/030823408x338701
Return to citation in text: [1] -
Castagnolo, D.; Contemori, L.; Maccari, G.; Avramova, S. I.; Neri, A.; Sgaragli, G.; Botta, M. ACS Med. Chem. Lett. 2010, 1, 416–421. doi:10.1021/ml100118k
Return to citation in text: [1] -
Nicolaou, K. C.; Yang, Z.; Liu, J.-J.; Nantermet, P. G.; Claiborne, C. F.; Renaud, J.; Guy, R. K.; Shibayama, K. J. Am. Chem. Soc. 1995, 117, 645–652. doi:10.1021/ja00107a008
Return to citation in text: [1] -
Ferri, F.; Brückner, R.; Herges, R. New J. Chem. 1998, 22, 531–546. doi:10.1039/a709205i
Return to citation in text: [1] -
Williams, D. R.; Heidebrecht, R. W. J. Am. Chem. Soc. 2003, 125, 1843–1850. doi:10.1021/ja0279803
Return to citation in text: [1] -
Nicolaou, K. C.; Sorensen, E. J.; Discordia, R.; Hwang, C.-K.; Minto, R. E.; Bharucha, K. N.; Bergman, R. G. Angew. Chem. 1992, 104, 1094–1096. doi:10.1002/ange.19921040838
Return to citation in text: [1] -
Comanita, B. M.; Heuft, M. A.; Rietveld, T.; Fallis, A. G. Isr. J. Chem. 2000, 40, 241–253. doi:10.1560/7j4f-d8bg-3fqr-e3td
Return to citation in text: [1] -
McMurry, J. E.; Miller, D. D. J. Am. Chem. Soc. 1983, 105, 1660–1661. doi:10.1021/ja00344a045
Return to citation in text: [1] -
Blair, M.; Andrews, P. C.; Fraser, B. H.; Forsyth, C. M.; Junk, P. C.; Massi, M.; Tuck, K. L. Synthesis 2007, 1523–1527. doi:10.1055/s-2007-966033
Return to citation in text: [1] -
Crystallographic Data Centre as supplementary publications no. CCDC-1853640. Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Masters, K.-S.; Flynn, B. L. J. Org. Chem. 2008, 73, 8081–8084. doi:10.1021/jo800682n
Return to citation in text: [1] -
Corey, E. J.; Tian, Y. Org. Lett. 2005, 7, 5535–5537. doi:10.1021/ol052476z
Return to citation in text: [1] -
Ambrose, M. G.; Binkley, R. W. J. Org. Chem. 1983, 48, 674–677. doi:10.1021/jo00153a011
Return to citation in text: [1] -
Netscher, T.; Bohrer, P. Tetrahedron Lett. 1996, 37, 8359–8362. doi:10.1016/0040-4039(96)01930-2
Return to citation in text: [1] -
Hendrickson, J. B.; Giga, A.; Wareing, J. J. Am. Chem. Soc. 1974, 96, 2275–2276. doi:10.1021/ja00814a061
Return to citation in text: [1] -
Braverman, S.; Pechenick, T.; Zafrani, Y. Tetrahedron Lett. 2001, 42, 1391–1393. doi:10.1016/s0040-4039(00)02255-3
Return to citation in text: [1]
| 1. | Bloor, S. J.; Schmitz, F. J.; Hossain, M. B.; Van der Helm, D. J. Org. Chem. 1992, 57, 1205–1216. doi:10.1021/jo00030a031 |
| 5. | Angulo-Preckler, C.; Genta-Jouve, G.; Mahajan, N.; de la Cruz, M.; de Pedro, N.; Reyes, F.; Iken, K.; Avila, C.; Thomas, O. P. J. Nat. Prod. 2016, 79, 1132–1136. doi:10.1021/acs.jnatprod.6b00040 |
| 7. | Rinkel, J.; Rabe, P.; Chen, X.; Köllner, T. G.; Chen, F.; Dickschat, J. S. Chem. – Eur. J. 2017, 23, 10501–10505. doi:10.1002/chem.201702704 |
| 10. | Nii, K.; Tagami, K.; Matsuoka, K.; Munakata, T.; Ooi, T.; Kusumi, T. Org. Lett. 2006, 8, 2957–2960. doi:10.1021/ol0608404 |
| 4. | Chen, Y.-H.; Tai, C.-Y.; Su, Y.-D.; Chang, Y.-C.; Lu, M.-C.; Weng, C.-F.; Su, J.-H.; Hwang, T.-L.; Wu, Y.-C.; Sung, P.-J. Mar. Drugs 2011, 9, 934–943. doi:10.3390/md9060934 |
| 11. | Al Batal, M.; Jones, P. G.; Lindel, T. Eur. J. Org. Chem. 2013, 2533–2536. doi:10.1002/ejoc.201300178 |
| 3. | Chen, B.-W.; Chao, C.-H.; Su, J.-H.; Tsai, C.-W.; Wang, W.-H.; Wen, Z.-H.; Huang, C.-Y.; Sung, P.-J.; Wu, Y.-C.; Sheu, J.-H. Org. Biomol. Chem. 2011, 9, 834–844. doi:10.1039/c0ob00351d |
| 11. | Al Batal, M.; Jones, P. G.; Lindel, T. Eur. J. Org. Chem. 2013, 2533–2536. doi:10.1002/ejoc.201300178 |
| 2. | Ortega, M. J.; Zubía, E.; Salvá, J. J. Nat. Prod. 1997, 60, 485–487. doi:10.1021/np970026c |
| 21. | Nicolaou, K. C.; Sorensen, E. J.; Discordia, R.; Hwang, C.-K.; Minto, R. E.; Bharucha, K. N.; Bergman, R. G. Angew. Chem. 1992, 104, 1094–1096. doi:10.1002/ange.19921040838 |
| 22. | Comanita, B. M.; Heuft, M. A.; Rietveld, T.; Fallis, A. G. Isr. J. Chem. 2000, 40, 241–253. doi:10.1560/7j4f-d8bg-3fqr-e3td |
| 10. | Nii, K.; Tagami, K.; Matsuoka, K.; Munakata, T.; Ooi, T.; Kusumi, T. Org. Lett. 2006, 8, 2957–2960. doi:10.1021/ol0608404 |
| 12. | Kato, N.; Kataoka, H.; Ohbuchi, S.; Tanaka, S.; Takeshita, H. J. Chem. Soc., Chem. Commun. 1988, 354–356. doi:10.1039/c39880000354 |
| 13. | Swindell, C. S.; Fan, W. J. Org. Chem. 1996, 61, 1109–1118. doi:10.1021/jo9519367 |
| 14. | Yamato, T.; Fujita, K.; Okuyama, K.-i.; Tsuzuki, H. New J. Chem. 2000, 24, 221–228. doi:10.1039/b001145m |
| 15. | Sergeeva, E. V.; Rozenberg, V. I.; Antonov, D. Y.; Vorontsov, E. V.; Starikova, Z. A.; Fedyanin, I. V.; Hopf, H. Chem. – Eur. J. 2005, 11, 6944–6961. doi:10.1002/chem.200500413 |
| 16. | Saisyo, T.; Shiino, M.; Shimizu, T.; Paudel, A.; Yamato, T. J. Chem. Res. 2008, 479–483. doi:10.3184/030823408x338701 |
| 17. | Castagnolo, D.; Contemori, L.; Maccari, G.; Avramova, S. I.; Neri, A.; Sgaragli, G.; Botta, M. ACS Med. Chem. Lett. 2010, 1, 416–421. doi:10.1021/ml100118k |
| 8. | Nicolaou, K. C.; Ohshima, T.; Hosokawa, S.; van Delft, F. L.; Vourloumis, D.; Xu, J. Y.; Pfefferkorn, J.; Kim, S. J. Am. Chem. Soc. 1998, 120, 8674–8680. doi:10.1021/ja9810639 |
| 9. |
Castoldi, D.; Caggiano, L.; Panigada, L.; Sharon, O.; Costa, A. M.; Gennari, C. Angew. Chem. 2005, 117, 594–597. doi:10.1002/ange.200461767
Angew. Chem., Int. Ed. 2005, 44, 588–591. doi:10.1002/anie.200461767 |
| 18. | Nicolaou, K. C.; Yang, Z.; Liu, J.-J.; Nantermet, P. G.; Claiborne, C. F.; Renaud, J.; Guy, R. K.; Shibayama, K. J. Am. Chem. Soc. 1995, 117, 645–652. doi:10.1021/ja00107a008 |
| 19. | Ferri, F.; Brückner, R.; Herges, R. New J. Chem. 1998, 22, 531–546. doi:10.1039/a709205i |
| 20. | Williams, D. R.; Heidebrecht, R. W. J. Am. Chem. Soc. 2003, 125, 1843–1850. doi:10.1021/ja0279803 |
| 7. | Rinkel, J.; Rabe, P.; Chen, X.; Köllner, T. G.; Chen, F.; Dickschat, J. S. Chem. – Eur. J. 2017, 23, 10501–10505. doi:10.1002/chem.201702704 |
| 6. | Pinto, A. C.; Pizzolatti, M. G.; de A. Epifanio, R.; Frankmölle, W.; Fenical, W. Tetrahedron 1997, 53, 2005–2012. doi:10.1016/s0040-4020(96)01183-0 |
| 11. | Al Batal, M.; Jones, P. G.; Lindel, T. Eur. J. Org. Chem. 2013, 2533–2536. doi:10.1002/ejoc.201300178 |
| 25. | Crystallographic Data Centre as supplementary publications no. CCDC-1853640. Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/data_request/cif. |
| 23. | McMurry, J. E.; Miller, D. D. J. Am. Chem. Soc. 1983, 105, 1660–1661. doi:10.1021/ja00344a045 |
| 24. | Blair, M.; Andrews, P. C.; Fraser, B. H.; Forsyth, C. M.; Junk, P. C.; Massi, M.; Tuck, K. L. Synthesis 2007, 1523–1527. doi:10.1055/s-2007-966033 |
| 31. | Braverman, S.; Pechenick, T.; Zafrani, Y. Tetrahedron Lett. 2001, 42, 1391–1393. doi:10.1016/s0040-4039(00)02255-3 |
| 29. | Netscher, T.; Bohrer, P. Tetrahedron Lett. 1996, 37, 8359–8362. doi:10.1016/0040-4039(96)01930-2 |
| 30. | Hendrickson, J. B.; Giga, A.; Wareing, J. J. Am. Chem. Soc. 1974, 96, 2275–2276. doi:10.1021/ja00814a061 |
| 28. | Ambrose, M. G.; Binkley, R. W. J. Org. Chem. 1983, 48, 674–677. doi:10.1021/jo00153a011 |
| 11. | Al Batal, M.; Jones, P. G.; Lindel, T. Eur. J. Org. Chem. 2013, 2533–2536. doi:10.1002/ejoc.201300178 |
| 26. | Masters, K.-S.; Flynn, B. L. J. Org. Chem. 2008, 73, 8081–8084. doi:10.1021/jo800682n |
© 2018 Frichert et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)