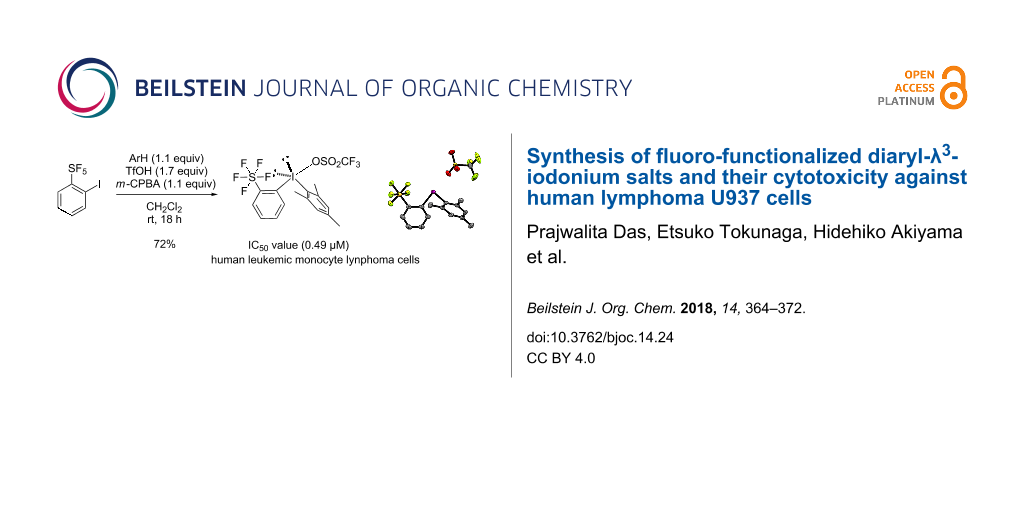
Abstract
Conscious of the potential bioactivity of fluorine, an investigation was conducted using various fluorine-containing diaryliodonium salts in order to study and compare their biological activity against human lymphoma U937 cells. Most of the compounds tested are well-known reagents for fluoro-functionalized arylation reactions in synthetic organic chemistry, but their biological properties are not fully understood. Herein, after initially investigating 18 fluoro-functionalized reagents, we discovered that the ortho-fluoro-functionalized diaryliodonium salt reagents showed remarkable cytotoxicity in vitro. These results led us to synthesize more compounds, previously unknown sterically demanding diaryliodonium salts having a pentafluorosulfanyl (SF5) functional group at the ortho-position, that is, unsymmetrical ortho-SF5 phenylaryl-λ3-iodonium salts. Newly synthesized mesityl(2-(pentafluoro-λ6-sulfanyl)phenyl)iodonium exhibited the greatest potency in vitro against U937 cells. Evaluation of the cytotoxicity of selected phenylaryl-λ3-iodonium salts against AGLCL (a normal human B cell line) was also examined.

Graphical Abstract
Introduction
There has been a surge in the number of reports about fluorine chemistry in recent decades. This is because fluorine is an extremely important element whose presence in a compound can completely change its original physical and chemical characteristics [1-3]. The chemical structures of various pharmaceuticals, agrochemicals and coatings contain fluorine or fluorinated functional groups [4-9]. Therefore, the development of efficient synthetic methodologies for organofluorine compounds has gained much attention [10-15]. Our research group has been actively working in this direction for decades [12,13,16-23]. Our primary goal has been to develop fluorinating and fluoro-functionalized reagents for fluorination [18,19], trifluoromethylation [13,18,19], trifluoromethylthiolation [12,21] and pentafluoroarylation [22,23]. Utilizing these reagents, we have successfully synthesized a wide variety of bioactive organofluorine compounds [24-30] including fluorinated thalidomide (antitumor) [24], fluorinated donepezil (cholinesterase inhibitor) [25], and fluorinated camptothecin (anticancer) [26]. During our research programs focused on the development of novel reagents for fluoro-functionalization [12,13,16-23], as well as the design and synthesis of biologically active fluorine-containing compounds [24-28], we noted that a series of fluoro-functionalization reagents could themselves be highly potential drug candidates. All of the reagents that we developed contain at least one fluorine atom in their structures, which may explain why they have potential biological activity [4-9]. In addition, examination of the successful records of heterocyclic compounds in the pharmaceutical history indicates that some of these reagents have a heterocyclic skeleton which makes them suitable as drug candidates [29-32]. Among these compounds, our group is interested in investigating the biological activity of hypervalent iodine-type reagents [33]. Hypervalent iodine compounds have been receiving a lot of attention lately due to their varied applications in organic synthesis [33-40]. A wide range of bioactive compounds make use of diaryliodonium reagents as a part of their synthesis [41-43]. On the other hand, there are only fragmented reports on the biological activity of diaryliodonium salts [44-49]. Goldstein et al. [45] and Doroshow et al. [46] reported that some diaryliodonium salts show effective antimicrobial and NOX inhibitor activity, respectively. Several aryliodonium salts, aryliodonium ylides, and (diacyloxyiodo)arenes were also examined for their antibacterial activities against ice nucleation active Pseudomonas syringae, and aryliodonium salts, especially those with electron-withdrawing groups, exhibit higher antibacterial activities [49]. Despite the long history of diaryliodonium salts, which exceeds 100 years, as Willgerodt's reagent [50], research on the biological effects of diaryliodonium salts is still undeveloped. Since there are iodine-containing pharmaceuticals such as a series of radiocontrast agents, levothyroxine, idoxuridine and amiodaron [51-54], we started to investigate a biological study of fluorine-containing hypervalent iodine compounds in vitro using U937 (a human histiocytic lymphoma cell line) [55-58]. The U937 cell line is maintained as replicative non-adherent cells having many of the biochemical and morphological characteristics of blood monocytes. This cell line was chosen due to the convenience with which it can be handled and its ease of growth [27]. Initially, 19 compounds [20-22,59-64] were examined for their potential cytotoxicity, and some of them showed potency, in particular ortho-fluoro-functionalized diaryliodonium salts. These findings led us to synthesize four more previously unknown diaryliodonium salts having a sterically demanding pentafluorosulfanyl (SF5) functional group at the ortho-position, that is, unsymmetrical ortho-SF5 phenylaryl-λ3-iodonium salts. Finally, one of the new compounds, namely mesityl(2-(pentafluoro-λ6-sulfanyl)phenyl)iodonium salt, exhibited the greatest potency in vitro against U937 cells with an IC50 value of 0.49 μM.
Results and Discussion
To begin our investigation related to bioactivity, we randomly selected some fluorinating reagents that we had already developed, including Shibata reagents I [20] and II [21] (trifluoromethylation reagent 1 and trifluoromethylthiolation reagent 2a, respectively), pentafluorophenylating reagent 2b and several hypervalent iodine reagents, i.e., diaryliodonium salts with a mesitylene ligand (3a–o) and a triisopropylphenyl ligand 4a [20-22,59-64] (Figure 1). We used the MuseTM Annexin V and Dead Cell Assay Kit (FITC), which is a common tool to detect the ability of compounds to induce cell death. U937, a human histiocytic lymphoma cell line (DS Pharma Biomedical EC85011440; Osaka, Japan) was used to examine the ability of our synthesized compounds to induce cell death.
![[1860-5397-14-24-1]](/bjoc/content/figures/1860-5397-14-24-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Compounds used for the biological study.
Figure 1: Compounds used for the biological study.
The investigation was initially carried out using 20 µM of several of the compounds, all of which showed strong cytotoxicity (Figure S1 in Supporting Information File 1). We therefore opted to examine this potency using lower concentrations (1 µM and 5 µM) of these compounds. The data (treated cells and untreated controls) were plotted together to compare the results (Figure 2). Shibata reagents 1 and 2a were not very cytotoxic at 5 μM. Perfluorinated phenylthio reagent 2b and perfluorinated phenyl reagent 3a showed similar, but unimpressive, results at both concentrations. Pyridinyl reagent 3b, having a pyridine-SF5 moiety, displayed high cytotoxicity (63.0% of annexin V-positive cells) at 5 μM, as did 3d with 58.6% cytotoxicity. However, another analogue of pyridine-SF5 reagent 3c showed weak (10.8%) cytotoxicity. Next, we investigated the various SF5-phenyl aryliodonium salts 3e–h. They all displayed potent cytotoxicities at 5 μM (3e, (86.2%); 3f (78.6%); 3g (94.0%); 3h (94.1%)) and 3h had the greatest result even at 1 μM (52.8%). Thereafter, we analysed SO2CF3-phenyl aryliodonium salts 3i–k and CF3-phenyl aryliodonium salts 3l,m. We observed that ortho-substituted phenyl aryliodonium salts 3k and 3m seemed to provide great potency at both concentrations (60.6% and 39.8% at 1 μM and 98.6% and 88.4% at 5 μM, respectively). The ortho-fluorophenyl aryliodonium salt 3n was also analysed, and its potency at 5 μM was found to be moderate (45.3%), but low at 1 μM (17.9%). The non-fluorinated diphenyl iodonium salt 3o, on the other hand, was weakly cytotoxic at both concentrations (1 μM, 7.5%; 5 μM, 10.1%). Analysis of the perfluorinated phenyl reagent 4a with a triisopropylphenyl ligand displayed strong cytotoxicity (93.7%) at 5 μM, but this value decreased considerably to 7.8% when used at 1 μM.
![[1860-5397-14-24-2]](/bjoc/content/figures/1860-5397-14-24-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Compounds 1–4 induced cell death in U937 cells. Briefly, U937 cells (1 × 104 cells/mL) were incubated with each test compound at 1 μM and 5 μM for 24 h. Cells were stained with annexin V and propidium iodide (PI). The data shown is the mean ± SD (n = 3).
Figure 2: Compounds 1–4 induced cell death in U937 cells. Briefly, U937 cells (1 × 104 cells/mL) were incubat...
Following this investigation, we found that the ortho-substituted diaryliodonium salts with an ortho-SO2CF3 group 3k and an ortho-CF3 group 3m displayed impressive results. A common feature of ortho-substitution on the aromatic group is steric demand, which allowed us to analyse the cell death-inducing potency of phenyl aryliodonium salts with a more sterically demanding fluoro-functional group of SF5 at the ortho-position. As we did not succeed in synthesizing ortho-SF5-substituted aryliodonium salts previously [59], we decided to proceed with a further investigation of the synthesis of ortho-SF5 phenyl aryliodonium salts.
Four ortho-SF5-substituted diaryliodonium salts were designed with different arenes as auxiliary groups, namely, electron-rich with sterically demanding mesitylene type 3p and triisopropylphenyl type 4b, electron-rich anisole type 5a and simple phenyl type 6a, which altered their electronic and steric properties [37]. First, pentafluoro-(2-iodophenyl)-λ6-sulfane (7) was synthesized from commercially available 2-(pentafluoro-λ6-sulfanyl)aniline (8), by completing a Sandmeyer reaction (Scheme 1). Fluoroboric acid and sodium nitrite were used to generate the diazonium ion and then KI was used to introduce iodide, providing the desired product 7 in 87% yield.
![[1860-5397-14-24-i1]](/bjoc/content/inline/1860-5397-14-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pentafluoro-(2-iodophenyl)-λ6-sulfane (7).
Scheme 1: Synthesis of pentafluoro-(2-iodophenyl)-λ6-sulfane (7).
With 7 in hand, the synthesis of target ortho-SF5 phenylaryl-λ3-iodonium salts 3p, 4b, 5a and 6a was carried out according to a previously reported method [65,66]. This synthesis was achieved by treating iodide 7 with the respective arene, m-CPBA, and trifluoromethanesulfonic acid at room temperature. The desired diaryliodonium salts 3p, 4b, 5a and 6a were obtained in good yields (72%, 86%, 81% and 92%, respectively) (Scheme 2).
![[1860-5397-14-24-i2]](/bjoc/content/inline/1860-5397-14-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of unsymmetrical ortho-SF5 diaryliodonium salts 3p, 4b, 5a and 6a.
Scheme 2: Synthesis of unsymmetrical ortho-SF5 diaryliodonium salts 3p, 4b, 5a and 6a.
The newly synthesized SF5-diaryliodonium salts 3p, 4b, 5a and 6a were characterized spectroscopically. The single crystal X-ray structure of 3p was also analysed. The SF5-diaryliodonium salt 3p has a T-shaped geometry at the iodine centre, consistent with the general structure of diaryliodonium salts [33] (Figure 3).
![[1860-5397-14-24-3]](/bjoc/content/figures/1860-5397-14-24-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystallographic structure of 3p drawn at 50% probability (CCDC 1573953).
Figure 3: X-ray crystallographic structure of 3p drawn at 50% probability (CCDC 1573953).
Following the synthesis of the ortho-SF5 phenyl aryliodonium salts, we selected salts 3p and 5a having mesitylene and anisole dummy ligands, respectively, and analysed their potential to induce cell death in U937 cells (Figure 4). Anisole type salt 5a showed moderate cytotoxicity (45.2%) at 5 μM but the value decreased to 6.8% at 1 μM. Mesitylene type salt 3p, on the other hand, displayed high potencies (98.9% and 66.9%) at 5 μM and 1 μM, respectively. To ensure that the cytotoxicity of 3p was not due to its decomposed fragments, we analysed pentafluoro-(2-iodophenyl)-λ6-sulfane (7) and pentafluoro(phenyl)-λ6-sulfane (SF5-C6H5, 9), which did not exhibit cytotoxicity.
![[1860-5397-14-24-4]](/bjoc/content/figures/1860-5397-14-24-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Ortho-SF5 phenyl iodonium salts 3p and 5a and their structural components 7 and 9 induced cell death in U937 cells. Briefly, U937 cells (1 × 104 cells/mL) were incubated with each test compound at 1 μM and 5 μM for 24 h. Cells were stained with annexin V and propidium iodide (PI). The data shown is the mean ± SD (n = 3).
Figure 4: Ortho-SF5 phenyl iodonium salts 3p and 5a and their structural components 7 and 9 induced cell deat...
From the above analysis, we selected a series of ortho-fluorinated diaryliodonium salts SO2CF3 type 3k, CF3 type 3m and SF5 type 3p and examined their IC50 values based on an MTT assay (Table 1). While 3k was more potent than 3m at both concentrations, i.e., 1 μM and 5 μM (Figure 2), 3m has a lower IC50 value of 0.68 μM than that of 3k (2.45 μM), as evaluated by the MTT assay. The best potency and IC50 value (0.49 μM) was obtained for SF5 type 3p, which is quite impressive when compared to the well-known antitumor drug cytosine arabinoside (ara-C), (0.16 μM) [27].
Table 1: Cytotoxicity of diaryliodonium salts 3k, 3m and 3p against a human histiocytic lymphoma cell line (U937).a
| diaryliodonium salt 3 | IC50 [µM] |
|---|---|
| 3k | 2.45 ± 0.24 |
| 3m | 0.68 ± 0.05 |
| 3p | 0.49 ± 0.05 |
aIC50 values were determined using an MTT assay; data represents the mean standard deviation of three independent experiments.
Since, 3k, 3m and 3p exhibited strong cytotoxicity against U937 cells, we finally evaluated their cytotoxicity against normal cells in vitro. AGLCL, a human normal B cell line (DS Pharma Biomedical EC89120566; Osaka, Japan) was chosen for the experiments and investigations were performed at 20 μM (Figure S2 in Supporting Information File 1) and 5 μM and 1 μM concentrations (Figure 5) of the compounds. Although 3k, 3m and 3p exhibited cytotoxicity even against AGLCL cells, a remarkable difference was observed. That is, moderate cytotoxicity at 5 μM (51.1%, 51.2% and 62.0%, respectively) and low cytotoxicity at 1 μM concentration (20.4%, 15.8% and 24.9%, respectively) against AGLCL cells were observed. It is noteworthy that the cytotoxicity displayed by 3k, 3m and 3p against U937 cells is much higher than those against AGLCL cells at both concentrations. These results strongly suggested that antitumor drug candidates could be designed by further structural modification of these compounds 3. Moreover, with 3p exhibiting the greatest potency against U937 cells with comparably lower toxicity against AGLCL cells, further biological studies using 3p including in vivo evaluation should be conducted. A mechanistic study that examines the structure–cytotoxicity relationships of a series of diaryliodonium salts 3 will also be conducted.
![[1860-5397-14-24-5]](/bjoc/content/figures/1860-5397-14-24-5.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 3k, 3m and 3p induced cell death in AGLCL, a human normal B cell line. Briefly, AGLCL cells (1 × 104 cells/mL) were incubated with each test compound at 1 μM and 5 μM for 24 h. Cells were stained with annexin V and propidium iodide (PI). The data shown is the mean ± SD (n = 3).
Figure 5: 3k, 3m and 3p induced cell death in AGLCL, a human normal B cell line. Briefly, AGLCL cells (1 × 104...
Conclusion
In conclusion, we have analysed a series of fluorinating reagents and diaryliodonium salts for their applicability in inducing cell death based on U937 (a human histiocytic lymphoma cell line). We have also successfully synthesized novel ortho-SF5 phenylaryl-λ3-iodonium salts. As expected, several fluorinated diaryliodonium salts exhibited cytotoxicity. Among the series, the newly synthesized ortho-SF5 salt 3p displayed the greatest potency. The ortho-fluorinated diaryliodonium salts 3k, 3m and 3p were also examined for comparison studies with AGLCL (a normal human B cell line). Although the values were rather low, selectivity was indeed observed against U937 cells. Also, even though the results are in a preliminary stage of biological evaluation, this is the first report to highlight the cytotoxicity of diaryliodonium salts against U937 cells. Since diaryliodonium salts are fundamentally oxidizing agents, there might be a stronger correlation between cytotoxicity and the oxidation potential of these salts. We will continue the biological investigation of 3 in this direction. From the view point of organic synthesis, the newly synthesized ortho-SF5-substituted unsymmetrical iodonium salts 3p, 4b, 5a and 6a have potential use as electrophilic SF5-phenylation reagents for a range of nucleophiles such as alcohols, amines, thiols, and active methylene nucleophiles [59-61]. The application of these ortho-SF5-substituted diaryliodonium salts in organic synthesis, as well as their detailed bioactive behaviour, will be reported in due course.
Experimental
Biological assay
Quantification of cytotoxicity by annexin V and propidium iodide (PI): Cytotoxicity was detected with the MuseTM Annexin V and Dead Cell Assay Kit (Merck Millipore Corp., Darmstadt, Germany) and Muse Cell Analyzer (Merck Millipore Corp.) according to the manufacturer’s protocols. Cells incubated in the presence or absence of the fluorinated compounds for 24 h were collected by centrifugation (2,000 rpm at 4 °C for 5 min). Cells were suspended in 100 μL of RPMI 1640 medium (Sigma-Aldrich, Steinheim, Germany), and incubated with 100 μL of annexin V reagent (in the kit) at room temperature for 20 min. These cells were measured by the Muse Cell Analyzer.
Statistical analysis: Data were analyzed using Excel software. Results are expressed as the mean ± SD of three independent replicates.
MTT assay: The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to evaluate cell viability by diaryliodonium salt compounds using the MTT cell proliferation assay kit (Cayman Chemical Company, Ann Arbor, USA). U937 cells were incubated in solutions containing the diaryliodonium salts (3k, 3m or 3p). After this treatment, the U937 cells were seeded in culture medium (100 μL) in a 96-well plate (Becton and Dickinson, at a density of 2 × 105 cells/well) and incubated at 37 °C for 24 h. MTT reagent (10 μL) was added to each well. After mixing gently, the cells were incubated for 3 h at 37 °C in a CO2 incubator. The culture medium was aspirated and the crystal-dissolving solution (100 μL) was added to each well and mixed. Finally, the optical density was measured (550 nm) using a microplate reader (BIO-RAD, Benchmark, Hercules, USA).
General information
All reactions were performed in oven-dried glassware under positive pressure of nitrogen or argon unless mentioned otherwise. Solvents were transferred via a syringe and were introduced into reaction vessels though a rubber septum. All reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Merck silica gel (60-F254). The TLC plates were visualized with UV light (254 nm). Column chromatography was carried out on columns packed with silica gel (60N spherical neutral size 63–210 μm). The 1H NMR (300 MHz), 19F NMR (282 MHz), and 13C NMR (125 MHz) spectra for solution in CDCl3 or (CD3)2CO were recorded on Varian Mercury 300 and Bruker Avance 500 spectrometers. Chemical shifts (δ) are expressed in ppm downfield from TMS (δ = 0.00) or C6F6 [δ = −162.2 (CDCl3) or −163.5 ((CD3)2CO)] as an internal standard. Mass spectra were recorded on a Shimadzu GCMS-QP5050A (EIMS) and Shimazu LCMS-2020 (ESIMS) spectrometer. The solvent CH2Cl2 was dried and distilled before use.
Preparation of pentafluoro(2-iodophenyl)-λ6-sulfane (7): The preparation of 7 was based on a modified procedure in the literature [67]. To 4 mL of HBF4 in a round bottomed flask, 1.1 g of 2-(pentafluoro-λ6-sulfanyl)aniline was added and heated until a clear solution formed. The solution was cooled to 0 °C and a cold solution of NaNO2 (380 mg in 2.5 mL distilled water) was added dropwise. The reaction was allowed to stir at 0 °C for 15 min after which it was added dropwise to a cold stirred solution of KI (1.33 g in 10 mL distilled water) at 0 °C. The reaction was allowed to warm to room temperature then stirred for 20 min. The reaction mixture was extracted with diethyl ether (3 × 20 mL). The combined organic extract was washed with NaHCO3 solution and Na2S2O3 solution and dried over Na2SO4. The solvent was concentrated under reduced pressure to give a crude product which was purified using silica gel column chromatography (9:1, hexane/ethyl acetate) to give 1.4 g of 7 as a yellow oil in 87% yield. HRMS (EI–TOF) m/z [M]+: calcd for C6H4F5SI, 329.8999; found, 329.9010; 1H NMR (CDCl3, 300 MHz) δ 7.11 (t, J = 9 Hz, 1H), 7.41–7.47 (m, 1H), 7.80 (dd, J = 9 Hz, 3 Hz, 1H), 8.14 (d, J = 9 Hz, 1H); 19F NMR (CDCl3, 282 MHz) δ 63.55 (d, J = 155.1 Hz, 4F), 83.56 (q, J = 155.1 Hz, 1F); 13C{1H}NMR (CDCl3, 126 MHz) δ 88.4, 128.1, 130.3 (t, J = 3.75 Hz), 132.4, 144.1, 158.57 (q, J = 15 Hz).
General procedure A: preparation of diaryliodonium salts I
These salts were prepared according to a modified procedure in the literature [64,65]. m-CPBA (assume 70 wt %, 1.1 equiv) was dried in vacuo at room temperature for 1 h before the addition of 7 (1.0 equiv) and CH2Cl2 (6.0 mL/mmol ArI) in a round-bottomed flask. The solution was cooled to 0 °C followed by the dropwise addition of TfOH (1.7 equiv). The resulting mixture was stirred at room temperature for 2 h. It was then cooled to 0 °C and the arene (1.1 equiv) was added dropwise. The mixture was warmed to room temperature and stirred for 18 h. The solvent was then removed under reduced pressure. The resulting crude product was precipitated by the addition of Et2O. The precipitate was filtered and dried in vacuo to give 3–6 as an off-white to white solid.
Mesityl(2-(pentafluoro-λ6-sulfanyl)phenyl)iodonium trifluoromethanesulfonate (3p): Following general procedure A, 7 (330 mg, 1 mmol), m-CPBA (271 mg, 1.1 mmol), TfOH (0.2 mL, 1.7 mmol) and mesitylene (0.15 mL, 1.1 mmol) in CH2Cl2 (6 mL) were used from 0 °C to room temperature for 18 h to give 3p as a white solid (430 mg) in 72% yield. mp: 163.7–164.7 °C; HRMS (ESI–TOF) m/z [M − OTf]+: calcd for C15H15F5SI, 448.9859; found, 448.9865; 1H NMR ((CD3)2CO), 300 MHz) δ 2.44 (s, 3H), 2.69 (s, 6H), 7.42 (s, 2H), 7.64 (d, J = 6 Hz, 1H), 7.73 (t, J = 9 Hz, 1H), 7.96 (t, J = 9 Hz, 1H), 8.36 (dd, J = 9 Hz, 1.5 Hz, 1H); 19F NMR ((CD3)2CO), 282 MHz) δ = –79.88 (s, 3F), 64.09 (d, J = 149.5 Hz, 4F), 81.34 (q, J = 149.5 Hz, 1F); 13C{1H}NMR ((CD3)2CO), 126 MHz) δ 21.2, 27.1, 106.6, 121.9 (q, J = 318.8 Hz), 123.0, 131.8, 132.5 (t, J = 5 Hz), 133.7, 135.4, 136.9, 144.5, 147.0, 154.1–154.7 (m).
(2-(Pentafluoro-λ6-sulfanyl)phenyl)(2,4,6-triisopropylphenyl)iodonium trifluoromethanesulfonate (4b): Following general procedure A, 7 (330 mg, 1 mmol), m-CPBA (271 mg, 1.1 mmol), TfOH (0.2 mL, 1.7 mmol) and triisopropylbenzene (0.26 mL, 1.1 mmol) in CH2Cl2 (6 mL) were used from 0 °C to room temperature for 18 h to give 4b as a white solid (604 mg) in 86% yield. mp: 106.6–107.9 °C; HRMS (ESI–TOF) m/z [M − OTf]+: calcd for C21H27F5SI, 533.0798; found, 533.0798; 1H NMR ((CD3)2CO, 300 MHz) δ 1.31 (t, J = 9 Hz, 18H), 3.14 (q, J = 9 Hz, 1H), 3.30 (q, J = 6 Hz, 2H), 7.45 (d, J = 9 Hz, 1H), 7.58 (s, 2H), 7.77 (t, J = 6 Hz, 1H), 7.96 (t, J = 9 Hz, 1H), 8.37 (dd, J = 9 Hz, 1.5 Hz, 1H); 19F NMR ((CD3)2CO, 282 MHz) δ −79.91 (s, 3F), 64.02 (d, J = 149.5 Hz, 4F), 81.31 (q, J = 149.5 Hz, 1F); 13C{1H}NMR ((CD3)2CO, 126 MHz) δ 23.8, 24.2, 34.9, 40.8, 107.7, 121.9 (q, J = 320 Hz), 123.5, 127.1, 132.7 (t, J = 5 Hz), 133.7, 134.5, 136.9, 153.8, 153.9–154.2 (m), 158.1.
(4-Methoxyphenyl)(2-(pentafluoro-λ6-sulfanyl)phenyl)iodonium trifluoromethanesulfonate (5a): Following general procedure A, 7 (416 mg, 1.26 mmol), m-CPBA (340 mg, 1.38 mmol), TfOH (0.24 mL, 2.14 mmol) and anisole (0.15 mL, 1.38 mmol) in CH2Cl2 (6 mL) were used from 0 °C to room temperature for 18 h to give 5a as a white solid (600 mg) in 81% yield. mp: 108.7–110.4 °C; HRMS (ESI–TOF) m/z [M − OTf]+: calcd for C13H11OF5SI, 436.9495; found, 436.9499; 1H NMR ((CD3)2CO, 300 MHz) δ 3.90 (s, 3H), 7.16 (d, J = 9 Hz, 2H), 7.86 (t, J = 9 Hz, 1H), 8.03 (t, J = 9 Hz, 1H), 8.29 (d, J = 9 Hz, 3H), 8.92 (d, J = 6 Hz, 1H); 19F NMR ((CD3)2CO, 282 MHz) δ −79.75 (s, 3F), 65.79 (d, J = 149.5 Hz, 4F), 81.73 (q, J = 152.3 Hz, 1F); 13C{1H}NMR ((CD3)2CO, 126 MHz) δ 56.4, 104.8, 109.8, 118.9, 131.7, 134.6, 136.9, 138.8, 142.2, 154.1–154.4 (m), 164.5.
(2-(Pentafluoro-λ6-sulfanyl)phenyl)(phenyl)iodonium trifluoromethanesulfonate (6a): Following general procedure A, 7 (330 mg, 1 mmol), m-CPBA (271 mg, 1.1 mmol), TfOH (0.2 mL, 1.7 mmol) and benzene (0.1 mL, 1.1 mmol) in CH2Cl2 (6 mL) were used from 0 °C to room temperature for 18 h to give 6a as a white solid (512 mg) in 92% yield. mp: 109.8–111.3 °C; HRMS (ESI–TOF) m/z [M–OTf]+: calcd for C12H9F5SI, 406.9390; found, 406.9385; 1H NMR ((CD3)2CO, 300 MHz) δ 7.61–7.68 (m, 2H), 7.76–7.82 (m, 1H), 7.89 (t, J = 9 Hz, 1H), 8.06 (t, J = 9 Hz, 1H), 8.31 (dd, J = 9 Hz, 3 Hz, 1H), 8.37 (d, J = 9 Hz, 2H), 9.02 (d, J = 6 Hz, 1H); 19F NMR ((CD3)2CO, 282 MHz) δ −79.81 (s, 3F), 65.83 (d, J = 149.5 Hz, 4F), 81.61 (q, J = 152.3 Hz, 1F); 13C{1H}NMR ((CD3)2CO, 126 MHz) δ 109.1, 116.9, 121.9 (q, J = 320 Hz), 131.8, 133.2, 134.1, 134.8, 136.3, 136.9, 142.9, 154.3–154.9 (m).
Supporting Information
| Supporting Information File 1: Cytotoxicity data, copies of 1H, 19F and 13C NMR spectra of 7, 3p, 4b, 5a and 6a and the ORTEP diagram of 3p. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C
Return to citation in text: [1] -
Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/S0022-1139(01)00375-X
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] [2] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] [2] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] [2] -
Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833
Return to citation in text: [1] [2] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] [2] -
Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell, 2009.
Return to citation in text: [1] [2] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386
Return to citation in text: [1] -
Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b
Return to citation in text: [1] [2] [3] [4] -
Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65
Return to citation in text: [1] [2] [3] [4] -
Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c
Return to citation in text: [1] -
Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/C6CS00351F
Return to citation in text: [1] -
Kawai, H.; Shibata, N. Chem. Rec. 2014, 14, 1024–1040. doi:10.1002/tcr.201402023
Return to citation in text: [1] [2] -
Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014
Return to citation in text: [1] [2] -
Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011
Return to citation in text: [1] [2] [3] [4] -
Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t
Return to citation in text: [1] [2] [3] [4] -
Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225
Return to citation in text: [1] [2] [3] [4] [5] -
Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045
Return to citation in text: [1] [2] [3] [4] [5] -
Saidalimu, I.; Suzuki, S.; Tokunaga, E.; Shibata, N. Chem. Sci. 2016, 7, 2106–2110. doi:10.1039/C5SC04208A
Return to citation in text: [1] [2] [3] -
Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Inoue, Y.; Hara, H.; Shibata, N. PLoS One 2017, 12, e0182152. doi:10.1371/journal.pone.0182152
Return to citation in text: [1] [2] [3] -
Maeno, M.; Kondo, H.; Tokunaga, E.; Shibata, N. RSC Adv. 2016, 6, 85058–85062. doi:10.1039/C6RA21253K
Return to citation in text: [1] [2] [3] -
Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810
Return to citation in text: [1] [2] [3] -
Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059
Return to citation in text: [1] [2] [3] [4] -
Nishimine, T.; Taira, H.; Mori, S.; Matsubara, O.; Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Shibata, N. Chem. Commun. 2017, 53, 1128–1131. doi:10.1039/C6CC08830A
Return to citation in text: [1] [2] -
Dinges, J.; Lamberth, C. Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012.
Return to citation in text: [1] [2] -
Gomtsyan, A. Chem. Heterocycl. Compd. 2012, 48, 7–10. doi:10.1007/s10593-012-0960-z
Return to citation in text: [1] [2] -
Kosjek, T.; Heath, E. Halogenated heterocycles as pharmaceuticals. In Halogenated Heterocycles; Iskra, J., Ed.; Springer-Verlag: Berlin, Heidelberg, 2011; Vol. 27, pp 219–246. doi:10.1007/7081_2011_61
Return to citation in text: [1] -
Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi, O. Org. Biomol. Chem. 2016, 14, 6611–6637. doi:10.1039/C6OB00936K
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] [2] [3] -
Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j
Return to citation in text: [1] -
Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651
Return to citation in text: [1] -
Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860
Return to citation in text: [1] [2] -
Ito, M.; Itani, I.; Toyoda, Y.; Morimoto, K.; Dohi, T.; Kita, Y. Angew. Chem., Int. Ed. 2012, 51, 12555–12558. doi:10.1002/anie.201206917
Return to citation in text: [1] -
Sokolovs, I.; Lubriks, D.; Suna, E. J. Am. Chem. Soc. 2014, 136, 6920–6928. doi:10.1021/ja502174d
Return to citation in text: [1] -
Phipps, R. J.; McMurray, L.; Ritter, S.; Duong, H. A.; Gaunt, M. J. J. Am. Chem. Soc. 2012, 134, 10773–10776. doi:10.1021/ja3039807
Return to citation in text: [1] -
Shimizu, I.; Matsumura, Y.; Inomata, Y. Preparation of 1,2-diarylethylene derivatives as drug intermediates. Jpn. Kokai Tokkyo Koho JP 63275533, Nov 14, 1988.
Return to citation in text: [1] -
Kuik, W.-J.; Kema, I. P.; Brouwers, A. H.; Zijlma, R.; Neumann, K. D.; Dierckx, R. A. J. O.; DiMagno, S. G.; Elsinga, P. H. J. Nucl. Med. 2015, 56, 106–112. doi:10.2967/jnumed.114.145730
Return to citation in text: [1] -
DeSolms, S. J.; Woltersdorf, O. W., Jr.; Cragoe, E. J., Jr.; Watson, L. S.; Fanelli, G. M., Jr. J. Med. Chem. 1978, 215, 437–443. doi:10.1021/jm00203a006
Return to citation in text: [1] -
Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+
Return to citation in text: [1] -
Goldstein, E. J. C.; Citron, D. M.; Warren, Y.; Merriam, C. V.; Tyrrell, K.; Fernandez, H.; Radhakrishnan, U.; Stang, P. J.; Conrads, G. Antimicrob. Agents Chemother. 2004, 48, 2766–2770. doi:10.1128/AAC.48.7.2766-2770.2004
Return to citation in text: [1] [2] -
Lu, J.; Risbood, P.; Kane, C. T., Jr.; Hossain, M. T.; Anderson, L.; Hill, K.; Monks, A.; Wu, Y.; Antony, S.; Juhasz, A.; Liu, H.; Jiang, G.; Harris, E.; Roy, K.; Meitzler, J. L.; Konaté, M.; Doroshow, J. H. Biochem. Pharmacol. 2017, 143, 25–38. doi:10.1016/j.bcp.2017.07.007
Return to citation in text: [1] [2] -
Wang, L.; Xu, S.; Qian, X.; Wu, X.; Han, J.; Qu, D.; Tian, H. Diaryliodonium salt compound as antibacterial agent. Faming Zhuanli Shenqing CN 106748940, May 31, 2017.
Return to citation in text: [1] -
Ding, Y.; Zhu, W.; Sun, R.; Yuan, G.; Zhang, D.; Fan, Y.; Sun, J. Oncol. Rep. 2015, 33, 1434–1442. doi:10.3892/or.2015.3726
Return to citation in text: [1] -
Menkissoglu-Spiroudi, U.; Karamanoli, K.; Spyroudis, S.; Constantinidou, H.-I. A. J. Agric. Food Chem. 2001, 49, 3746–3752. doi:10.1021/jf010293v
Return to citation in text: [1] [2] -
Willgerodt, C. J. Prakt. Chem. 1886, 33, 154–160. doi:10.1002/prac.18860330117
Return to citation in text: [1] -
Quader, M. A.; Sawmiller, C. J.; Sumpio, B. E. Radio Contrast Agents: History and Evolution. In Textbook of Angiology; Chang, J. B., Ed.; Springer: New York, 2000; pp 775–783. doi:10.1007/978-1-4612-1190-7_63
Return to citation in text: [1] -
Weissel, M. Acta Med. Austriaca 1991, 18, 6–11.
Return to citation in text: [1] -
Yuldasheva, G. A.; Zhidomirov, G. M.; Leszczynski, J.; Ilin, A. I. Iodine-containing drugs: complexes of molecular iodine and tri-iodide with bioorganic ligands and lithium halogenides in aqueous solutions. In Practical Aspects of Computational Chemistry IV; Leszczynski, J.; Shukla, M. K., Eds.; Springer: Boston, MA, 2016; pp 279–301. doi:10.1007/978-1-4899-7699-4_10
Return to citation in text: [1] -
Slater, S.; Numeroff, M. N. Engl. J. Med. 1961, 264, 449–450. doi:10.1056/NEJM196103022640908
Return to citation in text: [1] -
Akiyama, H.; Endo, M.; Matsui, T.; Katsuda, I.; Emi, N.; Kawamoto, Y.; Koike, T.; Beppu, H. Biochim. Biophys. Acta, Gen. Subj. 2011, 1810, 519–525. doi:10.1016/j.bbagen.2011.02.010
Return to citation in text: [1] -
Gao, H.; Jiang, Q.; Han, Y.; Peng, J.; Wang, C. Cell Biochem. Biophys. 2015, 71, 827–835. doi:10.1007/s12013-014-0270-4
Return to citation in text: [1] -
Kim, Y. H.; Park, C.; Lee, J. O.; Kim, G. Y.; Lee, W. H.; Choi, Y. H.; Ryu, C. H. Oncol. Rep. 2008, 19, 961–967. doi:10.3892/or.19.4.961
Return to citation in text: [1] -
Petsophonsakul, P.; Pompimon, W.; Banjerdpongchai, R. Asian Pac. J. Cancer Prev. 2013, 14, 2885–2889. doi:10.7314/APJCP.2013.14.5.2885
Return to citation in text: [1] -
Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323
Return to citation in text: [1] [2] [3] [4] -
Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. Chem. Commun. 2017, 53, 3850–3853. doi:10.1039/C7CC01043E
Return to citation in text: [1] [2] [3] -
Das, P.; Shibata, N. J. Org. Chem. 2017, 82, 11915–11924. doi:10.1021/acs.joc.7b01690
Return to citation in text: [1] [2] [3] -
Wang, J.; Jia, S.; Okuyama, K.; Huang, K.; Tokunaga, E.; Sumii, Y.; Shibata, N. J. Org. Chem. 2017, 82, 11939–11945. doi:10.1021/acs.joc.7b01908
Return to citation in text: [1] [2] -
Sinai, Á.; Mészáros, Á.; Gáti, T.; Kudar, V.; Palló, A.; Novák, Z. Org. Lett. 2013, 15, 5654–5657. doi:10.1021/ol402600r
Return to citation in text: [1] [2] -
Zhou, B.; Hou, W.; Yang, Y.; Feng, H.; Li, Y. Org. Lett. 2014, 16, 1322–1325. doi:10.1021/ol500033w
Return to citation in text: [1] [2] [3] -
Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a
Return to citation in text: [1] [2] -
Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373
Return to citation in text: [1] -
Diemer, V.; Leroux, F. R.; Colobert, F. Eur. J. Org. Chem. 2011, 2, 327–340. doi:10.1002/ejoc.201001217
Return to citation in text: [1]
| 20. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 22. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045 |
| 59. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 60. | Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. Chem. Commun. 2017, 53, 3850–3853. doi:10.1039/C7CC01043E |
| 61. | Das, P.; Shibata, N. J. Org. Chem. 2017, 82, 11915–11924. doi:10.1021/acs.joc.7b01690 |
| 62. | Wang, J.; Jia, S.; Okuyama, K.; Huang, K.; Tokunaga, E.; Sumii, Y.; Shibata, N. J. Org. Chem. 2017, 82, 11939–11945. doi:10.1021/acs.joc.7b01908 |
| 63. | Sinai, Á.; Mészáros, Á.; Gáti, T.; Kudar, V.; Palló, A.; Novák, Z. Org. Lett. 2013, 15, 5654–5657. doi:10.1021/ol402600r |
| 64. | Zhou, B.; Hou, W.; Yang, Y.; Feng, H.; Li, Y. Org. Lett. 2014, 16, 1322–1325. doi:10.1021/ol500033w |
| 20. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 1. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 2. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C |
| 3. | Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/S0022-1139(01)00375-X |
| 18. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 19. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 4. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 5. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527651351 |
| 6. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 7. | Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833 |
| 8. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 9. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell, 2009. |
| 59. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 60. | Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. Chem. Commun. 2017, 53, 3850–3853. doi:10.1039/C7CC01043E |
| 61. | Das, P.; Shibata, N. J. Org. Chem. 2017, 82, 11915–11924. doi:10.1021/acs.joc.7b01690 |
| 12. | Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b |
| 13. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 16. | Kawai, H.; Shibata, N. Chem. Rec. 2014, 14, 1024–1040. doi:10.1002/tcr.201402023 |
| 17. | Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014 |
| 18. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 19. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 20. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 22. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045 |
| 23. | Saidalimu, I.; Suzuki, S.; Tokunaga, E.; Shibata, N. Chem. Sci. 2016, 7, 2106–2110. doi:10.1039/C5SC04208A |
| 29. | Dinges, J.; Lamberth, C. Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. |
| 30. | Gomtsyan, A. Chem. Heterocycl. Compd. 2012, 48, 7–10. doi:10.1007/s10593-012-0960-z |
| 31. | Kosjek, T.; Heath, E. Halogenated heterocycles as pharmaceuticals. In Halogenated Heterocycles; Iskra, J., Ed.; Springer-Verlag: Berlin, Heidelberg, 2011; Vol. 27, pp 219–246. doi:10.1007/7081_2011_61 |
| 32. | Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi, O. Org. Biomol. Chem. 2016, 14, 6611–6637. doi:10.1039/C6OB00936K |
| 67. | Diemer, V.; Leroux, F. R.; Colobert, F. Eur. J. Org. Chem. 2011, 2, 327–340. doi:10.1002/ejoc.201001217 |
| 10. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 11. | Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386 |
| 12. | Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b |
| 13. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 14. | Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c |
| 15. | Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/C6CS00351F |
| 12. | Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b |
| 13. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 16. | Kawai, H.; Shibata, N. Chem. Rec. 2014, 14, 1024–1040. doi:10.1002/tcr.201402023 |
| 17. | Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014 |
| 18. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 19. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 20. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 22. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045 |
| 23. | Saidalimu, I.; Suzuki, S.; Tokunaga, E.; Shibata, N. Chem. Sci. 2016, 7, 2106–2110. doi:10.1039/C5SC04208A |
| 33. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 4. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 5. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527651351 |
| 6. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 7. | Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833 |
| 8. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 9. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell, 2009. |
| 24. | Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Inoue, Y.; Hara, H.; Shibata, N. PLoS One 2017, 12, e0182152. doi:10.1371/journal.pone.0182152 |
| 25. | Maeno, M.; Kondo, H.; Tokunaga, E.; Shibata, N. RSC Adv. 2016, 6, 85058–85062. doi:10.1039/C6RA21253K |
| 26. | Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810 |
| 27. | Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059 |
| 28. | Nishimine, T.; Taira, H.; Mori, S.; Matsubara, O.; Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Shibata, N. Chem. Commun. 2017, 53, 1128–1131. doi:10.1039/C6CC08830A |
| 27. | Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059 |
| 24. | Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Inoue, Y.; Hara, H.; Shibata, N. PLoS One 2017, 12, e0182152. doi:10.1371/journal.pone.0182152 |
| 25. | Maeno, M.; Kondo, H.; Tokunaga, E.; Shibata, N. RSC Adv. 2016, 6, 85058–85062. doi:10.1039/C6RA21253K |
| 26. | Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810 |
| 27. | Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059 |
| 28. | Nishimine, T.; Taira, H.; Mori, S.; Matsubara, O.; Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Shibata, N. Chem. Commun. 2017, 53, 1128–1131. doi:10.1039/C6CC08830A |
| 29. | Dinges, J.; Lamberth, C. Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. |
| 30. | Gomtsyan, A. Chem. Heterocycl. Compd. 2012, 48, 7–10. doi:10.1007/s10593-012-0960-z |
| 25. | Maeno, M.; Kondo, H.; Tokunaga, E.; Shibata, N. RSC Adv. 2016, 6, 85058–85062. doi:10.1039/C6RA21253K |
| 37. | Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860 |
| 22. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045 |
| 23. | Saidalimu, I.; Suzuki, S.; Tokunaga, E.; Shibata, N. Chem. Sci. 2016, 7, 2106–2110. doi:10.1039/C5SC04208A |
| 26. | Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810 |
| 65. | Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a |
| 66. | Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373 |
| 12. | Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 20. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 21. | Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782–8785. doi:10.1021/ja402455f |
| 22. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 233–237. doi:10.1002/open.201402045 |
| 59. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 60. | Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. Chem. Commun. 2017, 53, 3850–3853. doi:10.1039/C7CC01043E |
| 61. | Das, P.; Shibata, N. J. Org. Chem. 2017, 82, 11915–11924. doi:10.1021/acs.joc.7b01690 |
| 62. | Wang, J.; Jia, S.; Okuyama, K.; Huang, K.; Tokunaga, E.; Sumii, Y.; Shibata, N. J. Org. Chem. 2017, 82, 11939–11945. doi:10.1021/acs.joc.7b01908 |
| 63. | Sinai, Á.; Mészáros, Á.; Gáti, T.; Kudar, V.; Palló, A.; Novák, Z. Org. Lett. 2013, 15, 5654–5657. doi:10.1021/ol402600r |
| 64. | Zhou, B.; Hou, W.; Yang, Y.; Feng, H.; Li, Y. Org. Lett. 2014, 16, 1322–1325. doi:10.1021/ol500033w |
| 13. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 18. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 19. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 24. | Tokunaga, E.; Akiyama, H.; Soloshonok, V. A.; Inoue, Y.; Hara, H.; Shibata, N. PLoS One 2017, 12, e0182152. doi:10.1371/journal.pone.0182152 |
| 59. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 41. | Shimizu, I.; Matsumura, Y.; Inomata, Y. Preparation of 1,2-diarylethylene derivatives as drug intermediates. Jpn. Kokai Tokkyo Koho JP 63275533, Nov 14, 1988. |
| 42. | Kuik, W.-J.; Kema, I. P.; Brouwers, A. H.; Zijlma, R.; Neumann, K. D.; Dierckx, R. A. J. O.; DiMagno, S. G.; Elsinga, P. H. J. Nucl. Med. 2015, 56, 106–112. doi:10.2967/jnumed.114.145730 |
| 43. | DeSolms, S. J.; Woltersdorf, O. W., Jr.; Cragoe, E. J., Jr.; Watson, L. S.; Fanelli, G. M., Jr. J. Med. Chem. 1978, 215, 437–443. doi:10.1021/jm00203a006 |
| 33. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 64. | Zhou, B.; Hou, W.; Yang, Y.; Feng, H.; Li, Y. Org. Lett. 2014, 16, 1322–1325. doi:10.1021/ol500033w |
| 65. | Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a |
| 33. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 34. | Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j |
| 35. | Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 36. | Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651 |
| 37. | Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860 |
| 38. | Ito, M.; Itani, I.; Toyoda, Y.; Morimoto, K.; Dohi, T.; Kita, Y. Angew. Chem., Int. Ed. 2012, 51, 12555–12558. doi:10.1002/anie.201206917 |
| 39. | Sokolovs, I.; Lubriks, D.; Suna, E. J. Am. Chem. Soc. 2014, 136, 6920–6928. doi:10.1021/ja502174d |
| 40. | Phipps, R. J.; McMurray, L.; Ritter, S.; Duong, H. A.; Gaunt, M. J. J. Am. Chem. Soc. 2012, 134, 10773–10776. doi:10.1021/ja3039807 |
| 55. | Akiyama, H.; Endo, M.; Matsui, T.; Katsuda, I.; Emi, N.; Kawamoto, Y.; Koike, T.; Beppu, H. Biochim. Biophys. Acta, Gen. Subj. 2011, 1810, 519–525. doi:10.1016/j.bbagen.2011.02.010 |
| 56. | Gao, H.; Jiang, Q.; Han, Y.; Peng, J.; Wang, C. Cell Biochem. Biophys. 2015, 71, 827–835. doi:10.1007/s12013-014-0270-4 |
| 57. | Kim, Y. H.; Park, C.; Lee, J. O.; Kim, G. Y.; Lee, W. H.; Choi, Y. H.; Ryu, C. H. Oncol. Rep. 2008, 19, 961–967. doi:10.3892/or.19.4.961 |
| 58. | Petsophonsakul, P.; Pompimon, W.; Banjerdpongchai, R. Asian Pac. J. Cancer Prev. 2013, 14, 2885–2889. doi:10.7314/APJCP.2013.14.5.2885 |
| 27. | Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059 |
| 50. | Willgerodt, C. J. Prakt. Chem. 1886, 33, 154–160. doi:10.1002/prac.18860330117 |
| 51. | Quader, M. A.; Sawmiller, C. J.; Sumpio, B. E. Radio Contrast Agents: History and Evolution. In Textbook of Angiology; Chang, J. B., Ed.; Springer: New York, 2000; pp 775–783. doi:10.1007/978-1-4612-1190-7_63 |
| 52. | Weissel, M. Acta Med. Austriaca 1991, 18, 6–11. |
| 53. | Yuldasheva, G. A.; Zhidomirov, G. M.; Leszczynski, J.; Ilin, A. I. Iodine-containing drugs: complexes of molecular iodine and tri-iodide with bioorganic ligands and lithium halogenides in aqueous solutions. In Practical Aspects of Computational Chemistry IV; Leszczynski, J.; Shukla, M. K., Eds.; Springer: Boston, MA, 2016; pp 279–301. doi:10.1007/978-1-4899-7699-4_10 |
| 54. | Slater, S.; Numeroff, M. N. Engl. J. Med. 1961, 264, 449–450. doi:10.1056/NEJM196103022640908 |
| 46. | Lu, J.; Risbood, P.; Kane, C. T., Jr.; Hossain, M. T.; Anderson, L.; Hill, K.; Monks, A.; Wu, Y.; Antony, S.; Juhasz, A.; Liu, H.; Jiang, G.; Harris, E.; Roy, K.; Meitzler, J. L.; Konaté, M.; Doroshow, J. H. Biochem. Pharmacol. 2017, 143, 25–38. doi:10.1016/j.bcp.2017.07.007 |
| 49. | Menkissoglu-Spiroudi, U.; Karamanoli, K.; Spyroudis, S.; Constantinidou, H.-I. A. J. Agric. Food Chem. 2001, 49, 3746–3752. doi:10.1021/jf010293v |
| 44. | Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+ |
| 45. | Goldstein, E. J. C.; Citron, D. M.; Warren, Y.; Merriam, C. V.; Tyrrell, K.; Fernandez, H.; Radhakrishnan, U.; Stang, P. J.; Conrads, G. Antimicrob. Agents Chemother. 2004, 48, 2766–2770. doi:10.1128/AAC.48.7.2766-2770.2004 |
| 46. | Lu, J.; Risbood, P.; Kane, C. T., Jr.; Hossain, M. T.; Anderson, L.; Hill, K.; Monks, A.; Wu, Y.; Antony, S.; Juhasz, A.; Liu, H.; Jiang, G.; Harris, E.; Roy, K.; Meitzler, J. L.; Konaté, M.; Doroshow, J. H. Biochem. Pharmacol. 2017, 143, 25–38. doi:10.1016/j.bcp.2017.07.007 |
| 47. | Wang, L.; Xu, S.; Qian, X.; Wu, X.; Han, J.; Qu, D.; Tian, H. Diaryliodonium salt compound as antibacterial agent. Faming Zhuanli Shenqing CN 106748940, May 31, 2017. |
| 48. | Ding, Y.; Zhu, W.; Sun, R.; Yuan, G.; Zhang, D.; Fan, Y.; Sun, J. Oncol. Rep. 2015, 33, 1434–1442. doi:10.3892/or.2015.3726 |
| 49. | Menkissoglu-Spiroudi, U.; Karamanoli, K.; Spyroudis, S.; Constantinidou, H.-I. A. J. Agric. Food Chem. 2001, 49, 3746–3752. doi:10.1021/jf010293v |
| 45. | Goldstein, E. J. C.; Citron, D. M.; Warren, Y.; Merriam, C. V.; Tyrrell, K.; Fernandez, H.; Radhakrishnan, U.; Stang, P. J.; Conrads, G. Antimicrob. Agents Chemother. 2004, 48, 2766–2770. doi:10.1128/AAC.48.7.2766-2770.2004 |
© 2018 Das et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)