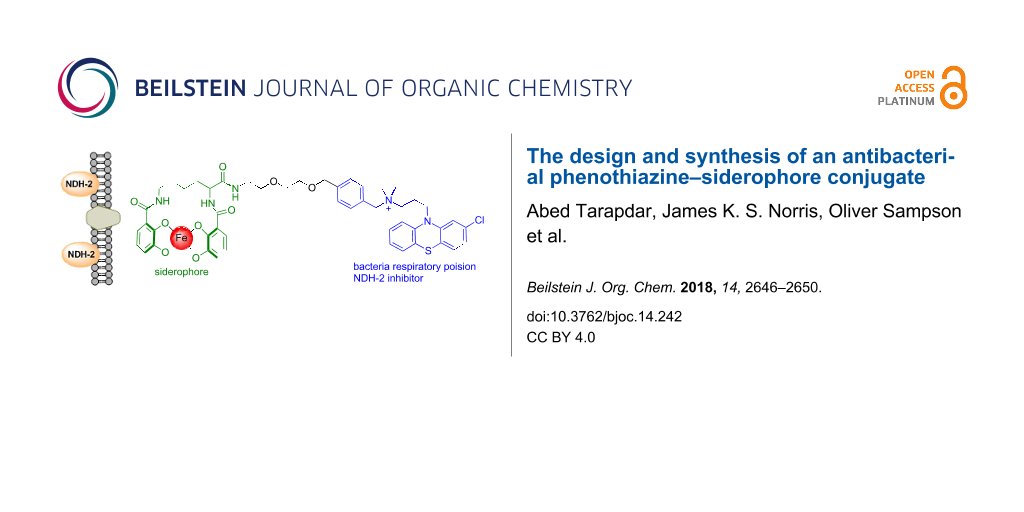
Abstract
Siderophore–antibiotic conjugates consist of an antibiotic covalently linked by a tether to a siderophore. Such conjugates can demonstrate enhanced uptake and internalisation to the bacterial cell resulting in significantly reduced MIC values and extended spectrum of activity. Phenothiazines are a class of small molecules that have been identified as a potential treatment for multidrug resistant tuberculosis and latent TB. Herein we report the design and synthesis of the first phenothiazine–siderophore conjugate. A convergent synthetic route was developed whereby the functionalised phenothiazine component was prepared in four steps and the siderophore component also prepared in four steps. In M. smegmatis the functionalised phenothiazine demonstrated an equipotent MIC value in direct comparison to the parent phenothiazine from which it was derived. The final conjugate was synthesised by amide bond formation between the two components and global deprotection of the PMB protecting groups to unmask the catechol iron chelating groups of the siderophore. The synthesis is readily amenable to the preparation of analogues whereby the siderophore component of the conjugate can be modified. The route will be used to prepare a library of siderophore–phenothiazine conjugates for full biological evaluation of much needed new antibacterial agents.

Graphical Abstract
Introduction
One of the biggest challenges facing the modern society is antibiotic resistance and the prospect of current antibiotics becoming near redundant against previously treatable infections [1]. To meet this challenge there is a desperate need for new antibiotics, antibiotic targets and strategies to enhance the efficacy of current antibiotics [2]. One novel strategy which is receiving significant interest is the manipulation of bacterial iron transport pathways to deliver antibiotics to the bacterial cell [3]. Iron is essential for bacterial survival and bacteria secrete high affinity iron chelating molecules to scavenge and solubilise Fe3+ from the extracellular environment [3]. The siderophore–Fe complex is recognised by specific receptor proteins on the outer membrane of the bacteria and internalised into the bacterium cell by active transport [4].
Siderophore–antibiotic conjugates consist of an antibiotic covalently linked by a ‘tether' to a siderophore. Such conjugates overcome the bacterial membrane permeability barrier and facilitate active transport of the antibiotic to its internal target. Siderophore–antibiotic conjugates can demonstrate significantly enhanced bacterial killing potencies and an extended spectrum of activity [5,6]. Although there has been success reported with a number of antibiotics with differing targets the most success to date has been achieved with beta-lactam-based siderophore conjugates targeting membrane associated penicillin binding proteins (PBPs) [7]. Cefiderocol (S-649266) is a beta-lactam–siderophore conjugate currently in phase III clinical trials which demonstrates enhanced potency against Gram-negative bacteria including multidrug resistant (MDR) Gram-negative pathogens [8]. One hypothesis for the success of siderophore conjugates targeting PBPs, in comparison to other antibiotic targets, is that PBPs are membrane associated and it is not necessary for the siderophore conjugate to cross into the bacterial cytoplasm [7].
Phenothiazines are a privileged scaffold in drug discovery most noted for their use as antipsychotic drugs including chlorpromazine, trifluoperazine, and thioridazine. However, such drugs have also long been noted for their significant antimicrobial activity particularly against Staphylococcus aureus and Mycobacterium tuberculosis [9,10]. The emergence of MDR-TB has led to structure–activity studies to enhance the antitubercular activity of phenothiazines leading to the identification of chlorpromazine analogue 1 (Figure 1) which demonstrates MIC values comparable to first-line TB drugs in vitro [11]. However, the potency of such phenothiazines, including 1, needs to be significantly increased to have more activity in vivo and direct clinical application [11]. A validated target of 1 has been identified as type II NADH dehydrogenase (NDH-2), a respiratory enzyme essential for growth in M. tuberculosis and other bacterial species [11]. NDH-2 is absent in mammalian cells and similar to PBPs is associated with the bacterial membrane [12]. Considering the significant antibacterial activity of phenothiazines, in particular the anti-TB activity of 1, and their membrane-associated NDH-2 target we hypothesised 1 may be an interesting candidate for siderophore conjugation.
![[1860-5397-14-242-1]](/bjoc/content/figures/1860-5397-14-242-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: NDH-2 is a validated target for 1 with an MIC of 1.1 µg/mL against M. tuberculosis.
Figure 1: NDH-2 is a validated target for 1 with an MIC of 1.1 µg/mL against M. tuberculosis.
Results and Discussion
Typically siderophore–antibiotic conjugates consist of a linker joining the siderophore and antibiotic components. As the target is membrane-associated NDH-2 we decided to functionalise our conjugate with a non-cleavable linker. A polyethylene glycol (PEG) linker was selected as PEG linkers demonstrate enhanced water solubility in comparison to alkyl chain linkers. We then had to make a decision on the position of attachment for the PEG linker to compound 1. For siderophore conjugates, it is crucial that the linker is attached to a position in 1 such that the antibacterial activity is not compromised. Based on previous structure–activity studies of 1 by Bate et al., whereby a methoxy group was positioned on the para-position of the phenyl ring of 1 without loss of activity, we hypothesised this may be a suitable position for PEG linker attachment [13].
From commercially available 2-(2-aminoethoxy)ethanol the amine functionality was Boc-protected under standard conditions to give compound 2 (Scheme 1). Under basic conditions 2 underwent an SN2 reaction with commercially available p-xylylene dichloride to give 3. Complete conversion of starting material was observed by 1H NMR, however, the isolated yield of 3 was poor possibly due to competing N-alkylation of the Boc group. Isolation of the O-alkyl product 3 was confirmed by 13C NMR. Despite the poor isolated yield of 3 the mass recovery was more than suitable to progress to the next steps. Initially, the reaction of 3 with chlorpromazine (free base) was attempted at room temperature; however, it was found refluxing was required to drive the reaction to completion to generate compound 4 in excellent yield. The final PEG-amine-functionalised phenothiazine 5 was isolated after removal of the Boc protecting group in TFA. Initial attempts at aqueous work-up conditions to isolate the free base resulted in lower isolated yields of 5 due to its high water solubility, and it was decided 5 would be progressed further as the TFA salt avoiding aqueous work-up.
![[1860-5397-14-242-i1]](/bjoc/content/inline/1860-5397-14-242-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of phenothiazine-PEG-amine component.
Scheme 1: Synthesis of phenothiazine-PEG-amine component.
To determine if the antibacterial activity of the derivatised phenothiazine was retained the MIC of compound 4 was determined in direct comparison to synthesised 1 against Mycobacterium smegmatis (see Supporting Information File 1). M. smegmatis is commonly used as a first assessment for antituberculosis activity. We were pleased to observe side by side compound 4 exhibited equimolar MIC values to 1 (6.25 μM, 1 and 4) against M. smegmatis.
Next our attention turned to the siderophore component of the conjugate. In our proof of concept study we chose to synthesise the bis-catechol siderophore azotochelin. Catechol-based siderophores can act as xenosiderophores and be recognised for uptake by Gram-negative bacteria and mycobacteria [14,15]. Most commonly benzyl protecting groups are used in the synthesis of catechol siderophores and cleaved in the final step by palladium catalysed hydrogenation. However, as our final conjugate contains an aromatic halide we wanted to avoid hydrogenation as the final step and we instead chose to use the para-methoxybenzyl (PMB) protecting group which can be removed under acidic conditions.
PMB-protected benzoic acid building block 7 was prepared following a literature procedure in two steps (Scheme 2) [16]. Building block 7 was coupled to commercially available L-lysine methyl ester dihydrochloride to yield 8 in moderate yield. The majority of the dicyclohexylurea byproduct could be removed by cooling a solution of the residue dissolved in acetonitrile; however, column chromatography was required for analytical pure material. Ester hydrolysis proceeded smoothly in excellent yield to give the protected siderophore component 9.
![[1860-5397-14-242-i2]](/bjoc/content/inline/1860-5397-14-242-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the azotochelin siderophore component.
Scheme 2: Synthesis of the azotochelin siderophore component.
Finally the phenothiazine component 5 and siderophore component 9 where coupled together by amide bond formation using HATU (Scheme 3). Although the isolated yield of 10 is poor the reaction proceeded with good conversion to the desired product; however, on purification an unknown contaminant was challenging to separate from 9 and we wanted to progress with only analytically pure material for the final deprotection step. We were also surprised to observe compound 10 had undergone racemisation under these conditions. The exact cause of racemisation is unknown, but may possibly be due to the four equivalents of DIPEA used to ensure 5 is converted to its free base. This will be investigated further for the synthesis of future conjugates. In the final PMB global deprotection step we were pleased to observe the formation of our desired final phenothiazine–sideophore conjugate in moderate yield. The addition of anisole to the reaction mixture was found to be essential to inhibit competing electrophilic substitution side reactions. A number of techniques where investigated for purification including standard chromatography, recrystallization and trituration as the crude 1H NMR revealed the majority of the desired product. However, purification by semi-preparative HPLC was required to obtain analytically pure material for biological evaluation.
![[1860-5397-14-242-i3]](/bjoc/content/inline/1860-5397-14-242-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Final conjugation and deprotection to yield a phenothiazine siderophore conjugate.
Scheme 3: Final conjugation and deprotection to yield a phenothiazine siderophore conjugate.
Conclusion
In conclusion we have developed a novel synthetic route to the first phenothiazine–siderophore conjugate. This was achieved by a convergent two component synthesis in a total of ten synthetic steps. The work extends research into antibacterial phenothiazines and siderophore-mediated antibiotic delivery. A library of mono-, bis- and tris-catechol phenothiazine–siderophore conjugates are currently being prepared using this route. Their synthesis and MIC values against pathogenic mycobacteria, Gram-negative bacteria and Gram-positive bacteria, along with compound 11, will be reported in due course.
Supporting Information
| Supporting Information File 1: Full experiential protocols, characterisation of compounds including 1H and 13C NMR spectra, and biological evaluation of compound 4. | ||
| Format: PDF | Size: 948.4 KB | Download |
Acknowledgements
We thank the Royal Society for funding this research. We would also like to acknowledge Mick Lee for HRMS data of final compounds, Gerald Griffith and Vanessa Timmermann for aid in NMR spectroscopy experiments of final compounds. We would like to thank Professor Paul Cullis for proof reading the manuscript.
References
-
Prestinaci, F.; Pezzotti, P.; Pantosti, A. Pathog. Global Health 2015, 109, 309–318. doi:10.1179/2047773215Y.0000000030
Return to citation in text: [1] -
O’Connell, K. M. G.; Hodgkinson, J. T.; Sore, H. F.; Welch, M.; Salmond, G. P. C.; Spring, D. R. Angew. Chem., Int. Ed. 2013, 52, 10706–10733. doi:10.1002/anie.201209979
Return to citation in text: [1] -
Schalk, I. J. J. Med. Chem. 2018, 61, 3842–3844. doi:10.1021/acs.jmedchem.8b00522
Return to citation in text: [1] [2] -
Rainesa, D. J.; Moroz, O. V.; Blagovab, E. V.; Turkenburgb, J. P.; Wilson, K. S.; Duhme-Klair, A.-K. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5850–5855. doi:10.1073/pnas.1520829113
Return to citation in text: [1] -
Zheng, T.; Nolan, E. M. J. Am. Chem. Soc. 2014, 136, 9677–9691. doi:10.1021/ja503911p
Return to citation in text: [1] -
Liu, R.; Miller, P. A.; Vakulenko, S. B.; Stewart, N. K.; Boggess, W. C.; Miller, M. J. J. Med. Chem. 2018, 61, 3845–3854. doi:10.1021/acs.jmedchem.8b00218
Return to citation in text: [1] -
Schalk, I. J.; Mislin, G. L. A. J. Med. Chem. 2017, 60, 4573–4576. doi:10.1021/acs.jmedchem.7b00554
Return to citation in text: [1] [2] -
Choi, J. J.; McCarthy, M. W. Expert Opin. Invest. Drugs 2018, 27, 193–197. doi:10.1080/13543784.2018.1426745
Return to citation in text: [1] -
Sharma, S.; Singh, A. Expert Opin. Invest. Drugs 2011, 20, 1665–1676. doi:10.1517/13543784.2011.628657
Return to citation in text: [1] -
Schurig-Briccio, L. A.; Yano, T.; Rubin, H.; Gennis, R. B. Biochim. Biophys. Acta 2014, 1837, 954–963. doi:10.1016/j.bbabio.2014.03.017
Return to citation in text: [1] -
Weinstein, E. A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A. A.; Duncan, K.; Lonsdale, J. T.; Rubin, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4548–4553. doi:10.1073/pnas.0500469102
Return to citation in text: [1] [2] [3] -
Sellamuthu, S.; Singh, M.; Kumar, A.; Singh, S. K. Expert Opin. Ther. Targets 2017, 21, 559–570. doi:10.1080/14728222.2017.1327577
Return to citation in text: [1] -
Bate, A. B.; Kalin, J. H.; Fooksman, E. M.; Amorose, E. L.; Price, C. M.; Williams, H. M.; Rodig, M. J.; Mitchell, M. O.; Cho, S. H.; Wang, Y.; Franzblau, S. G. Bioorg. Med. Chem. Lett. 2007, 17, 1346–1348. doi:10.1016/j.bmcl.2006.11.091
Return to citation in text: [1] -
Wittmann, S.; Heinisch, L.; Scherlitz-Hofmann, I.; Stoiber, T.; Ankel-Fuchs, D.; Möllmann, U. BioMetals 2004, 17, 53–64. doi:10.1023/A:1024409517626
Return to citation in text: [1] -
Schumann, G.; Möllmann, U. Antimicrob. Agents Chemother. 2001, 45, 1317–1322. doi:10.1128/AAC.45.5.1317-1322.2001
Return to citation in text: [1] -
Bergeron, R. J.; Bharti, N.; Singh, S.; McManis, J. S.; Wiegand, J.; Green, L. G. J. Med. Chem. 2009, 52, 3801–3813. doi:10.1021/jm900119q
Return to citation in text: [1]
| 16. | Bergeron, R. J.; Bharti, N.; Singh, S.; McManis, J. S.; Wiegand, J.; Green, L. G. J. Med. Chem. 2009, 52, 3801–3813. doi:10.1021/jm900119q |
| 1. | Prestinaci, F.; Pezzotti, P.; Pantosti, A. Pathog. Global Health 2015, 109, 309–318. doi:10.1179/2047773215Y.0000000030 |
| 4. | Rainesa, D. J.; Moroz, O. V.; Blagovab, E. V.; Turkenburgb, J. P.; Wilson, K. S.; Duhme-Klair, A.-K. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5850–5855. doi:10.1073/pnas.1520829113 |
| 13. | Bate, A. B.; Kalin, J. H.; Fooksman, E. M.; Amorose, E. L.; Price, C. M.; Williams, H. M.; Rodig, M. J.; Mitchell, M. O.; Cho, S. H.; Wang, Y.; Franzblau, S. G. Bioorg. Med. Chem. Lett. 2007, 17, 1346–1348. doi:10.1016/j.bmcl.2006.11.091 |
| 3. | Schalk, I. J. J. Med. Chem. 2018, 61, 3842–3844. doi:10.1021/acs.jmedchem.8b00522 |
| 14. | Wittmann, S.; Heinisch, L.; Scherlitz-Hofmann, I.; Stoiber, T.; Ankel-Fuchs, D.; Möllmann, U. BioMetals 2004, 17, 53–64. doi:10.1023/A:1024409517626 |
| 15. | Schumann, G.; Möllmann, U. Antimicrob. Agents Chemother. 2001, 45, 1317–1322. doi:10.1128/AAC.45.5.1317-1322.2001 |
| 3. | Schalk, I. J. J. Med. Chem. 2018, 61, 3842–3844. doi:10.1021/acs.jmedchem.8b00522 |
| 11. | Weinstein, E. A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A. A.; Duncan, K.; Lonsdale, J. T.; Rubin, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4548–4553. doi:10.1073/pnas.0500469102 |
| 2. | O’Connell, K. M. G.; Hodgkinson, J. T.; Sore, H. F.; Welch, M.; Salmond, G. P. C.; Spring, D. R. Angew. Chem., Int. Ed. 2013, 52, 10706–10733. doi:10.1002/anie.201209979 |
| 12. | Sellamuthu, S.; Singh, M.; Kumar, A.; Singh, S. K. Expert Opin. Ther. Targets 2017, 21, 559–570. doi:10.1080/14728222.2017.1327577 |
| 7. | Schalk, I. J.; Mislin, G. L. A. J. Med. Chem. 2017, 60, 4573–4576. doi:10.1021/acs.jmedchem.7b00554 |
| 11. | Weinstein, E. A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A. A.; Duncan, K.; Lonsdale, J. T.; Rubin, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4548–4553. doi:10.1073/pnas.0500469102 |
| 8. | Choi, J. J.; McCarthy, M. W. Expert Opin. Invest. Drugs 2018, 27, 193–197. doi:10.1080/13543784.2018.1426745 |
| 11. | Weinstein, E. A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A. A.; Duncan, K.; Lonsdale, J. T.; Rubin, H. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4548–4553. doi:10.1073/pnas.0500469102 |
| 7. | Schalk, I. J.; Mislin, G. L. A. J. Med. Chem. 2017, 60, 4573–4576. doi:10.1021/acs.jmedchem.7b00554 |
| 5. | Zheng, T.; Nolan, E. M. J. Am. Chem. Soc. 2014, 136, 9677–9691. doi:10.1021/ja503911p |
| 6. | Liu, R.; Miller, P. A.; Vakulenko, S. B.; Stewart, N. K.; Boggess, W. C.; Miller, M. J. J. Med. Chem. 2018, 61, 3845–3854. doi:10.1021/acs.jmedchem.8b00218 |
| 9. | Sharma, S.; Singh, A. Expert Opin. Invest. Drugs 2011, 20, 1665–1676. doi:10.1517/13543784.2011.628657 |
| 10. | Schurig-Briccio, L. A.; Yano, T.; Rubin, H.; Gennis, R. B. Biochim. Biophys. Acta 2014, 1837, 954–963. doi:10.1016/j.bbabio.2014.03.017 |
© 2018 Tarapdar et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)