Abstract
This account provides an overview of recent work, including our own contribution dealing with Suzuki–Miyaura cross coupling in combination with metathesis (or vice-versa). Several cyclophanes, polycycles, macrocycles, spirocycles, stilbenes, biaryls, and heterocycles have been synthesized by employing a combination of Suzuki cross-coupling and metathesis. Various popular reactions such as Diels–Alder reaction, Claisen rearrangement, cross-metathesis, and cross-enyne metathesis are used. The synergistic combination of these powerful reactions is found to be useful for the construction of complex targets and fulfill synthetic brevity.

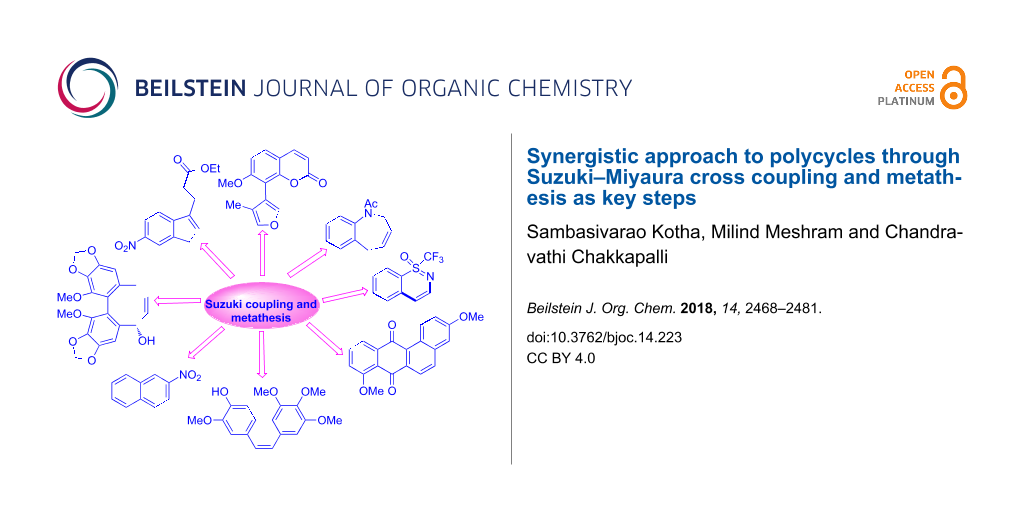
Graphical Abstract
Introduction
Transition-metal catalysts are used in metathesis and cross-coupling reactions. Such advances have opened the door for efficient construction of C–C bonds in organic synthesis. These catalysts tolerate diverse functional groups and the reaction occurs under mild reaction conditions. Among different metathetic processes, ring-closing metathesis (RCM) [1-6] is of a greater interest than cross-metathesis (CM). It is a widely used protocol for the synthesis of unsaturated cyclic systems [7]. Palladium-catalyzed Suzuki–Miyaura (SM) cross-coupling reaction is also considered as one of the most versatile methods for C–C bond formation [8-12]. Application of a wide range of organometallic reagents (e.g., organoboron reagents) are possible due to their commercial availability. Owing to the mild reaction conditions and ease of handling of organoboron reagents [13-17] have propelled the growth of the SM cross coupling. A synergistic combination of these two elegant methods (i.e., SM coupling and metathesis) [18] was found to increase the synthetic efficiency of complex targets (e.g., macrocycles [19-22], oligomers [23,24], polycyclic ethers [25], heterocycles [26], nonbenzenoid aromatics [27], and spirocycles [28,29]) by decreasing the number of steps. Different metathesis catalysts used in this study are shown in Figure 1.
![[1860-5397-14-223-1]](/bjoc/content/figures/1860-5397-14-223-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Various catalysts used for metathesis reactions.
Figure 1: Various catalysts used for metathesis reactions.
Review
Annulation
Grela and co-workers [30] demonstrated a useful protocol to build indene derivatives by employing SM coupling and RCM in sequence. To this end, the SM coupling of triflate 7 was accomplished by using pinacol boronic ester 8 in the presence of a palladium catalyst to give the cross-coupling product 9 (75%). Later on, exposure of the diolefinic precursor 9 to [Ru-2] catalyst 5 gave the ring-closure product 10 in quantitative yield (Scheme 1).
![[1860-5397-14-223-i1]](/bjoc/content/inline/1860-5397-14-223-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: SM coupling and RCM protocol to substituted indene derivative 10.
Scheme 1: SM coupling and RCM protocol to substituted indene derivative 10.
A sequential usage of SM cross coupling and RCM was responsible to construct various naphthalene derivatives such as 15 [31]. The SM coupling product 3,4-diallylbenzene derivative 13 (90%) was obtained from diiodobenzene 11 using allylboronate ester 12 via a SM-type allylation sequence [32]. Next, compound 13 was exposed to Grubbs 1st generation (G-I) catalyst 1 to effect the ring-closure to produce tetrahydronaphthalene derivative 14 (92%). Subsequently, aromatization of compound 14 was accomplished with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to generate nitronaphthalene 15 (60%, Scheme 2).
![[1860-5397-14-223-i2]](/bjoc/content/inline/1860-5397-14-223-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of polycycles via SM and RCM approach.
Scheme 2: Synthesis of polycycles via SM and RCM approach.
Due to their useful biological activity and intricate structural features of angucyclines such as 16–19 (Figure 2), several approaches have been reported for their assembly. In this context, de Koning and co-workers [33] demonstrated an efficient route for the construction of the benz[a]anthracene structural unit by employing SM cross coupling followed by RCM sequence. Treatment of the bromonaphthalene derivative 20 with (2-formyl-4-methoxyphenyl)boronic acid (21) in the presence of a palladium catalyst generated the cross-coupling product 22 (72%). Next, aldehyde 22 was subjected to Wittig olefination to provide the corresponding alkene 23 (69%), which on subsequent treatment with KOt-Bu in THF gave the isomerized product 24 (73%). Later, RCM of isomerized olefin 24 with the help of G-II catalyst offered the ring-closure product 25 (84%). Finally, CAN oxidation gave the desired tetracyclic compound 26 in 84% yield (Scheme 3).
![[1860-5397-14-223-2]](/bjoc/content/figures/1860-5397-14-223-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-223-i3]](/bjoc/content/inline/1860-5397-14-223-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: SM coupling and RCM protocol to the benz[a]anthracene skeleton 26.
Scheme 3: SM coupling and RCM protocol to the benz[a]anthracene skeleton 26.
Spirocycles
In another event, an efficient approach to spirocyclopentane derivatives has been described, where the combination of RCM and SM coupling was employed [34]. In this respect, the key building block 29 was derived by employing a sequential O-allylation and CR, then again O-allylation, and CR [35] starting with a commercially available 6-bromo-2-naphthol (27). Subsequently, the diallyl derivative 29 was exposed to G-II catalyst 2 to deliver a ring-closure product 30 (83%). Finally, the spiro compound 30 was subjected to the SM coupling using two different boronic acids to produce the aryl substituted spiro compounds such as 31 (96%) and 32 (79%) (Scheme 4).
![[1860-5397-14-223-i4]](/bjoc/content/inline/1860-5397-14-223-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of substituted spirocycles via RCM and SM sequence.
Scheme 4: Synthesis of substituted spirocycles via RCM and SM sequence.
Along similar lines, we have also demonstrated the synthesis of bis-spirocycles such as 37 by adopting a double RCM sequence followed by SM coupling [36]. The key precursor 34 was assembled from a commercially available tetralone 33 via tetraallylation sequence. Then, tetraallyl derivative 34 was subjected to RCM with the aid of the G-I catalyst 1 to furnish the bis-spirocyclic compound 35 (90%). Next, the cyclized product 35 was subjected to SM coupling using phenylboronic acid (36) to afford the cross-coupling product 37 (97%, Scheme 5).
![[1860-5397-14-223-i5]](/bjoc/content/inline/1860-5397-14-223-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of highly functionalized bis-spirocyclic derivative 37.
Scheme 5: Synthesis of highly functionalized bis-spirocyclic derivative 37.
In another instance, a simple synthetic approach to spiro-fluorene derivative 41 was described involving a serial usage of RCM and SM coupling [37]. To this end, bromofluorene 38 was reacted with allyl bromide (28) in the presence of 50% NaOH to deliver the expected 9,9′-diallylfluorene derivative 39 (90%). Next, diallyl compound 39 was subjected to RCM with the aid of the G-I catalyst 1 to furnish a ring-closure product, spirofluorene derivative 40 (93%). Later, the dibromide 40 was subjected to SM coupling in the presence of phenylboronic acid (36) to generate the new spirofluorene 41 (88%, Scheme 6).
![[1860-5397-14-223-i6]](/bjoc/content/inline/1860-5397-14-223-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of spirofluorene derivatives via RCM and SM coupling sequence.
Scheme 6: Synthesis of spirofluorene derivatives via RCM and SM coupling sequence.
Interestingly, highly substituted truxene derivatives 45–49 were also synthesized by applying the RCM and SM coupling protocol (Scheme 7).
![[1860-5397-14-223-i7]](/bjoc/content/inline/1860-5397-14-223-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of truxene derivatives via RCM and SM coupling.
Scheme 7: Synthesis of truxene derivatives via RCM and SM coupling.
Heterocycles
Couture and co-workers [38] demonstrated an elegant approach to highly substituted isoquinolones (e.g., 57a–d, Scheme 8) by employing a SM coupling followed by RCM. To this end, they started with o-vinylbenzoic acid and it was transformed to the benzamide derivatives 50 by employing a four-step synthetic sequence. Later, compound 50 was treated with KHMDS in THF at −78 °C to produce enolate 51. Further, it was reacted with diphenyl chlorophosphate to generate vinyl phosphate 52, which was subjected to SM coupling in the presence of different 2-formylboronic acids 53 with the aid of the Pd(PPh3)4 catalyst to provide the respective coupling products 54a–d (72–87%). Next, exposure of the diolefins 54a–d to G-II catalyst 2 delivered ring-closure products, isoquinolones 55a–d (76–88%). Finally, the cyclized products 55a–d were converted into the corresponding indeno[1,2-c]isoquinolin-5,11-diones 57a–d (73–85%) through cyclization with the aid of HCl followed by pyridinium dichromate (PDC) oxidation (Scheme 8).
![[1860-5397-14-223-i8]](/bjoc/content/inline/1860-5397-14-223-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of substituted isoquinoline derivative via SM and RCM protocol.
Scheme 8: Synthesis of substituted isoquinoline derivative via SM and RCM protocol.
Schmidt and co-workers [39] described an efficient route involving RCM and SM coupling towards the synthesis of 8-aryl-substituted coumarin 64, a natural product isolated from the plant Galipea panamensis. To this end, aldehydes 58a,b were subjected to a Wittig olefination followed by condensation with acryloyl chloride (60) to generate the corresponding diolefinic substrates such as 61a (70%) and 61b (65%). Later, these diolefins 61a,b were subjected to RCM with the aid of G-II catalyst 2 to furnish the respective ring-closure products 62a (98%) and 62b (97%). Finally, SM coupling of 8-halo-7-methoxycoumarins 62a,b with (4-methylfuran-3-yl)boronic acid (63) delivered the cross-coupling product 64 (Scheme 9).
![[1860-5397-14-223-i9]](/bjoc/content/inline/1860-5397-14-223-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis to 8-aryl substituted coumarin 64 via RCM and SM sequence.
Scheme 9: Synthesis to 8-aryl substituted coumarin 64 via RCM and SM sequence.
In another event, Magnier and co-workers [40] described a simple synthetic route to sulfoximines by adopting SM coupling and RCM as key steps. In this respect, SM coupling of sulfoximine 65 with potassium vinyltrifluoroborate (66) in the presence of a palladium catalyst produced vinyl sulfoximine derivative 67 (73%). Next, N-alkenylation of sulfoximine 67 was accomplished with Z-vinyl bromide (68) to generate diolefinic substrate 69 (86%). Finally, diolefin 69 was exposed to Hoveyda–Grubbs 2nd generation catalyst (HG-II) 3 to deliver the cyclic sulfoximine 70 in 98% yield (Scheme 10).
![[1860-5397-14-223-i10]](/bjoc/content/inline/1860-5397-14-223-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of cyclic sulfoximine 70 via SM and RCM as key steps.
Scheme 10: Synthesis of cyclic sulfoximine 70 via SM and RCM as key steps.
Additionally, we also demonstrated a sequential usage of SM coupling and the RCM protocol to construct 1-benzazepine derivative 75 [41]. To this end, iodoacetanilide 71 was subjected to SM coupling in the presence of allyboronate ester 12 to give ortho-allylacetanilide (72), which was further modified by N-allylation with allyl bromide (28) to offer a mixture of diallyl compound 73a (82%) and isomerized product 73b (8%). Next, exposure of the diallyl derivative 73a to G-II catalyst 2 yielded the cyclized product 74 (72%). Eventually, hydrogenation of the RCM product 74 was achieved with H2, Pd/C conditions to give the saturated 2,3,4,5-tetrahydro-1-benzazepine 75 in 81% yield (Scheme 11).
![[1860-5397-14-223-i11]](/bjoc/content/inline/1860-5397-14-223-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of 1-benzazepine derivative 75 via SM and RCM as key steps.
Scheme 11: Synthesis of 1-benzazepine derivative 75 via SM and RCM as key steps.
Naphthoxepine derivatives play an important role as cosmetics and as pharmaceutical ingredients. Therefore, we conceived a simple approach, where the SM coupling and RCM were employed as critical steps [42,43]. Our journey begin with O-allylation of β-naphthol 76 by using allyl bromide (28) to give O-allyl derivative 77. Then, Claisen rearrangement (CR) of 77 under microwave irradiation (MWI) conditions on a silica gel support followed by O-allylation of the resulting CR product furnished diallyl compound 78. Treatment of diallyl compound 78 with G-I catalyst 1 delivered the expected naphthoxepine derivative 79 (96%). Next, Suzuki coupling of 79 with diverse arylboronic acids (e.g., phenylboronic acid (36)) gave a highly substituted naphthoxepine derivative 80 (90%) (Scheme 12).
![[1860-5397-14-223-i12]](/bjoc/content/inline/1860-5397-14-223-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of naphthoxepine derivative 79 via RCM followed by SM coupling.
Scheme 12: Synthesis of naphthoxepine derivative 79 via RCM followed by SM coupling.
Stilbene derivatives
Hoveyda and co-workers [44] reported the synthesis of Z-(pinacolato)allylboron and Z-(pinacolato)alkenylboron derivatives via CM by using Mo complex 6. In this regard, they assembled stilbene derivative 85 as an antitumor agent by a two-step strategy that involve catalytic CM and SM coupling. To this end, the Z-selective CM of a styrene derivative (e.g., 81) with vinyl-B(pin) 82 was realized in the presence of Mo complex 6 to provide a highly substituted vinyl-B(pin) 83 (73%) with excellent selectivity (96:4 Z:E). Further, vinylboron compound 83 was subjected to SM coupling with a suitable partner (e.g., 84) to afford the stilbene derivative 85 (96:4 Z:E) in 74% yield (Scheme 13).
![[1860-5397-14-223-i13]](/bjoc/content/inline/1860-5397-14-223-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Sequential CM and SM coupling approach to Z-stilbene derivative 85.
Scheme 13: Sequential CM and SM coupling approach to Z-stilbene derivative 85.
Majchrzak and co-workers [45] demonstrated a synergistic approach involving SM cross coupling and CM to synthesize various substituted trans-stilbene derivatives 89–95 stereoselectively. In this context, 4-vinylphenylboronic acid (86) was subjected to SM coupling using diverse bromoarenes 87a–g in the presence of [Pd(η2-dba){P(o-tolyl)3}2] catalyst to obtain the cross-coupling products 88a–g (81–96%). Finally, exposure of olefins 88a–g to G-II catalyst 2 in CH2Cl2 led to the formation of the respective trans-stilbene derivatives 89–95 in high yields (Scheme 14). It is worth mentioning that the loading of only 0.0001 mol % catalyst can effect a CM in an efficient manner.
![[1860-5397-14-223-i14]](/bjoc/content/inline/1860-5397-14-223-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of substituted trans-stilbene derivatives via SM coupling and RCM.
Scheme 14: Synthesis of substituted trans-stilbene derivatives via SM coupling and RCM.
Biaryl derivatives
In view of the interesting properties of biaryl derivatives, we have identified a three-step sequence, which involve cross-enyne metathesis (CEM), DA reaction followed by SM coupling [46]. To this end, acetylene derivatives 96a,b were subjected to CEM with G-I catalyst 1 under ethylene, which resulted in the formation of the dienes 97a (63%) and 97b (83%, Scheme 15). Further, treatment of dienes 97a,b with dimethyl acetylenedicarboxylate (DMAD, 98) separately delivered the corresponding cycloadducts. Subsequently, aromatization was achieved by using DDQ to give biaryl products 99a,b. Further, aryl halides 99a,b were subjected to SM coupling by employing various boronic acids (e.g., 4-formylphenylboronic acid (100) to produce biaryl derivative 101 (80% from 99a and 74% from 99b).
![[1860-5397-14-223-i15]](/bjoc/content/inline/1860-5397-14-223-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of biaryl derivatives via sequential EM, DA followed by SM coupling.
Scheme 15: Synthesis of biaryl derivatives via sequential EM, DA followed by SM coupling.
Very recently, Suresh Babu and co-workers [47] demonstrated a new route to construct the dibenzocyclooctadiene lignan core of the natural product schisandrene via SM coupling and RCM as key steps. In this context, the SM reaction of boronic acid 102 with bromoaldehyde 103 in the presence of Pd2(dba)3 and the S-Phos ligand provided the cross-coupling product 104 (82%). Later, it was transformed into the allyl substrate 105 by following a three-step sequence. Afterwards, the aldehyde 105 was treated with vinylmagnesium bromide (106) to furnish diallyl derivative 107 (85%). Next, diolefinic substrate 107 was exposed to G-II catalyst 2 to furnish the ring-closure product 108 (89%). Then, MnO2 oxidation of compound 108 offered the keto derivative in 90% yield. Corey–Bakshi–Shibata (CBS) reduction of the resulting keto derivative produced the hydroxy compound 109 (85%, ee 98%). Eventually, hydroxy olefin 109 was subjected to Sharpless asymmetric epoxidation to generate the corresponding epoxide 110. Unfortunately, generation of epoxide was not realized (Scheme 16).
![[1860-5397-14-223-i16]](/bjoc/content/inline/1860-5397-14-223-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of the dibenzocyclooctadiene core of schisandrene.
Scheme 16: Synthesis of the dibenzocyclooctadiene core of schisandrene.
Macrocycles
To develop new synthetic strategies to various cyclophanes, we conceived a sequential usage of the SM coupling and RCM as key steps [48,49]. In this context, the required dialdehyde 113 (80%) was prepared via a SM coupling of the dibromo compound 112 with 4-formylphenylboronic acid (100). Treatment of dialdehyde 113 with allyl bromide (28) in the presence of indium powder furnished the RCM precursor 114. Under the influence of the G-II catalyst 2 RCM of diolefinic compound 114 was realized. Then, the cyclized product was subjected to the oxidation sequence with pyridinium chlorochromate (PCC) to generate cylophane derivative 115 in 75% yield (Scheme 17).
![[1860-5397-14-223-i17]](/bjoc/content/inline/1860-5397-14-223-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of cyclophane 115 via SM coupling and RCM as key steps.
Scheme 17: Synthesis of cyclophane 115 via SM coupling and RCM as key steps.
Similarly, treatment of dialdehyde 113 with a freshly prepared Grignard reagent derived from 4-bromobut-1-ene (116) afforded dialkenyl substrate 117, which was subjected to RCM with the aid of G-II catalyst 2 to produce a mixture of products 119 and 121 in combined 47% yield. It should be noted that the resulting product 121 was obtained through isomerization of the terminal double bond followed by RCM. Later, oxidation of diols 119 and 121 was accomplished with PCC to provide the corresponding diones 120 (79%) and 122 (76%) with trans geometry. The stereochemistry was confirmed on the basis of the coupling constant (J = 15.0 Hz, 1H NMR spectrum) of the olefinic protons (Scheme 18).
![[1860-5397-14-223-i18]](/bjoc/content/inline/1860-5397-14-223-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of cyclophane 120 and 122 via SM coupling and RCM as key steps.
Scheme 18: Synthesis of cyclophane 120 and 122 via SM coupling and RCM as key steps.
A variety of macrocycles were synthesized through SM cross coupling followed by RCM as key steps [50]. To this end, dibromo compound 123 was subjected to diallylation by using allylboronate ester 12 to form the diallyl derivative 124 (73%). Treatment of compound 124 with G-I catalyst 1 gave unsaturated dimer 126 (30%) and monomer 125 (15%). Subsequently, hydrogenation of compounds 126 and 125 was accomplished with H2 under Pd/C catalysis conditions to afford the respective saturated macrocyclic products 127 (80%) and 128 (90%). Since the small ring cyclophane is highly strained, compound 125 was formed as a minor product (Scheme 19).
![[1860-5397-14-223-i19]](/bjoc/content/inline/1860-5397-14-223-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of cyclophanes via SM and RCM.
Scheme 19: Synthesis of cyclophanes via SM and RCM.
Recently, Li et al. [51] disclosed an elegant synthesis of MK-6325 (141) through a sequential usage of RCM and SM coupling as key steps. In this respect, the required RCM precursor 130 was derived from 129 by employing a six-step synthesis sequence. Next, the alkene derivative 130 was subjected to RCM under the influence of Zhan-1B catalyst 4 to deliver the cyclized product 131 (91%). Later, TFA-mediated deprotection of cyclized product 131 gave amine 132 (97%). Treatment of chloro derivative 132 with boronate ester 133 provided the SM coupling precursor 134 (77%). Later, an intramolecular SM coupling of Bpin derivative 134 was realized in the presence of a Pd(OAc)2 catalyst with the aid of the ligand cataCXium A (135) to generate the macrocyclic product 136. Eventually, synthesis of MK-6325 (141) was achieved by adopting saponification followed by amidation (Scheme 20).
![[1860-5397-14-223-i20]](/bjoc/content/inline/1860-5397-14-223-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of MK-6325 (141) via RCM and SM coupling.
Scheme 20: Synthesis of MK-6325 (141) via RCM and SM coupling.
Conclusion
In this review, we have summarized various approaches to a wide range of carbocycles and heterocycles that deals with a strategic utilization of SM coupling and metathesis as key steps. Interestingly, application of these two powerful methods in combination for a C–C bond formation process shorten the synthesis sequence for the assembly of the target molecules and thus enhances the ease of preparation of various functional molecules. These processes are considered as “green” because of atom economy and synthetic brevity [52] involved in these reactions [12,53,54]. Additionally, several methods are available to remove palladium and ruthenium impurities in minor amounts from the reaction mixture. This aspect is also important in the pharmaceutical industry [4,55].
Biography of the Authors
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-223-i21.png?max-width=637&scale=1.18182)
Sambasivarao Kotha graduated with M.Sc. degree in Chemistry from the University of Hyderabad and obtained his Ph.D. in Organic Chemistry from the University of Hyderabad in 1985. Later, he moved to UMIST Manchester, UK and the University of Wisconsin, USA as a research associate. Subsequently, he was appointed as a visiting scientist at Cornell University and as a research chemist at Hoechst Celanese Texas prior to joining IIT Bombay in 1994 as an assistant professor. Later, in 2001, he was promoted to Professor. He has published 250 publications in peer-reviewed journals and elected fellow of various academies (FNASc, FASc, FRSC and FNA). He was also associated with editorial advisory boards of several journals. His research interests include: organic synthesis, green chemistry, development of new synthetic methods for unusual amino acids, peptide modifications, cross-coupling reactions, and metathesis. Currently, he occupies the Pramod Chaudhari Chair Professor in Green Chemistry.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-223-i22.png?max-width=637&scale=1.18182)
Milind P. Meshram was born in Amravati, Maharashtra, India. He obtained his M.Sc. degree in Chemistry from the Amravati University. He joined the Department of Chemistry, IIT Bombay in 2007 and graduated with Ph.D. degree in 2014 (Organic Chemistry) under the supervision of Prof. S. Kotha. Later, he worked with Prof. Dr. Van der Eycken as a Post-Doctoral Fellow at the KU Leuven, Belgium under the EMINTE programme. During post-doctoral work his research work was related to organic synthesis under microwave reaction conditions. Presently, he is Research Associate with Prof. S. Kotha. His research interests include various transition-metal-catalyzed reactions and their applications in organic synthesis.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-223-i23.png?max-width=637&scale=1.18182)
Chandravathi Chakkapalli obtained her B.Sc. and M.Sc. in Chemistry from Andhra University, Andhra Pradesh, India. She completed her Ph.D. under the supervision of Dr. J. S. Yadav from IICT, Hyderabad in 2016. Her research interests are in the area of organic synthesis and green chemistry.
Acknowledgements
We thank the Council of Scientific and Industrial Research (CSIR), New Delhi [02(0272)/16/EMR-II] for the financial support. SK thanks the Department of Science and Technology (DST), New Delhi for the award of a J. C. Bose fellowship (SR/S2/JCB-33/2010), and Praj Industries, Pune for the Pramod Chaudhari Chair Professorship (Green Chemistry).
References
-
Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2015; Vol. 1. doi:10.1002/9783527674107
Return to citation in text: [1] -
Ogba, O. M.; Warner, N. C.; O’Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a
Return to citation in text: [1] -
Kotha, S.; Lahiri, K. Synlett 2007, 2767–2784. doi:10.1055/s-2007-990954
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397–421. doi:10.1016/j.tet.2011.10.018
Return to citation in text: [1] [2] -
Kotha, S.; Sreenivasachary, N. Indian J. Chem. 2001, 40B, 763–780.
Return to citation in text: [1] -
Kotha, S.; Misra, S.; Sreevani, G.; Babu, B. Curr. Org. Chem. 2013, 17, 2776–2795. doi:10.2174/13852728113179990118
Return to citation in text: [1] -
Carey, F. A.; Sunburg, R. J. Reactions Involving Transition Metals. Advanced Organic Chemistry: Reaction and Synthesis, 5th ed.; Springer: New York, NY, U.S.A., 2007. doi:10.1007/978-0-387-71481-3
Return to citation in text: [1] -
Suzuki, A.; Miyaura, N.; Yamada, K. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/s0040-4039(01)95429-2
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633–9695. doi:10.1016/s0040-4020(02)01188-2
Return to citation in text: [1] -
Chatterjee, A.; Ward, T. R. Catal. Lett. 2016, 146, 820–840. doi:10.1007/s10562-016-1707-8
Return to citation in text: [1] [2] -
Lennox, A. J. J.; Lloyd-Jones, G. C. Chem. Soc. Rev. 2014, 43, 412–443. doi:10.1039/c3cs60197h
Return to citation in text: [1] -
Molander, G. A. J. Org. Chem. 2015, 80, 7837–7848. doi:10.1021/acs.joc.5b00981
Return to citation in text: [1] -
Hall, D. G., Ed. Boronic Acids, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2011. doi:10.1002/9783527639328
Return to citation in text: [1] -
Hemelaere, R.; Carreaux, F.; Carboni, B. J. Org. Chem. 2013, 78, 6786–6792. doi:10.1021/jo400872x
Return to citation in text: [1] -
Topolovčan, N.; Panov, I.; Kotora, M. Org. Lett. 2016, 18, 3634–3637. doi:10.1021/acs.orglett.6b01682
Return to citation in text: [1] -
Kotha, S.; Mandal, K. Chem. – Asian J. 2009, 4, 354–362. doi:10.1002/asia.200800244
Return to citation in text: [1] -
Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.-H.; Fürstner, A. Tetrahedron 2010, 66, 6421–6428. doi:10.1016/j.tet.2010.05.043
Return to citation in text: [1] -
Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Angew. Chem., Int. Ed. 2016, 55, 13263–13266. doi:10.1002/anie.201606679
Return to citation in text: [1] -
Reck, L. M.; Haberhauer, G.; Lüning, U. Eur. J. Org. Chem. 2016, 1119–1131. doi:10.1002/ejoc.201501289
Return to citation in text: [1] -
Sakamoto, K.; Hakamata, A.; Tsuda, M.; Fuwa, H. Angew. Chem., Int. Ed. 2018, 57, 3801–3805. doi:10.1002/anie.201800507
Return to citation in text: [1] -
Inoue, M.; Komori, T.; Iwanaga, T.; Toyota, S. Chem. Lett. 2017, 46, 1836–1838. doi:10.1246/cl.170884
Return to citation in text: [1] -
Kuttner, J. R.; Hilt, G. Macromolecules 2014, 47, 5532–5541. doi:10.1021/ma5012446
Return to citation in text: [1] -
Sasaki, M.; Ishikawa, M.; Fuwa, H.; Tachibana, K. Tetrahedron 2002, 58, 1889–1911. doi:10.1016/s0040-4020(02)00045-5
Return to citation in text: [1] -
Essig, S.; Schmalzbauer, B.; Bretzke, S.; Scherer, O.; Koeberle, A.; Werz, O.; Müller, R.; Menche, D. J. Org. Chem. 2016, 81, 1333–1357. doi:10.1021/acs.joc.5b02844
Return to citation in text: [1] -
Arican, D.; Braukmüller, S.; Brückner, R. Chem. – Eur. J. 2017, 23, 4537–4541. doi:10.1002/chem.201700622
Return to citation in text: [1] -
Maitra, S.; Bodugam, M.; Javed, S.; Hanson, P. R. Org. Lett. 2016, 18, 3094–3097. doi:10.1021/acs.orglett.6b01248
Return to citation in text: [1] -
Kotha, S.; Panguluri, N. R.; Ali, R. Eur. J. Org. Chem. 2017, 5316–5342. doi:10.1002/ejoc.201700439
Return to citation in text: [1] -
Jana, A.; Misztal, K.; Żak, A.; Grela, K. J. Org. Chem. 2017, 82, 4226–4234. doi:10.1021/acs.joc.7b00200
Return to citation in text: [1] -
Kotha, S.; Shah, V. R.; Mandal, K. Adv. Synth. Catal. 2007, 349, 1159–1172. doi:10.1002/adsc.200600469
Return to citation in text: [1] -
Kotha, S.; Behera, M.; Shah, V. R. Synlett 2005, 1877–1880. doi:10.1055/s-2005-871569
Return to citation in text: [1] -
Johnson, M. M.; Ngwira, K. J.; Rousseau, A. L.; Lemmerer, A.; de Koning, C. B. Tetrahedron 2018, 74, 12–18. doi:10.1016/j.tet.2017.11.023
Return to citation in text: [1] -
Kotha, S.; Ali, R.; Srinivas, V.; Krishna, N. G. Tetrahedron 2015, 71, 129–138. doi:10.1016/j.tet.2014.11.024
Return to citation in text: [1] -
Kotha, S.; Mandal, K. Tetrahedron Lett. 2004, 45, 1391–1394. doi:10.1016/j.tetlet.2003.12.075
Return to citation in text: [1] -
Kotha, S.; Ali, R. Turk. J. Chem. 2015, 39, 1190–1198. doi:10.3906/kim-1502-116
Return to citation in text: [1] -
Ali, R. Diversity oriented approach to spirocycles and heterocycles via olefin metathesis, cyloaddition reaction, Fischer indolization, and Suzuki–Miyaura cross-coupling reaction as key steps. Ph.D. Thesis, IIT, Bombay, India, 2015.
Return to citation in text: [1] -
Lebrun, S.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron Lett. 2011, 52, 1481–1484. doi:10.1016/j.tetlet.2011.01.113
Return to citation in text: [1] -
Schmidt, B.; Krehl, S.; Kelling, A.; Schilde, U. J. Org. Chem. 2012, 77, 2360–2367. doi:10.1021/jo2026564
Return to citation in text: [1] -
Barthelemy, A.-L.; Prieto, A.; Diter, P.; Hannedouche, J.; Toffano, M.; Anselmi, E.; Magnier, E. Eur. J. Org. Chem. 2018, 3764–3770. doi:10.1002/ejoc.201800324
Return to citation in text: [1] -
Kotha, S.; Shah, V. R. Eur. J. Org. Chem. 2008, 1054–1064. doi:10.1002/ejoc.200700921
Return to citation in text: [1] -
Kotha, S.; Mandal, K.; Tiwari, A.; Mobin, S. M. Chem. – Eur. J. 2006, 12, 8024–8038. doi:10.1002/chem.200600540
Return to citation in text: [1] -
Kotha, S.; Srinivas, V.; Krishna, N. G. Heterocycles 2012, 86, 1555–1563. doi:10.3987/com-12-s(n)89
Return to citation in text: [1] -
Kiesewetter, E. T.; O’Brien, R. V.; Yu, E. C.; Meek, S. J.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2013, 135, 6026–6029. doi:10.1021/ja403188t
Return to citation in text: [1] -
Majchrzak, M.; Wilkowski, G.; Kubicki, M. Eur. J. Org. Chem. 2017, 4291–4299. doi:10.1002/ejoc.201700602
Return to citation in text: [1] -
Kotha, S.; Seema, V. Synlett 2011, 2329–2334. doi:10.1055/s-0030-1260315
Return to citation in text: [1] -
Venkanna, A.; Poornima, B.; Siva, B.; Babu, B. H.; Babu, K. S. Synlett 2018, 29, 908–911. doi:10.1055/s-0036-1591539
Return to citation in text: [1] -
Kotha, S.; Mandal, K.; Arora, K. K.; Pedireddi, V. R. Adv. Synth. Catal. 2005, 347, 1215–1218. doi:10.1002/adsc.200404373
Return to citation in text: [1] -
Kotha, S.; Mandal, K. Eur. J. Org. Chem. 2006, 5387–5393. doi:10.1002/ejoc.200600549
Return to citation in text: [1] -
Kotha, S.; Chavan, A. S.; Shaikh, M. J. Org. Chem. 2012, 77, 482–489. doi:10.1021/jo2020714
Return to citation in text: [1] -
Li, H.; Scott, J. P.; Chen, C.-y.; Journet, M.; Belyk, K.; Balsells, J.; Kosjek, B.; Baxter, C. A.; Stewart, G. W.; Wise, C.; Alam, M.; Song, Z. J.; Tan, L. Org. Lett. 2015, 17, 1533–1536. doi:10.1021/acs.orglett.5b00418
Return to citation in text: [1] -
Hudlicky, T.; Reed, J. W. The Way of Synthesis; Wiley-VCH: Weinheim, Germany, 2007; p 98.
Return to citation in text: [1] -
Hughes, D.; Wheeler, P.; Ene, D. Org. Process Res. Dev. 2017, 21, 1938–1962. doi:10.1021/acs.oprd.7b00319
Return to citation in text: [1] -
Náray-Szabó, G.; Mika, L. T. Green Chem. 2018, 20, 2171–2191. doi:10.1039/c8gc00514a
Return to citation in text: [1] -
Szczepaniak, G.; Ruszczyńska, A.; Kosiński, K.; Bulska, E.; Grela, K. Green Chem. 2018, 20, 1280–1289. doi:10.1039/c7gc03324a
Return to citation in text: [1]
| 47. | Venkanna, A.; Poornima, B.; Siva, B.; Babu, B. H.; Babu, K. S. Synlett 2018, 29, 908–911. doi:10.1055/s-0036-1591539 |
| 48. | Kotha, S.; Mandal, K.; Arora, K. K.; Pedireddi, V. R. Adv. Synth. Catal. 2005, 347, 1215–1218. doi:10.1002/adsc.200404373 |
| 49. | Kotha, S.; Mandal, K. Eur. J. Org. Chem. 2006, 5387–5393. doi:10.1002/ejoc.200600549 |
| 50. | Kotha, S.; Chavan, A. S.; Shaikh, M. J. Org. Chem. 2012, 77, 482–489. doi:10.1021/jo2020714 |
| 1. | Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2015; Vol. 1. doi:10.1002/9783527674107 |
| 2. | Ogba, O. M.; Warner, N. C.; O’Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a |
| 3. | Kotha, S.; Lahiri, K. Synlett 2007, 2767–2784. doi:10.1055/s-2007-990954 |
| 4. | Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397–421. doi:10.1016/j.tet.2011.10.018 |
| 5. | Kotha, S.; Sreenivasachary, N. Indian J. Chem. 2001, 40B, 763–780. |
| 6. | Kotha, S.; Misra, S.; Sreevani, G.; Babu, B. Curr. Org. Chem. 2013, 17, 2776–2795. doi:10.2174/13852728113179990118 |
| 18. | Kotha, S.; Mandal, K. Chem. – Asian J. 2009, 4, 354–362. doi:10.1002/asia.200800244 |
| 33. | Johnson, M. M.; Ngwira, K. J.; Rousseau, A. L.; Lemmerer, A.; de Koning, C. B. Tetrahedron 2018, 74, 12–18. doi:10.1016/j.tet.2017.11.023 |
| 13. | Lennox, A. J. J.; Lloyd-Jones, G. C. Chem. Soc. Rev. 2014, 43, 412–443. doi:10.1039/c3cs60197h |
| 14. | Molander, G. A. J. Org. Chem. 2015, 80, 7837–7848. doi:10.1021/acs.joc.5b00981 |
| 15. | Hall, D. G., Ed. Boronic Acids, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2011. doi:10.1002/9783527639328 |
| 16. | Hemelaere, R.; Carreaux, F.; Carboni, B. J. Org. Chem. 2013, 78, 6786–6792. doi:10.1021/jo400872x |
| 17. | Topolovčan, N.; Panov, I.; Kotora, M. Org. Lett. 2016, 18, 3634–3637. doi:10.1021/acs.orglett.6b01682 |
| 34. | Kotha, S.; Ali, R.; Srinivas, V.; Krishna, N. G. Tetrahedron 2015, 71, 129–138. doi:10.1016/j.tet.2014.11.024 |
| 8. | Suzuki, A.; Miyaura, N.; Yamada, K. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/s0040-4039(01)95429-2 |
| 9. | Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866 |
| 10. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 11. | Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633–9695. doi:10.1016/s0040-4020(02)01188-2 |
| 12. | Chatterjee, A.; Ward, T. R. Catal. Lett. 2016, 146, 820–840. doi:10.1007/s10562-016-1707-8 |
| 31. | Kotha, S.; Shah, V. R.; Mandal, K. Adv. Synth. Catal. 2007, 349, 1159–1172. doi:10.1002/adsc.200600469 |
| 7. | Carey, F. A.; Sunburg, R. J. Reactions Involving Transition Metals. Advanced Organic Chemistry: Reaction and Synthesis, 5th ed.; Springer: New York, NY, U.S.A., 2007. doi:10.1007/978-0-387-71481-3 |
| 32. | Kotha, S.; Behera, M.; Shah, V. R. Synlett 2005, 1877–1880. doi:10.1055/s-2005-871569 |
| 26. | Essig, S.; Schmalzbauer, B.; Bretzke, S.; Scherer, O.; Koeberle, A.; Werz, O.; Müller, R.; Menche, D. J. Org. Chem. 2016, 81, 1333–1357. doi:10.1021/acs.joc.5b02844 |
| 28. | Maitra, S.; Bodugam, M.; Javed, S.; Hanson, P. R. Org. Lett. 2016, 18, 3094–3097. doi:10.1021/acs.orglett.6b01248 |
| 29. | Kotha, S.; Panguluri, N. R.; Ali, R. Eur. J. Org. Chem. 2017, 5316–5342. doi:10.1002/ejoc.201700439 |
| 12. | Chatterjee, A.; Ward, T. R. Catal. Lett. 2016, 146, 820–840. doi:10.1007/s10562-016-1707-8 |
| 53. | Hughes, D.; Wheeler, P.; Ene, D. Org. Process Res. Dev. 2017, 21, 1938–1962. doi:10.1021/acs.oprd.7b00319 |
| 54. | Náray-Szabó, G.; Mika, L. T. Green Chem. 2018, 20, 2171–2191. doi:10.1039/c8gc00514a |
| 25. | Sasaki, M.; Ishikawa, M.; Fuwa, H.; Tachibana, K. Tetrahedron 2002, 58, 1889–1911. doi:10.1016/s0040-4020(02)00045-5 |
| 30. | Jana, A.; Misztal, K.; Żak, A.; Grela, K. J. Org. Chem. 2017, 82, 4226–4234. doi:10.1021/acs.joc.7b00200 |
| 4. | Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397–421. doi:10.1016/j.tet.2011.10.018 |
| 55. | Szczepaniak, G.; Ruszczyńska, A.; Kosiński, K.; Bulska, E.; Grela, K. Green Chem. 2018, 20, 1280–1289. doi:10.1039/c7gc03324a |
| 23. | Inoue, M.; Komori, T.; Iwanaga, T.; Toyota, S. Chem. Lett. 2017, 46, 1836–1838. doi:10.1246/cl.170884 |
| 24. | Kuttner, J. R.; Hilt, G. Macromolecules 2014, 47, 5532–5541. doi:10.1021/ma5012446 |
| 51. | Li, H.; Scott, J. P.; Chen, C.-y.; Journet, M.; Belyk, K.; Balsells, J.; Kosjek, B.; Baxter, C. A.; Stewart, G. W.; Wise, C.; Alam, M.; Song, Z. J.; Tan, L. Org. Lett. 2015, 17, 1533–1536. doi:10.1021/acs.orglett.5b00418 |
| 19. | Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.-H.; Fürstner, A. Tetrahedron 2010, 66, 6421–6428. doi:10.1016/j.tet.2010.05.043 |
| 20. | Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Angew. Chem., Int. Ed. 2016, 55, 13263–13266. doi:10.1002/anie.201606679 |
| 21. | Reck, L. M.; Haberhauer, G.; Lüning, U. Eur. J. Org. Chem. 2016, 1119–1131. doi:10.1002/ejoc.201501289 |
| 22. | Sakamoto, K.; Hakamata, A.; Tsuda, M.; Fuwa, H. Angew. Chem., Int. Ed. 2018, 57, 3801–3805. doi:10.1002/anie.201800507 |
| 27. | Arican, D.; Braukmüller, S.; Brückner, R. Chem. – Eur. J. 2017, 23, 4537–4541. doi:10.1002/chem.201700622 |
| 52. | Hudlicky, T.; Reed, J. W. The Way of Synthesis; Wiley-VCH: Weinheim, Germany, 2007; p 98. |
| 37. | Ali, R. Diversity oriented approach to spirocycles and heterocycles via olefin metathesis, cyloaddition reaction, Fischer indolization, and Suzuki–Miyaura cross-coupling reaction as key steps. Ph.D. Thesis, IIT, Bombay, India, 2015. |
| 35. | Kotha, S.; Mandal, K. Tetrahedron Lett. 2004, 45, 1391–1394. doi:10.1016/j.tetlet.2003.12.075 |
| 36. | Kotha, S.; Ali, R. Turk. J. Chem. 2015, 39, 1190–1198. doi:10.3906/kim-1502-116 |
| 45. | Majchrzak, M.; Wilkowski, G.; Kubicki, M. Eur. J. Org. Chem. 2017, 4291–4299. doi:10.1002/ejoc.201700602 |
| 42. | Kotha, S.; Mandal, K.; Tiwari, A.; Mobin, S. M. Chem. – Eur. J. 2006, 12, 8024–8038. doi:10.1002/chem.200600540 |
| 43. | Kotha, S.; Srinivas, V.; Krishna, N. G. Heterocycles 2012, 86, 1555–1563. doi:10.3987/com-12-s(n)89 |
| 44. | Kiesewetter, E. T.; O’Brien, R. V.; Yu, E. C.; Meek, S. J.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2013, 135, 6026–6029. doi:10.1021/ja403188t |
| 40. | Barthelemy, A.-L.; Prieto, A.; Diter, P.; Hannedouche, J.; Toffano, M.; Anselmi, E.; Magnier, E. Eur. J. Org. Chem. 2018, 3764–3770. doi:10.1002/ejoc.201800324 |
| 41. | Kotha, S.; Shah, V. R. Eur. J. Org. Chem. 2008, 1054–1064. doi:10.1002/ejoc.200700921 |
| 38. | Lebrun, S.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron Lett. 2011, 52, 1481–1484. doi:10.1016/j.tetlet.2011.01.113 |
| 39. | Schmidt, B.; Krehl, S.; Kelling, A.; Schilde, U. J. Org. Chem. 2012, 77, 2360–2367. doi:10.1021/jo2026564 |
© 2018 Kotha et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)