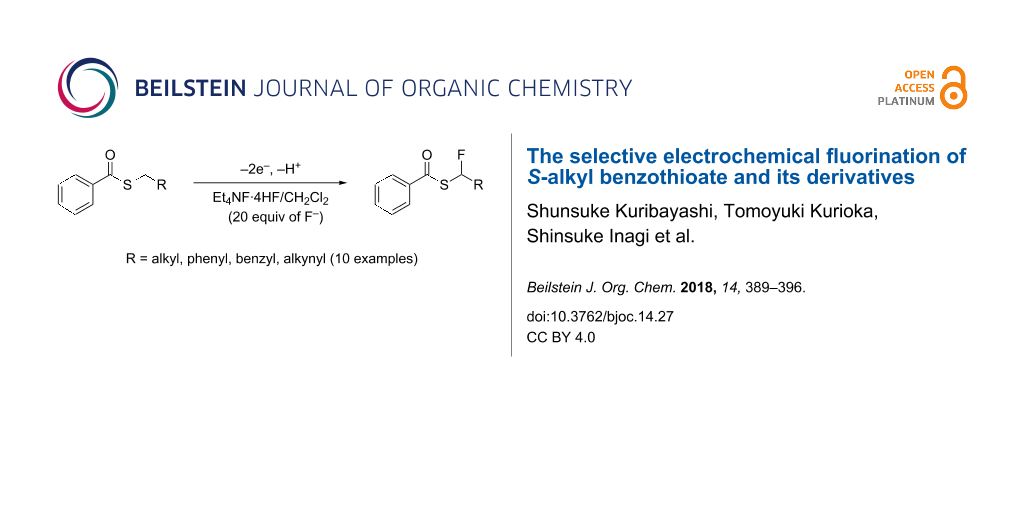
Abstract
We herein report that the regioselective anodic fluorination of S-alkyl benzothioate and its derivatives in various aprotic solvents using Et3N·nHF (n = 3–5) and Et4NF·nHF (n = 3–5) as supporting electrolyte and a fluorine source successfully provided the corresponding α-fluorinated products in moderate yields. Dichloromethane containing Et4NF·4HF was found to be the most suitable combination as electrolytic solvent and supporting salt as well as fluorine source for the anodic fluorination. The electrochemical fluorination of cyclic benzothioates such as benzothiophenone was also achieved.

Graphical Abstract
Introduction
Due to the interesting properties of fluorine atoms and carbon–fluorine bonds, organofluorine compounds are widely used in various fields like pharmaceutical chemistry, agrochemistry, and materials sciences [1,2]. Therefore, the selective fluorination of organic compounds is highly useful for the development of novel organofluorine compounds. Although a number of fluorination reagents have been developed so far, they have still some problems, i.e., they are costly, difficult to handle, and explosive [3,4]. On the other hand, Rozhkov and Laurent reported an electrochemical partial fluorination of naphthalene and olefins about 30 years ago [5,6]. However, at that time, there has been no report on the anodic fluorination of heteroatom-containing compounds. At almost the same time, we found that the anodic α-methoxylation of organosulfur and amino compounds was generally enhanced by the presence of an α-electron-withdrawing group (EWG) such as a CF3 group. Here, the deprotonation of an anodically generated radical cation intermediate is accelerated by an EWG [7,8]. Based on these facts, we successfully achieved the first anodic fluorination of sulfides having various EWGs at their α-position using Et3N·3HF [9,10]. Since then, we have systematically studied the electrochemical fluorination of numerous organic compounds, heterocycles, and macromolecules by using various fluoride salts such as Et3N·nHF (n = 3–5) and Et4NF·nHF (n = 3–5) [11-19].
On the other hand, Simonet and co-workers reported the anodic fluorination of alkyl phenyl sulfides having an EWG on the phenyl group in Et3N·3HF/MeCN to provide α-monofluorinated products in moderate yields [20]. We also achieved the anodic fluorination of benzyl and ethyl thiocyanates as well as O-methyl S-aralkyl thiocarbonates by using the anodically stable Et3N·5HF and Et4NF·4HF [21,22]. In both cases, an EWG attached to the phenyl group and the electron-withdrawing SCN group both contribute to the generation of the cation resulting in a regioselective α-fluorination. Based on these findings, we anticipated that the α-cationic intermediate could also be generated anodically from S-alkyl benzothioates. Moreover, we previously successfully carried out an anodic fluorodesulfurization of S-aryl thiobenzoates, and found that the indirect electrolysis using a triarylamine mediator gave much better yields of benzoyl fluorides compared to the direct electrolysis [23]. Therefore, we became interested in the anodic behavior of S-alkyl benzothioates in the presence of fluoride ions. With this in mind and in order to provide an additional application of our electrochemical fluorination, we have studied the anodic fluorination of S-alkyl benzothioate and its derivatives as well as its cyclic analogues such as benzothiophenone.
Results and Discussion
Oxidation potentials of S-butyl benzothioates
At first, the anodic oxidation potentials of S-butyl benzothioate (1a), S-butyl p-chlorobenzothioate (1b), and S-butyl p-fluorobenzothioate (1c) were determinded by cyclic voltammetry (CV). The compounds did not exhibit clear oxidation peaks, however, discharge started at around +2.3 V to +2.4 V vs SCE as exemplified in Figure 1 (CV curves of 1a). Thus, it was found that these compounds have rather high oxidation potentials. DFT calculation of 1a indicated that the highest occupied molecular orbital (HOMO) is located at the sulfur atom (Figure 2). Although sulfur atoms are easily oxidized, the oxidation potential of 1a was found to be extremely high, that is due to the strongly electron-withdrawing benzoyl group attached to the sulfur atom. Benzyl thiocyanate is also known to be oxidized at a high potential which is similar to that of 1a [21].
![[1860-5397-14-27-1]](/bjoc/content/figures/1860-5397-14-27-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cyclic voltammograms of 0.1 M Bu4NBF4/MeCN with a Pt disk working electrode in the absence (brown line) and presence of 5.0 mM (blue line) and 10.0 mM 1a (red line). Scan rate of 100 mV/s.
Figure 1: Cyclic voltammograms of 0.1 M Bu4NBF4/MeCN with a Pt disk working electrode in the absence (brown l...
![[1860-5397-14-27-2]](/bjoc/content/figures/1860-5397-14-27-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Anodic fluorination of S-butyl benzothioates
Next, we carried out the anodic fluorination of 1a as a model compound under various electrolytic conditions. Electrolysis was performed at a constant current (40 mA) with platinum electrodes (2 cm × 2 cm) in several solvents containing various fluoride salts in an undivided cell at room temperature and current was passed basically until 1a was consumed. The results are summarized in Table 1.
Table 1: Electrochemical fluorination of S-butyl benzothioate derivatives.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-27-i7.svg?max-width=637&scale=1.0)
|
|||||||
| entry | compound | R | supporting electrolyte | solvent | electricity (F/mol) | yield (%)a | |
|---|---|---|---|---|---|---|---|
| 2 | 3 | ||||||
| 1 | 1a | H | Et3N·3HF | CH2Cl2 | 4.5 | 55 | − |
| 2 | 1a | H | Et3N·3HF | CH3CN | 7.0 | 26 | − |
| 3 | 1a | H | Et3N·3HF | DME | 2.0 | 0b | − |
| 4 | 1a | H | Et3N·3HF | CH3NO2 | 7.0 | 29 | − |
| 5 | 1a | H | Et3N·4HF | CH2Cl2 | 4.5 | 56 | − |
| 6 | 1a | H | Et3N·5HF | CH2Cl2 | 4.0 | 43 | − |
| 7 | 1a | H | Et4NF·3HF | CH2Cl2 | 5.4 | 57 | − |
| 8 | 1a | H | Et4NF·4HF | CH2Cl2 | 4.0 | 55 | − |
| 9 | 1a | H | Et4NF·5HF | CH2Cl2 | 4.0 | 38 | − |
| 10c | 1a | H | Et4NF·4HF | CH2Cl2 | 4.0 | 67 (61) | 12 |
| 11c | 1b | Cl | Et4NF·4HF | CH2Cl2 | 6.0 | 51 (45) | 20 |
| 12c | 1c | F | Et4NF·4HF | CH2Cl2 | 4.0 | 60 (50) | 9 |
aDetermined by 19F NMR. Isolated yield is shown in parentheses. bSubstrate 1a was mostly recovered. cTwenty equiv of the fluoride source were used.
Regardless of the electrolytic conditions, the anodic fluorination proceeded to provide the desired fluorinated product 2a except for the electrolysis performed in dimethoxyethane (DME) as the solvent (Table 1, entry 3). The oxidation potential of 1a is rather high, while that of DME is relatively low [24]. Therefore, DME seems to be oxidized prior to 1a resulting in no formation of 2a, and 1a was mostly recovered even when passing the theoretical amount of electricity (2 F/mol). Among the solvents tested, CH2Cl2 was found to be the best and the desired fluorinated product 2a was obtained in 55% yield (Table 1, entry 1). In this case, the starting 1a was consumed completely at 4.5 F/mol. It is well known that the fluoride salt, Et3N·3HF is easily oxidized (2.0 V vs Ag/Ag+) because it contains a considerable amount of free Et3N [25]. However, the fluorination proceeded well particularly in a CH2Cl2 solution containing Et3N·3HF. In contrast, when MeCN and MeNO2 were used as an electrolytic solvent, the yield was decreased considerably (Table 1, entries 2 and 4). This is because of a low conversion of 1a in these solvents and indeed a large amount of 1a was recovered.
Subsequently the effect of different fluoride salts on the fluorination was investigated similarly (Table 1, entries 5–9). Regardless of the fluoride salts, product 2a was obtained in moderate yield, however, using Et4NF·4HF gave the best current efficiency for the formation of 2a (Table 1, entry 8). Increasing the amount of Et4NF·4HF from 10 to 20 equiv, the product yield was also increased from 55% to 67% (Table 1, entry 10). However, in this case, C–S bond cleavage also took place to form benzoyl fluoride (3a) as a byproduct in considerable amounts. In all cases, no fluorination at the phenyl group took place.
Then, this anodic fluorination was extended to p-chloro- and p-fluorobenzothioates, (1b) and (1c), using 20 equiv of Et4NF·4HF as the supporting electrolyte (Table 1, entries 11 and 12). In both cases, a fluorine atom was introduced selectively to the α-position to provide 2b and 2c in moderate yields. However, the corresponding benzoyl fluorides 3b and 3c were also formed similarly to the case of 1a.
Anodic fluorination of S-(ω-substituted alkyl) benzylthioates
Initially, the oxidation potentials of various S-(substituted alkyl) benzylthioates 1d–j were determined by CVs in 0.1 M Bu4NBF4/MeCN similarly to the case of 1a and the results are collected in Table 2. Unexpectedly, no relationship between the substituents and the oxidation potentials could be observed. Next, the anodic fluorination of 1d–j was carried out at a constant current in Et4NF·4HF/CH2Cl2 and the results are summarized also in Table 2.
Table 2: Oxidation potentials and electrochemical fluorination of S-substituted alkyl benzothioates.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-27-i8.svg?max-width=637&scale=1.0)
|
||||||
| entry | compound | R |
oxidation potential
(V vs SCE) |
electricity (F/mol) | yield (%)a | |
|---|---|---|---|---|---|---|
| 2 | 3 | |||||
| 1 | 1a | n-Pr- | 2.3 | 4.0 | 67 (61) | 12 |
| 2 | 1d | NCCH2CH2- | 2.1 | 6.0 | 43 (35) | 12 |
| 3 | 1e | MeOCH2CH2- | 2.0 | 6.0 | 33 (29) | 19 |
| 4 | 1f | EtOOC- | 2.2 | 6.0 | 36 (29) | 12 |
| 5 | 1g | HC≡C- | 2.3 | 8.0 | 40 (37) | 10 |
| 6 | 1h | Ph- | 2.1 | 4.0 | 56 (46) | 5 |
| 7 | 1i | PhCH2- | 2.2 | 6.0 | 10 (7) | 5 |
| 8 | 1j | p-CF3C6H4CH2- | 2.1 | 6.0 | 48 (40) | 12 |
aDetermined by 19F NMR. Isolated yields are shown in parentheses.
Regardless of the substituents, the anodic fluorination took place to afford the corresponding α-fluorinated products 2 in moderate to reasonable yields along with benzoyl fluoride as byproduct (Table 2). Generally, no fluorination of the phenyl ring was observed. In the case of 1h, electron transfer seems to take place from both the sulfur atom and the α-phenyl group, which was suggested by DFT calculation (Figure 3). However, regardless of the discharging sites, the same cationic intermediate is generated through two-electron oxidation and deprotonation (in α position to both the sulfur atom and phenyl group), which forms the α-fluorinated product 2h selectively. In contrast, the electron transfer of 1i seems to take place mainly from the β-phenyl group as indicated by DFT calculation (Figure 3). As shown in Scheme 1, an electron transfer from the β-phenyl group in 1i followed by deprotonation and an additional electron transfer generates the benzylic cationic intermediate A, which affords the benzylic fluorinated product. It is known that benzylic fluorinated compounds are known to be generally prone to lose a fluoride anion [26]. On the other hand, intermediate A may undergo also elimination of a β-proton due to the electron-withdrawing benzothioate group prior to the reaction with fluoride as reported before [27]. As a consequence the expected α-fluorinated product 2i was formed in an only low yield of 10%.
![[1860-5397-14-27-3]](/bjoc/content/figures/1860-5397-14-27-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Calculated HOMO diagrams of 1h, 1i and 1j.
Figure 3: Calculated HOMO diagrams of 1h, 1i and 1j.
![[1860-5397-14-27-i1]](/bjoc/content/inline/1860-5397-14-27-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Plausible reaction paths of the anodic oxidation of 1i in Et4NF·4HF/CH2Cl2.
Scheme 1: Plausible reaction paths of the anodic oxidation of 1i in Et4NF·4HF/CH2Cl2.
In order to suppress the discharge from the β-phenyl group of 1i, a strongly electron-withdrawing CF3 group was introduced in the para-position of the phenyl group. The HOMO of 1j was found to be located mainly at the sulfur atom as determined by DFT calculation (Figure 3). As expected, the anodic fluorination proceeded to provide the α-fluorinated product 2j in a moderate yield of 48%.
Next, the anodic fluorination was performed with the benzothioate derivative 1k having a camphor group as a chiral auxiliary. The anodic fluorination proceeded to afford the corresponding α-fluoro products as a diastereoisomeric mixture (43% de) in a reasonable yield 42% along with benzoyl fluoride as byproduct (Scheme 2).
![[1860-5397-14-27-i2]](/bjoc/content/inline/1860-5397-14-27-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
We next also carried out the anodic fluorination of a cyclic benzothioate namely benzothiophenone 1l. In this case, the fluorination took place predominantly at the benzylic position to afford the fluorinated product 2l in 60% isolated yield (Scheme 3). With this substrate, neither C–S bond cleavage nor benzene ring fluorination took place at all.
![[1860-5397-14-27-i3]](/bjoc/content/inline/1860-5397-14-27-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Anodic fluorination of cyclic derivative 1l.
Scheme 3: Anodic fluorination of cyclic derivative 1l.
Finally, the anodic fluorination of benzothioates having a γ and δ-carboxyl group, 1m and 1n, was examined. The anodic oxidation of 1m and 1n proceeded; however, α-fluorination did not occur. In both cases, an anodic intramolecular cyclization took place to give the corresponding lactone derivatives in reasonable yields as shown in Scheme 4.
![[1860-5397-14-27-i4]](/bjoc/content/inline/1860-5397-14-27-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Anodic oxidation of 1m and 1n in Et4NF·4HF/CH2Cl2.
Scheme 4: Anodic oxidation of 1m and 1n in Et4NF·4HF/CH2Cl2.
Reaction mechanism
In the cases of open-chain benzothioate derivatives, the anodic fluorination is initiated by an electron transfer occurring mainly from the sulfur atom of the benzothioates following two pathways (Scheme 5). The main pathway comprises the elimination of an α-proton, which is facilitated by the presence of an electron-withdrawing benzoyl group followed by another one-electron oxidation to generate a cationic intermediate which upon reaction with fluoride forms the α-fluorinated products. On the other hand, a minor pathway involves a C–S bond cleavage to form benzoyl fluoride as is observed in the case of the anodic oxidation of S-aryl benzothioates [23].
![[1860-5397-14-27-i5]](/bjoc/content/inline/1860-5397-14-27-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: General reaction mechanism for the anodic fluorination of 1.
Scheme 5: General reaction mechanism for the anodic fluorination of 1.
In the case of S-alkyl benzothioates bearing a carboxyl group at the γ and δ-position with respect to the sulfur atom, after generation of an α-cationic intermediate, the intramolecular cyclization seems to take place faster than combination with a fluoride ion as shown in Scheme 6. In support of this, we have reported a fluoride ion-promoted anodic cyclization of α-(phenylthio)-N-phenyl- and α-(phenylthio)-N-benzylacetamides [28] as well as 2-(tert-butoxycarbonyl)oxy-3,3,3-trifluoropropyl phenyl sulfide [29].
![[1860-5397-14-27-i6]](/bjoc/content/inline/1860-5397-14-27-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction mechanism for the anodic oxidation of carboxylic acids 1m and 1n in the presence of a fluoride source.
Scheme 6: Reaction mechanism for the anodic oxidation of carboxylic acids 1m and 1n in the presence of a fluo...
Conclusion
In summary, we have achieved the regioselective anodic fluorination of various S-alkyl and S-substituted alkyl benzothioate derivatives in Et4NF·4HF/CH2Cl2 and a fluorine atom was selectively introduced in the α-position to the sulfur atom. Under the conditions, a camphor-substituted analogue was anodically fluorinated with moderate diastereoselectivity. Moreover, the anodic fluorination of a cyclic benzothioate such as benzothiophenone was also successfully demonstrated. In contrast, the anodic fluorination of S-(ω-carboxy)alkyl benzothioates afforded intramolecular cyclization products like lactones instead of the corresponding α-fluorinated products.
Experimental
General information
1H, 13C and 19F NMR spectra were recorded on a JEOL JNM EX-270 (1H: 270 MHz, 13C: 67.8 MHz, 19F: 254.05 MHz) spectrometer in CDCl3. The chemical shifts for 1H, 13C and 19F NMR spectra are given in δ (ppm) from internal TMS, CDCl3 and monofluorobenzene, respectively. Cyclic voltammetry was performed using an ALS Instrument model 600A. Preparative electrolysis experiments were carried out with Metronnix Corp. (Tokyo) constant current power supply model 5944 by monitoring electricity with a Hokutodenko Coulomb/Ampere-hour meter HF-201.
Cyclic voltammetry measurements
Cyclic voltammetry was carried out in 0.1 M Bu4NBF4/MeCN using a glass cell. A platinum disk electrode (![[Graphic 3]](/bjoc/content/inline/1860-5397-14-27-i9.svg?max-width=637&scale=1.18182) = 0.8 mm) was used as a working electrode. A platinum plate (1 cm × 1 cm) was used as a counter electrode and a saturated calomel electrode was used as a reference electrode. Electrolyte solutions for cyclic voltammetry were deoxygenated with bubbling N2 gas before use.
= 0.8 mm) was used as a working electrode. A platinum plate (1 cm × 1 cm) was used as a counter electrode and a saturated calomel electrode was used as a reference electrode. Electrolyte solutions for cyclic voltammetry were deoxygenated with bubbling N2 gas before use.
Materials
The starting materials were prepared according to the literature procedures [21,23,30,31]. The known compounds, 1a,1b [32], 1c [33], 1f–1h [34], 1i [35], 1k [36], 1l [31], 1m [37] and 1n [38] were characterized by comparison of the spectral data with those reported in the literature.
General procedure for the anodic fluorination
Analogous as described in [17], the anodic oxidation of a substrate (1 mmol) was carried out in a plastic undivided cell equipped with platinum anode and cathode (2 cm × 2 cm) containing 10 mL of HF salt (20 equiv of F− to substrate/solvent) at room temperature. A constant current (40 mA) was passed until the starting material was mostly consumed (monitored by TLC). After electrolysis, the electrolytic solution was passed through a short column filled with silica gel using EtOAc as an eluent to remove the HF salt. The fluorinated product was further purified by silica gel column or preparative thin layer chromatography using a solution of hexane/EtOAc (20:1 to 1:1) as an eluent. The yield of the fluorinated products were estimated by 19F NMR using monofluorobenzene as an internal standard. The known fluorinated products, benzoyl fluoride [39], p-chlorobenzoyl fluoride [40] and p-fluorobenzoyl fluoride [41] were identified by comparison with 19F NMR and MS spectral data of the authentic samples.
Supporting Information
| Supporting Information File 1: Characterization data. | ||
| Format: PDF | Size: 111.7 KB | Download |
References
-
Purser, S.; Moore, P. R.; Swallow, S.; Gouvermeur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Boschet, F.; Ameduri, B. Chem. Rev. 2014, 114, 927–980. doi:10.1021/cr2002933
Return to citation in text: [1] -
Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h
Return to citation in text: [1] -
Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133, 11482–11484. doi:10.1021/ja2048072
Return to citation in text: [1] -
Rozhkov, I. N. In Organic Electrochemistry, 2nd ed.; Baizer, M. M.; Lund, H., Eds.; chapter 24; Marcel Decker: New York, 1983.
Return to citation in text: [1] -
Laurent, E.; Marquet, B.; Tardivel, R.; Thiebault, H. Bull. Soc. Chim. Fr. 1986, 955–964.
Return to citation in text: [1] -
Fuchigami, T.; Nakagawa, Y.; Nonaka, T. Tetrahedron Lett. 1986, 27, 3869–3872. doi:10.1016/S0040-4039(00)83902-7
Return to citation in text: [1] -
Fuchigami, T.; Nakagawa, Y.; Nonaka, T. J. Org. Chem. 1987, 52, 5489–5491. doi:10.1021/jo00233a043
Return to citation in text: [1] -
Fuchigami, T.; Shimojo, M.; Konno, A.; Nakagawa, K. J. Org. Chem. 1990, 55, 6074–6075. doi:10.1021/jo00312a006
Return to citation in text: [1] -
Konno, A.; Nakagawa, K.; Fuchigami, T. J. Chem. Soc., Chem. Commun. 1991, 1027–1029. doi:10.1039/C39910001027
Return to citation in text: [1] -
Fuchigami, T. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; chapter 25; Marcel Dekker: New York, 2001.
Return to citation in text: [1] -
Noel, M.; Suryanarayanan, V. J. Appl. Electrochem. 2004, 34, 357–369. doi:10.1023/B:JACH.0000016670.97434.c7
Return to citation in text: [1] -
Dawood, K. M. Tetrahedron 2004, 60, 1435–1451. doi:10.1016/j.tet.2003.11.017
Return to citation in text: [1] -
Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/C1CC12414E
Return to citation in text: [1] -
Fuchigami, T.; Inagi, S. In Organic Electrochemistry,, 5th ed.; Hammerich, O.; Speiser, B., Eds.; chapter 20; CRC Press: Boca Raton, FL, 2015.
Return to citation in text: [1] -
Fuchigami, T.; Inagi, S. Organic Electrosynthesis. In Fundamentals and Applications of Organic Electrochemistry; Fuchigami,, T.; Atobe, M.; Inagi, S., Eds.; chapter 5; Wiley: West Sussex, 2015. doi:10.1002/9781118670750.ch05
Return to citation in text: [1] -
Kuribayashi, S.; Shida, N.; Inagi, S.; Fuchigami, T. Tetrahedron 2016, 72, 5343–5349. doi:10.1016/j.tet.2016.07.016
Return to citation in text: [1] [2] -
Tanigawa, M.; Kuriyama, Y.; Inagi, S.; Fuchigami, T. Electrochim. Acta 2016, 199, 314–318. doi:10.1016/j.electacta.2016.02.051
Return to citation in text: [1] -
Suzuki, J.; Shida, N.; Inagi, S.; Fuchigami, T. Electroanalysis 2016, 28, 2797–2801. doi:10.1002/elan.201600130
Return to citation in text: [1] -
Boroux, P.; Tardivel, R.; Simonet, J. J. Electrochem. Soc. 1997, 144, 841–847. doi:10.1149/1.1837498
Return to citation in text: [1] -
Hou, Y.; Higashiya, S.; Fuchigami, T. Electrochim. Acta 2000, 45, 3005–3010. doi:10.1016/S0013-4686(00)00379-0
Return to citation in text: [1] [2] [3] -
Cao, Y.; Fuchigami, T. J. Electroanal. Chem. 2006, 587, 25–30. doi:10.1016/j.jelechem.2005.10.010
Return to citation in text: [1] -
Shen, Y.; Suzuki, K.; Atobe, M.; Fuchigami, T. J. Electroanal. Chem. 2003, 540, 189–194. doi:10.1016/S0022-0728(02)01297-4
Return to citation in text: [1] [2] [3] -
Hou, Y.; Fuchigami, T. J. Electrochem. Soc. 2000, 147, 4567–4572. doi:10.1149/1.1394102
Return to citation in text: [1] -
Chen, S.-Q.; Hatakeyama, T.; Fukuhara, T.; Hara, S.; Yoneda, N. Electrochim. Acta 1997, 42, 1951–1960. doi:10.1016/S0013-4686(97)85466-7
Return to citation in text: [1] -
Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037
Return to citation in text: [1] -
Erian, A. W.; Konno, A.; Fuchigami, T. J. Org. Chem. 1995, 60, 7654–7659. doi:10.1021/jo00128a044
Return to citation in text: [1] -
Shen, Y.; Atobe, M.; Fuchigami, T. Org. Lett. 2004, 6, 2441–2444. doi:10.1021/ol049152f
Return to citation in text: [1] -
Isokawa, M.; Sano, M.; Kubota, K.; Suzuki, K.; Inagi, S.; Fuchigami, T. J. Electrochem. Soc. 2017, 164, G121–G127. doi:10.1149/2.1601713jes
Return to citation in text: [1] -
Riyadh, S. M.; Ishii, H.; Fuchigami, T. Tetrahedron 2002, 58, 5877–5883. doi:10.1016/S0040-4020(02)00559-8
Return to citation in text: [1] -
Lee, E.-S.; Choi, B.-W.; Jung, D.-I.; Hwang, H.-J.; Han, J.-T.; Lee, B.-H. Bull. Korean Chem. Soc. 2003, 24, 243–245. doi:10.5012/bkcs.2003.24.2.243
Return to citation in text: [1] [2] -
Qiao, Z.; Jiang, X. Org. Lett. 2016, 18, 1550–1553. doi:10.1021/acs.orglett.6b00324
Return to citation in text: [1] -
Ogawa, K. A.; Boydston, A. J. Org. Lett. 2014, 16, 1928–1931. doi:10.1021/ol500459x
Return to citation in text: [1] -
Lu, G.-p.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059
Return to citation in text: [1] -
Moon, H. K.; Sung, G. H.; Kim, B. R.; Park, J. K.; Yoon, Y.-J.; Yoon, H. J. Adv. Synth. Catal. 2016, 358, 1725–1730. doi:10.1002/adsc.201501177
Return to citation in text: [1] -
Montenegro, E.; Echarri, R.; Claver, C.; Castillón, S.; Moyano, A.; Pericàs, M. A.; Riera, A. Tetrahedron: Asymmetry 1996, 7, 3553–3558. doi:10.1016/S0957-4166(96)00463-6
Return to citation in text: [1] -
Oya, M.; Baba, T.; Kato, E.; Kawashima, Y.; Watanabe, T. Chem. Pharm. Bull. 1982, 30, 440–461. doi:10.1248/cpb.30.440
Return to citation in text: [1] -
Beckwith, A. L. J.; Duggan, S. A. M. J. Chem. Soc., Perkin Trans. 2 1994, 1509–1518. doi:10.1039/P29940001509
Return to citation in text: [1] -
Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; LaFlamme, F.; L’Heureux, A. Org. Lett. 2009, 11, 5050–5053. doi:10.1021/ol902039q
Return to citation in text: [1] -
Bildsoe, H.; Schaumburg, K. J. Magn. Reson. 1974, 14, 223–234. doi:10.1016/0022-2364(74)90275-3
Return to citation in text: [1] -
Birrell, J. A.; Desrosiers, J.-N.; Jacobsen, E. N. J. Am. Chem. Soc. 2011, 133, 13872–13875. doi:10.1021/ja205602j
Return to citation in text: [1]
| 39. | Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; LaFlamme, F.; L’Heureux, A. Org. Lett. 2009, 11, 5050–5053. doi:10.1021/ol902039q |
| 40. | Bildsoe, H.; Schaumburg, K. J. Magn. Reson. 1974, 14, 223–234. doi:10.1016/0022-2364(74)90275-3 |
| 41. | Birrell, J. A.; Desrosiers, J.-N.; Jacobsen, E. N. J. Am. Chem. Soc. 2011, 133, 13872–13875. doi:10.1021/ja205602j |
| 1. | Purser, S.; Moore, P. R.; Swallow, S.; Gouvermeur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 2. | Boschet, F.; Ameduri, B. Chem. Rev. 2014, 114, 927–980. doi:10.1021/cr2002933 |
| 9. | Fuchigami, T.; Shimojo, M.; Konno, A.; Nakagawa, K. J. Org. Chem. 1990, 55, 6074–6075. doi:10.1021/jo00312a006 |
| 10. | Konno, A.; Nakagawa, K.; Fuchigami, T. J. Chem. Soc., Chem. Commun. 1991, 1027–1029. doi:10.1039/C39910001027 |
| 23. | Shen, Y.; Suzuki, K.; Atobe, M.; Fuchigami, T. J. Electroanal. Chem. 2003, 540, 189–194. doi:10.1016/S0022-0728(02)01297-4 |
| 7. | Fuchigami, T.; Nakagawa, Y.; Nonaka, T. Tetrahedron Lett. 1986, 27, 3869–3872. doi:10.1016/S0040-4039(00)83902-7 |
| 8. | Fuchigami, T.; Nakagawa, Y.; Nonaka, T. J. Org. Chem. 1987, 52, 5489–5491. doi:10.1021/jo00233a043 |
| 28. | Shen, Y.; Atobe, M.; Fuchigami, T. Org. Lett. 2004, 6, 2441–2444. doi:10.1021/ol049152f |
| 5. | Rozhkov, I. N. In Organic Electrochemistry, 2nd ed.; Baizer, M. M.; Lund, H., Eds.; chapter 24; Marcel Decker: New York, 1983. |
| 6. | Laurent, E.; Marquet, B.; Tardivel, R.; Thiebault, H. Bull. Soc. Chim. Fr. 1986, 955–964. |
| 26. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 3. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 4. | Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133, 11482–11484. doi:10.1021/ja2048072 |
| 27. | Erian, A. W.; Konno, A.; Fuchigami, T. J. Org. Chem. 1995, 60, 7654–7659. doi:10.1021/jo00128a044 |
| 23. | Shen, Y.; Suzuki, K.; Atobe, M.; Fuchigami, T. J. Electroanal. Chem. 2003, 540, 189–194. doi:10.1016/S0022-0728(02)01297-4 |
| 24. | Hou, Y.; Fuchigami, T. J. Electrochem. Soc. 2000, 147, 4567–4572. doi:10.1149/1.1394102 |
| 21. | Hou, Y.; Higashiya, S.; Fuchigami, T. Electrochim. Acta 2000, 45, 3005–3010. doi:10.1016/S0013-4686(00)00379-0 |
| 22. | Cao, Y.; Fuchigami, T. J. Electroanal. Chem. 2006, 587, 25–30. doi:10.1016/j.jelechem.2005.10.010 |
| 25. | Chen, S.-Q.; Hatakeyama, T.; Fukuhara, T.; Hara, S.; Yoneda, N. Electrochim. Acta 1997, 42, 1951–1960. doi:10.1016/S0013-4686(97)85466-7 |
| 20. | Boroux, P.; Tardivel, R.; Simonet, J. J. Electrochem. Soc. 1997, 144, 841–847. doi:10.1149/1.1837498 |
| 11. | Fuchigami, T. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; chapter 25; Marcel Dekker: New York, 2001. |
| 12. | Noel, M.; Suryanarayanan, V. J. Appl. Electrochem. 2004, 34, 357–369. doi:10.1023/B:JACH.0000016670.97434.c7 |
| 13. | Dawood, K. M. Tetrahedron 2004, 60, 1435–1451. doi:10.1016/j.tet.2003.11.017 |
| 14. | Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/C1CC12414E |
| 15. | Fuchigami, T.; Inagi, S. In Organic Electrochemistry,, 5th ed.; Hammerich, O.; Speiser, B., Eds.; chapter 20; CRC Press: Boca Raton, FL, 2015. |
| 16. | Fuchigami, T.; Inagi, S. Organic Electrosynthesis. In Fundamentals and Applications of Organic Electrochemistry; Fuchigami,, T.; Atobe, M.; Inagi, S., Eds.; chapter 5; Wiley: West Sussex, 2015. doi:10.1002/9781118670750.ch05 |
| 17. | Kuribayashi, S.; Shida, N.; Inagi, S.; Fuchigami, T. Tetrahedron 2016, 72, 5343–5349. doi:10.1016/j.tet.2016.07.016 |
| 18. | Tanigawa, M.; Kuriyama, Y.; Inagi, S.; Fuchigami, T. Electrochim. Acta 2016, 199, 314–318. doi:10.1016/j.electacta.2016.02.051 |
| 19. | Suzuki, J.; Shida, N.; Inagi, S.; Fuchigami, T. Electroanalysis 2016, 28, 2797–2801. doi:10.1002/elan.201600130 |
| 21. | Hou, Y.; Higashiya, S.; Fuchigami, T. Electrochim. Acta 2000, 45, 3005–3010. doi:10.1016/S0013-4686(00)00379-0 |
| 32. | Qiao, Z.; Jiang, X. Org. Lett. 2016, 18, 1550–1553. doi:10.1021/acs.orglett.6b00324 |
| 29. | Isokawa, M.; Sano, M.; Kubota, K.; Suzuki, K.; Inagi, S.; Fuchigami, T. J. Electrochem. Soc. 2017, 164, G121–G127. doi:10.1149/2.1601713jes |
| 21. | Hou, Y.; Higashiya, S.; Fuchigami, T. Electrochim. Acta 2000, 45, 3005–3010. doi:10.1016/S0013-4686(00)00379-0 |
| 23. | Shen, Y.; Suzuki, K.; Atobe, M.; Fuchigami, T. J. Electroanal. Chem. 2003, 540, 189–194. doi:10.1016/S0022-0728(02)01297-4 |
| 30. | Riyadh, S. M.; Ishii, H.; Fuchigami, T. Tetrahedron 2002, 58, 5877–5883. doi:10.1016/S0040-4020(02)00559-8 |
| 31. | Lee, E.-S.; Choi, B.-W.; Jung, D.-I.; Hwang, H.-J.; Han, J.-T.; Lee, B.-H. Bull. Korean Chem. Soc. 2003, 24, 243–245. doi:10.5012/bkcs.2003.24.2.243 |
| 38. | Beckwith, A. L. J.; Duggan, S. A. M. J. Chem. Soc., Perkin Trans. 2 1994, 1509–1518. doi:10.1039/P29940001509 |
| 17. | Kuribayashi, S.; Shida, N.; Inagi, S.; Fuchigami, T. Tetrahedron 2016, 72, 5343–5349. doi:10.1016/j.tet.2016.07.016 |
| 31. | Lee, E.-S.; Choi, B.-W.; Jung, D.-I.; Hwang, H.-J.; Han, J.-T.; Lee, B.-H. Bull. Korean Chem. Soc. 2003, 24, 243–245. doi:10.5012/bkcs.2003.24.2.243 |
| 37. | Oya, M.; Baba, T.; Kato, E.; Kawashima, Y.; Watanabe, T. Chem. Pharm. Bull. 1982, 30, 440–461. doi:10.1248/cpb.30.440 |
| 35. | Moon, H. K.; Sung, G. H.; Kim, B. R.; Park, J. K.; Yoon, Y.-J.; Yoon, H. J. Adv. Synth. Catal. 2016, 358, 1725–1730. doi:10.1002/adsc.201501177 |
| 36. | Montenegro, E.; Echarri, R.; Claver, C.; Castillón, S.; Moyano, A.; Pericàs, M. A.; Riera, A. Tetrahedron: Asymmetry 1996, 7, 3553–3558. doi:10.1016/S0957-4166(96)00463-6 |
| 33. | Ogawa, K. A.; Boydston, A. J. Org. Lett. 2014, 16, 1928–1931. doi:10.1021/ol500459x |
| 34. | Lu, G.-p.; Cai, C. Adv. Synth. Catal. 2013, 355, 1271–1276. doi:10.1002/adsc.201201059 |
© 2018 Kuribayashi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)