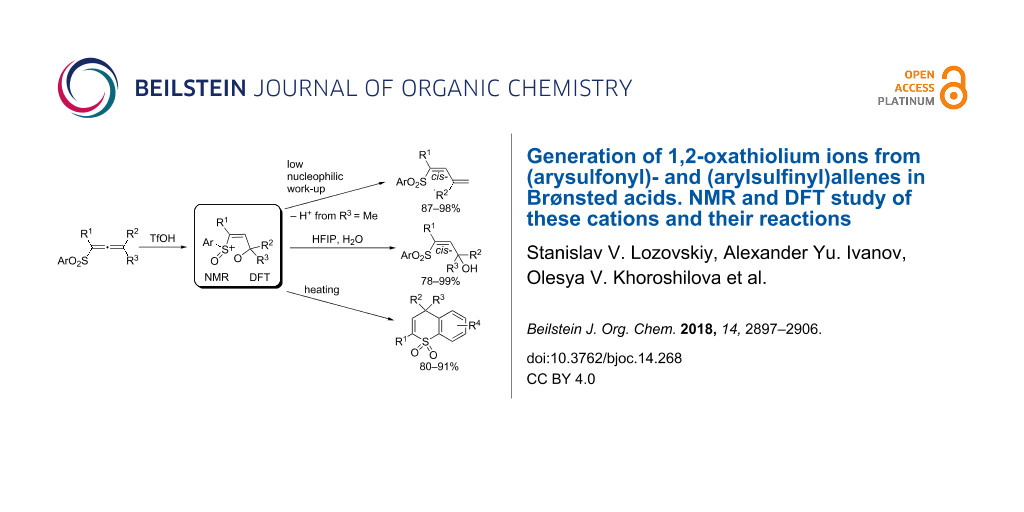
Abstract
In strong Brønsted acids (CF3SO3H, FSO3H, D2SO4), (arysulfonyl)allenes (ArSO2–CR1=C=CR2R3) and (arylsulfinyl)allenes (ArSO–CR1=C=CR2R3) undergo cyclization into the corresponding stable 1,2-oxathiolium ions, which were studied by means of NMR and DFT calculations. Quenching of solutions of these cations with low nucleophilic media, aqueous HCl, leads to their deprotonation with a stereoselective formation of (arysulfonyl)butadienes (for instance, ArSO2–CR1=C–C(Me)=CH2, for R2 = R3 = Me, yields of 87–98%). Reactions of (arysulfonyl)allenes in the system TfOH (0.1 equiv)–HFIP (hexafluoropropan-2-ol) followed by hydrolysis give rise to allyl alcohols (ArSO2–CR1=CH–C(OH)R2R3, yields of 78–99%). Reflux of solutions of (arysulfonyl)allenes in the presence of TfOH (1 equiv) in 1,2-dichlorobenzene leads to the cyclization into thiochromene 1,1-dioxides in high yields. Under the action of TfOH or AlX3 (X = Cl, Br) followed by hydrolysis of reaction mixtures, (arylsulfinyl)allenes give allyl alcohols (ArSO2–CR1=CH–C(OH)R2R3). Plausible reaction mechanisms have been proposed for all studied reactions.

Graphical Abstract
Introduction
Allenes are widely explored in organic synthesis for the construction of various molecules [1-7]. In particular, arylsulfonyl (ArSO2) allenes are usefull building blocks in miscellaneous transformations. For instance, addition of such allenes to Michael acceptors leading to terminal acetylenes has been recently shown [8]. These allenes give rise to pyrrolidines [9], pyrroles [10], chromenes [11], benzoazepinones [12], macrolides [13], and some other carbo- and heterocycles [14-16]. It should be specially emphasized that many compounds containing SO2 groups are drugs, such as, dapson [17], oxicams [18], or amisulpride [19]. Substantial contribution in this area was made by Harmata et al. [20-24]. However, to the best of our knowledge, electrophilic reactions of (arylsulfonyl)allenes have not been widely studied yet. It has been shown by Ma et al. that ArSO2-allenes take part into halogenohydroxylation (Hal = I, Br) or addition–elemination of bromine (forming bromobutadienes) with high stereoselectivity [25,26]. Apart from that, reactions of sulfur containing allenes were studied in acidic media [27,28]. Despite promising results, there was no further research in this area.
Based on our recent work on transformations of phosphonoallenes under the action of strong Brønsted or Lewis acids [29-32], we undertook a special study on reactions of (arylsulfonyl)allenes 2a–j and (arylsulfinyl)allenes 1a,b (Scheme 1).
![[1860-5397-14-268-i1]](/bjoc/content/inline/1860-5397-14-268-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (Arylsulfinyl)allenes 1 and (arylsulfonyl)allenes 2 used in this study.
Scheme 1: (Arylsulfinyl)allenes 1 and (arylsulfonyl)allenes 2 used in this study.
The reaction between proparylic alcohols and arylsulfanyl chlorides followed by acetylene–allene rearrangement was used to prepare (arylsulfinyl)allenes 1 according to the literature procedure [24,25]. The latter were in situ oxidized to (arylsulfonyl)allenes 2 (see X-ray structure of 2h in Figure 1). Allenes 1a,b were specially isolated to compare their reactivity with allenes 2.
![[1860-5397-14-268-1]](/bjoc/content/figures/1860-5397-14-268-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structures of compounds 2h (CCDC 1843276), 3e (CCDC 1843277), 5c (CCDC 1580895), 7b (CCDC 1843239); ellipsoid contours of probability levels are 50%.
Figure 1: X-ray crystal structures of compounds 2h (CCDC 1843276), 3e (CCDC 1843277), 5c (CCDC 1580895), 7b (...
The main goals of this work were the investigation of reactions of sulfur-containing allenes 1 and 2 under electrophilic activation with Brønsted or Lewis (super)acids, and the study on cationic intermediates of these reactions by means of NMR and DFT calculations.
Results and Discussion
First, the behavior of allenes 1a,b and 2a–h in Brønsted acids (TfOH, D2SO4) was studied by means of NMR (Table 1). Dissolving these allenes in TfOH or D2SO4 directly in NMR tubes at room temperature gave intensively colored red solutions of cationic species, which were stable for a long time. The NMR data, including 1H, 13C, DEPT, COSY, and HSQC spectra (see Supporting Information File 1), demonstrated unambiguously that compounds 1a,b and 2a–h underwent cyclization into the corresponding ions Aa,b and Ba–h via protonation (deuteration for D2SO4, Bd-d) of the central carbon allenic triad followed by nucleophilic attack of oxygen of the SO2 group (for Ba–h) or SO group (for Aa,b) onto the carbocationic center. The similar cyclization was observed for phosphonoallenes (see P1, P2, Table 1) by us previously [30,32]. A new signal of the attached proton H4 at δ 8.05–6.83 ppm range appeared in 1H NMR spectra of species A, B. The comparison of 13C NMR spectra of oxathiolylium A, B and oxaphospholium P1, P2 ions shows that for the former species the signal of carbon C5 is about 10–15 ppm downfield shifted relatively the same signal in the cations P1, P2 (Table 1). This reveals that carbon C5 bears a rather large positive charge in cations A, B. For dication Bh, different signals were detected for quaternary carbons C5 and C5', and vinyl carbons C4 and C4', etc., that, probably, indicates formation of two diastereomers (one meso-form and one pair of enantiomers) due to the stereogenic sulfur centers.
Table 1: Selected 1H and 13C NMR data for cations Aa,b and Ba–h, P1, P2 derived at the protonation of the corresponding allenes at room temperature in TfOH and D2SO4.
| initial allene | cation | acid | 1H NMR, δ, ppm | 13C NMR, δ, ppm | |||
|---|---|---|---|---|---|---|---|
| H3 | H4 | C3 | C4 | C5 | |||
| 1a |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-268-i8.svg?max-width=637&scale=1.0)
Aa |
TfOH | 6.85 d (J = 5.7 Hz) | 7.47 d (J = 5.7 Hz) | 117.2 | 150.2 | 112.4 |
| 1b |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-268-i9.svg?max-width=637&scale=1.0)
Ab |
TfOH | 6.83 d (J = 6.1 Hz) | 7.51 d (J = 6.1 Hz) | 117.2 | 150.6 | 112.8 |
| 2a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-268-i10.svg?max-width=637&scale=1.0)
Ba |
TfOH | 7.16 d (J = 6.2 Hz) | 8.05 d (J = 6.2 Hz) | 121.0 | 158.7 | 112.0 |
| 2b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-268-i11.svg?max-width=637&scale=1.0)
Bb |
TfOH | 7.17 d (J = 6.0 Hz) | 8.07 d (J = 6.0 Hz) | 122.4 | 159.6 | 113.0 |
| 2c |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-268-i12.svg?max-width=637&scale=1.0)
Bc |
TfOH | 7.12 d (J = 6.2 Hz) | 8.01 d (J = 6.2 Hz) | 121.3 | 157.1 | 110.2 |
| 2d |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-268-i13.svg?max-width=637&scale=1.0)
Bd |
TfOH | – | 7.95 s | 109.9 | 153.0 | 112.7 |
| 2d |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-268-i14.svg?max-width=637&scale=1.0)
Bd-d |
D2SO4 | – | – | 109.0 | 153.0 | 113.0 |
| 2e |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-268-i15.svg?max-width=637&scale=1.0)
Be |
TfOH | – | 7.91 | 123.4 | 154.1 | 110.0 |
| 2f |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-268-i16.svg?max-width=637&scale=1.0)
Bf 2 isomers in a ratio of 5:1a |
TfOH | – | 8.02 | 115.6 | 154.1 | 119.3 |
| 2g |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-268-i17.svg?max-width=637&scale=1.0)
Bg |
TfOH | 7.11 d (J = 6.2 Hz) | 8.02 d (J = 6.2 Hz) | 122.6 | 157.7 | 114.7 |
| 2h |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-268-i18.svg?max-width=637&scale=1.0)
Bh |
TfOH | – | 8.35 and 8.09 | 120.9 and 120.7 |
157.9
and 157.1 |
112.8 and 112.2 |
|
Data from
ref. [30] |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-268-i19.svg?max-width=637&scale=1.0)
P1 |
TfOH | – | 7.78 d (JHP = 28 Hz) | 102.0 | 164.3 | 103.6 |
| Data from ref. [32] |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-268-i20.svg?max-width=637&scale=1.0)
P2 |
TfOH | – | 7.84 d (JHP = 46 Hz) | 96.0 | 169.1 | 102.8 |
aNMR data for major isomer.
In the case of the cation Bf, the signals of two isomers were found in the spectra in a ratio of 5 to 1. These isomers appear due to cis-, trans-orientation of t-Bu and ArS groups in the five-membered ring.
To the best of our knowledge, this is one of the first examples of full NMR characterization of such broad series of cyclic sulfur containing cations Aa,b and Ba–h. Allenes 2i,j did not react with acids at room temperature, however, they react with TfOH at higher temperature (see below).
To estimate the charge distribution in species Aa, Ba we carried out DFT calculations (Table 2). The calculations confirm the experimental NMR data (Table 1) and prove that C5 does have a large positive charge 0.25 e, which should make this carbon a highly reactive electrophilic center. Another electrophilic center is the sulfur atom, which also bears a large positive charge (1.21–2.06 e). Apart from that, the atomic coefficient of contribution in the LUMO for sulfur is much higher than for C5. Thus, the electrophilic reactivity of sulfur may be explained by both charge and orbital control. Also, ortho-carbons in the phenyl group bear a negative charge −0.17 to −0.16 e; this means that intramolecular cyclization on these atoms is possible.
Table 2: Selected electronic characteristics of cations Aa, Ba generated from allenes 1a, 2a, correspondingly (DFT calculations).
| cation | q(S)a e | q(C3)a e | q(C4)a e | q(C5)a e | q(Co-Ph)a e | kLUMOb % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | C3 | C4 | C5 | ∑CPh (ortho + para + ipso) | ||||||
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-268-i21.svg?max-width=637&scale=1.0)
Aa |
1.21 | −0.38 | −0.13 | 0.25 | −0.17 | 35 | 4 | 0 | 2 | 38 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-268-i22.svg?max-width=637&scale=1.0)
Ba |
2.06 | −0.43 | −0.10 | 0.25 | −0.16 | 11 | 14 | 25 | 1 | 47 |
aNatural charges. bContribution of atomic orbital into the molecular orbital.
To conclude the study on electronic characteristics of 1,2-oxathiolylium ions A, B by means of NMR and DFT calculations, one should expect that these species may react in several pathways. First, they may undergo nucleophilic attack on sulfur or on carbon C5, due to a high positive charge on it. Another pathway may be an electrophilic cyclization at the ortho-carbon in the S-phenyl ring.
Then, the preparative reactions of allene 2a under the action of different electrophilic reagents were conducted. Transformations of 2a using an excess of various Brønsted acids followed by aqueous quenching of the reaction mixture are shown in Table 3. These reactions resulted in the formation of three different products, Z-3a, Z-4a and 5a, depending on the reaction conditions. At room temperature in CF3SO3H or H2SO4 for a short time, 10 min or 1 h, respectively, butadiene Z-3a and alcohol Z-4a were formed (Table 3, entries 1 and 3). Increasing the reaction temperature to 60 °C and the time to 8 h in CF3SO3H led to the formation of thiochromene 1,1-dioxide 5a (Table 3, entry 2). Decreasing the reaction temperature down to −60 °C in FSO3H with work-up of the superacidic reaction solution with a low nucleophilic medium (frozen aqueous HCl at −60 °C) gave almost quantitatively butadiene Z-3a with a small admixture of its E-isomer (Table 3, entry 4). Weaker acids, CF3CO2H or aqueous HCl, did not activate allene 2a, no reactions took place (Table 3, entries 5 and 6). Apart from that, Lewis acids of various strength (AlCl3, AlBr3, FeCl3, CeCl3, BF3-Et2O, In(OTf)3) were found to be ineffective for this transformation, no reactions of allenes 2 occurred with them.
Table 3: Reactions of allene 2a under the action of various Brønsted acids.
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-268-i23.svg?max-width=637&scale=1.0)
|
||||||
| entry | reaction conditions | reaction products, yield, % | ||||
| acid (equiv) | temperature, °C | time | Z-3a | Z-4a | 5a | |
| 1 | TfOH (40) | rt | 10 min | 18 | 81 | – |
| 2 | TfOH (40) | 60 | 8 h | – | – | 40 |
| 3 | H2SO4 (40) | rt | 1 h | 20 | 20 | – |
| 4 | FSO3H (40)a | −60 | 1 h | 90 (+ E-3a, 9%) | – | – |
| 5 | CF3CO2H (40)b | 50 | 24 h | – | – | – |
| 6 | HClaq (40)b | rt | 24 h | – | – | – |
aWork-up with frozen aqueous HCl at −60 °C. bQuantitative recovery of starting 2a.
Taking into account the data on the formation of cations B (Table 1), their electronic characteristics (Table 2), and reactions of allene 2a in Brønsted acids (Table 3), one may propose a plausible mechanism for the transformation of 2a (Scheme 2). Protonation of the allene system gives cation C, which is cyclized into stable species Ba. Upon work-up of the acidic reaction solution, the fate of the cation Ba strongly depends on the nucleophilicity of the quenching medium. Under the conditions of low nucleophilic work-up with aqueous HCl (Table 3, entry 4), the deprotonation takes place leading to butadiene Z-3a. The predominant formation of the Z-isomer of 3a may reveal that cation Ba undergoes deprotonation and recyclization, rather than species C. Quenching of species Ba with water (high nucleophilicity) affords alcohol Z-4a. The formation of compound 4a in exclusively Z-configuration may indicate that cation Ba reacts with H2O in SN2 manner, keeping in mind that carbon C5 in Ba possesses a large positive charge (see data in Table 1 and Table 2). An alternative mechanism of the formation of alcohol 4a includes the attack of H2O on the sulfur electrophilic center giving intermediate D, which is rearranged into alcohol Z-4a. Preparation of sulfur heterocycle 5a at high reaction temperature (Table 3, entry 2) shows that the intramolecular cyclization to the ortho-carbon of phenyl ring occurs, most probably, through cation C (Scheme 2). And this reaction has a high activation barrier, analogously to the similar cyclization of phosphonoallenes to the corresponding phosphonoheterocycles [30].
![[1860-5397-14-268-i2]](/bjoc/content/inline/1860-5397-14-268-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanisms of transformations of allene 2a in Brønsted acids.
Scheme 2: Plausible reaction mechanisms of transformations of allene 2a in Brønsted acids.
We decided to achieve the selective formation of each of these different products, butadienes 3, adducts with nucleophiles 4, and thiochromene 1,1-dioxides 5, from allenes 2. The preparation of compounds 3a–h was done by the following method (Scheme 3). Reactions of 2a–h were carried out in CH2Cl2 with 1 equivalent of TfOH to generate the corresponding cations Ba–h. Then, the reaction mixture was cooled down to −35 °C and quenched under very mild and low nucleophilic conditions with frozen aqueous HCl at −60 °C, that finally led quantitatively to butadienes 3a–h (see X-ray structure of 3e in Figure 1). Worth noting, that compounds 3a–h have strictly cis-configuration of SO2Ar group and a vinyl substituent at C2 carbon. It should be mentioned that palladium-catalyzed isomerization of such (arylsulfonyl)allenes 2 into trans-butadienes 3 was described recently [23]. Herein, we have developed a novel metal-free approach for the synthesis of cis-isomers of 3.
![[1860-5397-14-268-i3]](/bjoc/content/inline/1860-5397-14-268-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Selective formation of butadienes 3a–h from allenes 2a–h.
Scheme 3: Selective formation of butadienes 3a–h from allenes 2a–h.
Then, we tried to get selectively products of the nucleophilic attack onto cations B (like structure Z-4a in Scheme 2) by quenching of the acidic reaction solutions (in TfOH) with various nucleophiles (water, methanol, benzene, acetonitrile). But, in all cases, these reactions were unselective. For instance, for allene 2a, mixtures of butadiene Z-3a and alcohol Z-4a were obtained. To overcome this obstacle we decided to use 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP), which was known to form the corresponding ether for further substitution reactions [33]. Indeed, the use of HFIP and a catalytic amount of TfOH (0.1 equiv) followed by hydrolysis allowed to achieve an exclusive formation of allyl alcohols Z-4a,b and E-4c from allenes 2a,b,d, respectively, in high yields (Scheme 4). The most probably, this reaction proceeds through intermediate formation of ethers E from the corresponding species B. The ethers E are hydrolyzed to compounds 4. Reactions of allenes 2 with other nucleophiles (methanol, benzene, acetonitrile) led to the formation of complex mixtures of reaction products. It must be noted that no reaction proceeded in HFIP without TfOH.
![[1860-5397-14-268-i4]](/bjoc/content/inline/1860-5397-14-268-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of allenes 2 in the system HFIP/TfOH followed by interaction with nucleophiles leading to allyl alcohols 4.
Scheme 4: Reactions of allenes 2 in the system HFIP/TfOH followed by interaction with nucleophiles leading to...
The assignment of the cis-configuration of the ArSO2 group and the C3-substituent in compounds 3 and 4 was based on the low spin–spin interaction constant of 8.0–11.8 Hz between the vinyl protons in the 1H NMR spectrum (see Supporting Information File 1) and on comparison with the known trans-isomers of 3 [23].
Compounds Z-3a and Z-4a could be interconverted in acids through species Ba (Scheme 2). Thus, both Z-3a and Z-4a give cation Ba upon dissolving in TfOH. Then, a different quenching of solution of the cation affords Z-3a (Scheme 3) or Z-4a (Scheme 5). Heating of the solution of Ba leads to 5a (see Scheme 5).
![[1860-5397-14-268-i5]](/bjoc/content/inline/1860-5397-14-268-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Formation of thiochromene 1,1-dioxides 5a–c from allenes 2a,c,d.
Scheme 5: Formation of thiochromene 1,1-dioxides 5a–c from allenes 2a,c,d.
The exclusive formation of thiochromene 1,1-dioxides 5a–c was obtained by running the reaction of 2a,c,d with 1 equivalent of CF3SO3H at high temperature (reflux in ortho-dichlorobenzene at 144 °C) for 2 h (Scheme 5, see X-ray structure of 5c in Figure 1). It was found by H,H-NOESY and COSY correlations, that there was a [6,7]-shift of the methyl group in the thiochromene system of 5b obtained from allene 2c, which, at first, should give 6-methyl substituted thiochromene. This shift is caused by the action of superacid at high reaction temperature. Phenyl-substituted allenes 2e,f did not afford the corresponding thiochromenes, due to oligomerization under the harsh reaction conditions. Other allenes 2g,h gave the corresponding heterocycles 5 in very poor yields (<4%, by GC–MS and 1H NMR data).
As it was mentioned above, unsubsituted allenes 2i,j did not react with TfOH at room temperature (see discussion on NMR of cations B, Table 1). Under the heating in TfOH at 100 °C for 0.5 h, these allenes afforded (arylsulfonyl)acetones 6a,b, which may be formed under the hydrolysis of the formed vinyl triflates F (Scheme 6).
![[1860-5397-14-268-i6]](/bjoc/content/inline/1860-5397-14-268-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of (arylsulfonyl)acetones 6a,b from allenes 2h,j in TfOH (100 °C, 0.5 h) followed by hydrolysis of superacidic reaction solutions.
Scheme 6: Formation of (arylsulfonyl)acetones 6a,b from allenes 2h,j in TfOH (100 °C, 0.5 h) followed by hydr...
Finally, in this study, we carried out reactions of (arylsulfinyl)allenes 1a,b under superelectrophilic activation with TfOH or AlX3 (X = Cl, Br). The corresponding cations Aa,b generated from 1a,b in TfOH were subjected to quenching with various nucleophiles followed by hydrolysis (Scheme 7). In all the cases, allyl alcohols 7a,b were isolated (see X-ray structure of 7b in Figure 1). The same alcohols were obtained in reactions of 1a,b with AlX3 (X = Cl, Br) after the hydrolysis of reaction solutions. In these reactions, presumably, intermediate adducts G, which are formed upon interaction of species Aa,b with nucleophiles, are easily hydrolazible and give rise to alcohols 7a,b.
![[1860-5397-14-268-i7]](/bjoc/content/inline/1860-5397-14-268-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reactions of (arylsulfinyl)allenes 1a,b under superelectrophilic activation.
Scheme 7: Reactions of (arylsulfinyl)allenes 1a,b under superelectrophilic activation.
Conclusion
Transformations of (arylsulfonyl)- and (arylsulfinyl)allenes under the action of the Brønsted superacid TfOH, or strong Lewis acids AlX3 (X = Cl, Br) have been studied. Under electrophilic conditions, these allenes form the corresponding 1,2-oxathiolium ions, which have been studied by NMR and DFT calculations. Depending on electrophilic activator, reaction conditions (temperature, time), and nucleophilicity of media for quenching of solutions of 1,2-oxathiolium ions, these species may undergo various transformations leading to the selective formation of one of the reaction products: conjugated dienes, allyl alcohols, or thiochromene 1,1-dioxides. These reactions open new opportunities for organic synthesis based on electrophilic activation of sulfur containing allenes.
Supporting Information
| Supporting Information File 1: Copies of 1H and 13C NMR spectra of compounds and cations, X-ray data, and data of DFT calculations. | ||
| Format: PDF | Size: 4.3 MB | Download |
Acknowledgements
This work was supported by Russian Scientific Foundation (grant no. 18-13-00008). Spectral studies were performed at Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research, and Research Center for X-ray Diffraction Studies of Saint Petersburg State University, Saint Petersburg, Russia.
References
-
Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, 2004; Vol. 1 and 2. doi:10.1002/9783527619573
Return to citation in text: [1] -
Brummond, K. M.; DeForrest, J. E. Synthesis 2007, 795–818. doi:10.1055/s-2007-965963
Return to citation in text: [1] -
Bates, R. W.; Satcharoen, V. Chem. Soc. Rev. 2002, 31, 12–21. doi:10.1039/b103904k
Return to citation in text: [1] -
Ma, S. Aldrichimica Acta 2007, 40, 91–102.
Return to citation in text: [1] -
Hassan, H. Curr. Org. Synth. 2007, 4, 413–439. doi:10.2174/157017907782408798
Return to citation in text: [1] -
Back, T. G.; Clary, K. N.; Gao, D. Chem. Rev. 2010, 110, 4498–4553. doi:10.1021/cr1000546
Return to citation in text: [1] -
Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460
Return to citation in text: [1] -
Martzel, T.; Lohier, J.-F.; Gaumont, A.-C.; Brière, J.-F.; Perrio, S. Adv. Synth. Catal. 2017, 359, 96–106. doi:10.1002/adsc.201600929
Return to citation in text: [1] -
Lin, T.-Y.; Zhu, C.-Z.; Zhang, P.; Wang, Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Angew. Chem., Int. Ed. 2016, 55, 10844–10848. doi:10.1002/anie.201605530
Return to citation in text: [1] -
Undeela, S.; Thadkapally, S.; Nanubolu, J. B.; Singarapu, K. K.; Menon, R. S. Chem. Commun. 2015, 51, 13748–13751. doi:10.1039/c5cc04871k
Return to citation in text: [1] -
Kumar, A.; Thadkapally, S.; Menon, R. S. J. Org. Chem. 2015, 80, 11048–11056. doi:10.1021/acs.joc.5b02324
Return to citation in text: [1] -
Mo, D.-L.; Wink, D. J.; Anderson, L. L. Chem. – Eur. J. 2014, 20, 13217–13225. doi:10.1002/chem.201403268
Return to citation in text: [1] -
Xiong, Z.; Hale, K. J. Org. Lett. 2016, 18, 4254–4257. doi:10.1021/acs.orglett.6b02002
Return to citation in text: [1] -
Mukai, C.; Kobayashi, M.; Kubota, S.; Takahashi, Y.; Kitagaki, S. J. Org. Chem. 2004, 69, 2128–2136. doi:10.1021/jo035729f
Return to citation in text: [1] -
Mukai, C.; Yamashita, H.; Hanaoka, M. Org. Lett. 2001, 3, 3385–3387. doi:10.1021/ol0101842
Return to citation in text: [1] -
Mukai, C.; Ukon, R.; Kuroda, N. Tetrahedron Lett. 2003, 44, 1583–1586. doi:10.1016/s0040-4039(03)00067-4
Return to citation in text: [1] -
Zhu, Y. I.; Stiller, M. J. J. Am. Acad. Dermatol. 2001, 45, 420–434. doi:10.1067/mjd.2001.114733
Return to citation in text: [1] -
Xu, S.; Rouzer, C. A.; Marnett, L. J. IUBMB Life 2014, 66, 803–811. doi:10.1002/iub.1334
Return to citation in text: [1] -
Noble, S.; Benfield, P. CNS Drugs 1999, 12, 471–483. doi:10.2165/00023210-199912060-00005
Return to citation in text: [1] -
Denmark, S. E.; Harmata, M. A.; White, K. S. J. Org. Chem. 1987, 52, 4031–4042. doi:10.1021/jo00227a017
Return to citation in text: [1] -
Harmata, M.; Cai, Z.; Huang, C. Org. Synth. 2011, 88, 309–316. doi:10.15227/orgsyn.088.0309
Return to citation in text: [1] -
Tata, R. R.; Hampton, C. S.; Harmata, M. Adv. Synth. Catal. 2017, 359, 1232–1241. doi:10.1002/adsc.201600986
Return to citation in text: [1] -
Hampton, C. S.; Harmata, M. Org. Lett. 2014, 16, 1256–1259. doi:10.1021/ol500259m
Return to citation in text: [1] [2] [3] -
Hampton, C. S.; Harmata, M. J. Org. Chem. 2016, 81, 4807–4822. doi:10.1021/acs.joc.6b00880
Return to citation in text: [1] [2] -
Ma, S.; Ren, H.; Wei, Q. J. Am. Chem. Soc. 2003, 125, 4817–4830. doi:10.1021/ja034039q
Return to citation in text: [1] [2] -
Zhou, C.; Fu, C.; Ma, S. Tetrahedron 2007, 63, 7612–7616. doi:10.1016/j.tet.2007.05.039
Return to citation in text: [1] -
Zhou, C.; Fang, Z.; Fu, C.; Ma, S. J. Org. Chem. 2009, 74, 2887–2890. doi:10.1021/jo802755k
Return to citation in text: [1] -
Fang, Z.; Zhou, C.; Fu, C.; Ma, S. Org. Biomol. Chem. 2010, 8, 4554–4561. doi:10.1039/c0ob00007h
Return to citation in text: [1] -
Bogachenkov, A. S.; Dogadina, A. V.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2015, 13, 1333–1338. doi:10.1039/c4ob02269f
Return to citation in text: [1] -
Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j
Return to citation in text: [1] [2] [3] [4] -
Lozovskiy, S. V.; Bogachenkov, A. S.; Dogadina, A. V.; Vasilyev, A. V. Tetrahedron Lett. 2016, 57, 3167–3170. doi:10.1016/j.tetlet.2016.06.026
Return to citation in text: [1] -
Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637
Return to citation in text: [1] [2] [3] -
Trillo, P.; Baeza, A.; Nájera, C. J. Org. Chem. 2012, 77, 7344–7354. doi:10.1021/jo301049w
Return to citation in text: [1]
| 32. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 30. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 32. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 30. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 1. | Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, 2004; Vol. 1 and 2. doi:10.1002/9783527619573 |
| 2. | Brummond, K. M.; DeForrest, J. E. Synthesis 2007, 795–818. doi:10.1055/s-2007-965963 |
| 3. | Bates, R. W.; Satcharoen, V. Chem. Soc. Rev. 2002, 31, 12–21. doi:10.1039/b103904k |
| 4. | Ma, S. Aldrichimica Acta 2007, 40, 91–102. |
| 5. | Hassan, H. Curr. Org. Synth. 2007, 4, 413–439. doi:10.2174/157017907782408798 |
| 6. | Back, T. G.; Clary, K. N.; Gao, D. Chem. Rev. 2010, 110, 4498–4553. doi:10.1021/cr1000546 |
| 7. | Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460 |
| 11. | Kumar, A.; Thadkapally, S.; Menon, R. S. J. Org. Chem. 2015, 80, 11048–11056. doi:10.1021/acs.joc.5b02324 |
| 29. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2015, 13, 1333–1338. doi:10.1039/c4ob02269f |
| 30. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 31. | Lozovskiy, S. V.; Bogachenkov, A. S.; Dogadina, A. V.; Vasilyev, A. V. Tetrahedron Lett. 2016, 57, 3167–3170. doi:10.1016/j.tetlet.2016.06.026 |
| 32. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 10. | Undeela, S.; Thadkapally, S.; Nanubolu, J. B.; Singarapu, K. K.; Menon, R. S. Chem. Commun. 2015, 51, 13748–13751. doi:10.1039/c5cc04871k |
| 24. | Hampton, C. S.; Harmata, M. J. Org. Chem. 2016, 81, 4807–4822. doi:10.1021/acs.joc.6b00880 |
| 25. | Ma, S.; Ren, H.; Wei, Q. J. Am. Chem. Soc. 2003, 125, 4817–4830. doi:10.1021/ja034039q |
| 9. | Lin, T.-Y.; Zhu, C.-Z.; Zhang, P.; Wang, Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Angew. Chem., Int. Ed. 2016, 55, 10844–10848. doi:10.1002/anie.201605530 |
| 25. | Ma, S.; Ren, H.; Wei, Q. J. Am. Chem. Soc. 2003, 125, 4817–4830. doi:10.1021/ja034039q |
| 26. | Zhou, C.; Fu, C.; Ma, S. Tetrahedron 2007, 63, 7612–7616. doi:10.1016/j.tet.2007.05.039 |
| 8. | Martzel, T.; Lohier, J.-F.; Gaumont, A.-C.; Brière, J.-F.; Perrio, S. Adv. Synth. Catal. 2017, 359, 96–106. doi:10.1002/adsc.201600929 |
| 27. | Zhou, C.; Fang, Z.; Fu, C.; Ma, S. J. Org. Chem. 2009, 74, 2887–2890. doi:10.1021/jo802755k |
| 28. | Fang, Z.; Zhou, C.; Fu, C.; Ma, S. Org. Biomol. Chem. 2010, 8, 4554–4561. doi:10.1039/c0ob00007h |
| 17. | Zhu, Y. I.; Stiller, M. J. J. Am. Acad. Dermatol. 2001, 45, 420–434. doi:10.1067/mjd.2001.114733 |
| 19. | Noble, S.; Benfield, P. CNS Drugs 1999, 12, 471–483. doi:10.2165/00023210-199912060-00005 |
| 33. | Trillo, P.; Baeza, A.; Nájera, C. J. Org. Chem. 2012, 77, 7344–7354. doi:10.1021/jo301049w |
| 14. | Mukai, C.; Kobayashi, M.; Kubota, S.; Takahashi, Y.; Kitagaki, S. J. Org. Chem. 2004, 69, 2128–2136. doi:10.1021/jo035729f |
| 15. | Mukai, C.; Yamashita, H.; Hanaoka, M. Org. Lett. 2001, 3, 3385–3387. doi:10.1021/ol0101842 |
| 16. | Mukai, C.; Ukon, R.; Kuroda, N. Tetrahedron Lett. 2003, 44, 1583–1586. doi:10.1016/s0040-4039(03)00067-4 |
| 20. | Denmark, S. E.; Harmata, M. A.; White, K. S. J. Org. Chem. 1987, 52, 4031–4042. doi:10.1021/jo00227a017 |
| 21. | Harmata, M.; Cai, Z.; Huang, C. Org. Synth. 2011, 88, 309–316. doi:10.15227/orgsyn.088.0309 |
| 22. | Tata, R. R.; Hampton, C. S.; Harmata, M. Adv. Synth. Catal. 2017, 359, 1232–1241. doi:10.1002/adsc.201600986 |
| 23. | Hampton, C. S.; Harmata, M. Org. Lett. 2014, 16, 1256–1259. doi:10.1021/ol500259m |
| 24. | Hampton, C. S.; Harmata, M. J. Org. Chem. 2016, 81, 4807–4822. doi:10.1021/acs.joc.6b00880 |
| 23. | Hampton, C. S.; Harmata, M. Org. Lett. 2014, 16, 1256–1259. doi:10.1021/ol500259m |
| 13. | Xiong, Z.; Hale, K. J. Org. Lett. 2016, 18, 4254–4257. doi:10.1021/acs.orglett.6b02002 |
| 30. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 12. | Mo, D.-L.; Wink, D. J.; Anderson, L. L. Chem. – Eur. J. 2014, 20, 13217–13225. doi:10.1002/chem.201403268 |
| 18. | Xu, S.; Rouzer, C. A.; Marnett, L. J. IUBMB Life 2014, 66, 803–811. doi:10.1002/iub.1334 |
| 23. | Hampton, C. S.; Harmata, M. Org. Lett. 2014, 16, 1256–1259. doi:10.1021/ol500259m |
© 2018 Lozovskiy et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)