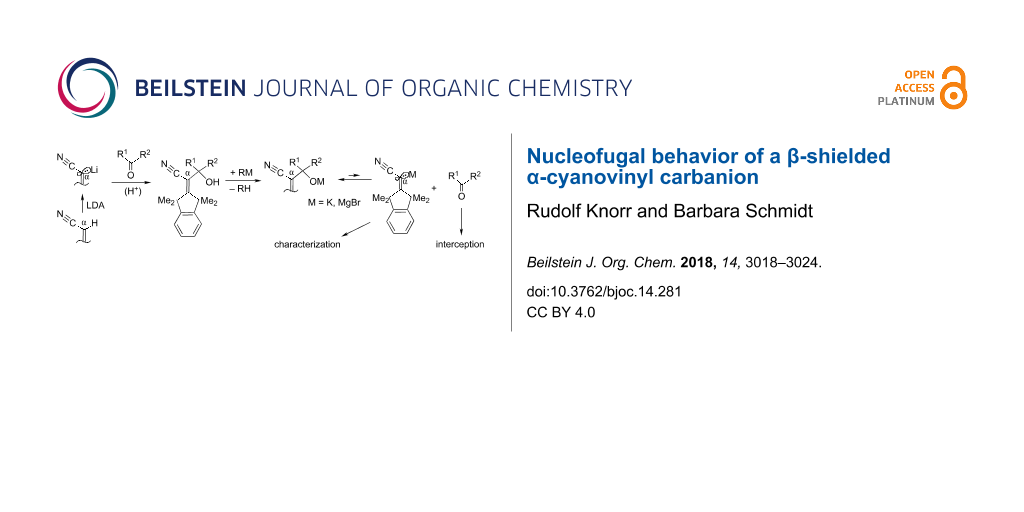
Abstract
Sterically well-shielded against unsolicited Michael addition and polymerization reactions, α-metalated α-(1,1,3,3-tetramethylindan-2-ylidene)acetonitriles added reversibly to three small aldehydes and two bulky ketones at room temperature. Experimental conditions were determined for transfer of the nucleofugal title carbanion unit between different carbonyl compounds. These readily occurring retro-additions via C–C(O) bond fission may also be used to generate different metal derivatives of the nucleofugal anions as equilibrium components. Fluoride-catalyzed, metal-free desilylation admitted carbonyl addition but blocked the retro-addition.

Graphical Abstract
Introduction
A carbanionic fragment shows nucleofugal behavior on breaking its single-bond connection with a developing electrophilic center. Apart from deprotonation reactions or formation of a separated ion pair and other trivial examples, the (at least formally) heterolytic cleavage of C–C single bonds can provide cases of interest if it generates organometallic compounds under unusual conditions. The well-known cases of alkoxide fission [1-3] (top line of Scheme 1) may be viewed as a reversed formation of an alkoxide A1M1 from an organometallic C–M1 and a carbonyl compound (R1)2C=O. Such a C–C fission (retro-addition reaction) can occur already near room temperature (rt) if the nucleofugal carbanion proves to be an electronically stabilized N≡C–CH2– [1] or allylic [2] species or a short-lived equilibrium component [3].
![[1860-5397-14-281-i1]](/bjoc/content/inline/1860-5397-14-281-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The nucleofugal carbanion unit C escapes from the alkoxides A1M1 or A1M2 (M = metal), generating the organometallics C–M1 or C–M2, respectively. Route (a): C–M1 can be trapped by (R2)2C=O to form the new alkoxide A2M1. Route (b): Trapping (*) of (R1)2C=O might accumulate C–M1 or C–M2. Route (c): A basic reagent BM2 replaces M1 by M2 and may generate C–M2. Achiral alkoxides (A) are depicted for simplicity.
Scheme 1: The nucleofugal carbanion unit C escapes from the alkoxides A1M1 or A1M2 (M = metal), generating th...
A cleavable alkoxide A1M1 can be employed in at least three ways: (a) its equilibrium component C–M1 may be trapped by a different carbonyl compound (R2)2C=O to give the new alkoxide A2M1 (and from there the alcohol A2H) as evidence for the intermediacy of a short-lived species C–M1. If A2M1 is also cleavable, the two alkoxides A1M1 and A2M1 may be obtained under thermodynamic control. (b) Trapping of the other equilibrium component (R1)2C=O by a nucleophile might accumulate the organometallic compound C–M1. (c) A1M1 may be used to replace M1 by M2 with intent to study a different organometallic C–M2 (bottom part of Scheme 1). For this purpose, A1M1 should be demetalated through work-up to give, for instance, the alcohol A1H; the subsequent deprotonation of A1H by an M2-containing base BM2 must be complete before the generated alkoxide A1M2 begins to release C–M2 that would otherwise be protonated by any unconsumed portion of A1H. We had used this technique [3] to generate the short-lived, NMR-invisible pivaloylmetals (H3C)3C–COM from the overburdened alkoxides of tri-tert-butylacyloin that were readily cleaved with relief of internal strain as a driving force. We now report here on the above-mentioned options (a) and (c) with the focus on some nucleofugal traits of the carbanion unit of the stable [4], β-shielded α-metalated acrylonitriles.
Results and Discussion
The α-cyanoalkenyllithium 2Li (top line of Scheme 2) may be prepared [4] either by LDA-mediated deprotonation (LDA = iPr2NLi) of the β-shielded acrylonitrile derivative 1 or by an Sn/Li transmetalation reaction of n-butyllithium (“n-BuLi”) with the α-stannyl derivative 3. Due to intermolecular LiN coordination, 2Li was deduced [4] to form a clustered ground state that showed no obvious rate anomaly with previously [4] studied electrophiles; this should also hold true here for pivalaldehyde t-BuCH=O (4, “t-Bu” = tert-butyl). The chiral adduct 7 of 4 exhibited four different NMR signals for the four methyl groups at the 1- and 3-positions. Analytically pure 7 was obtained only through distillation as a crystalline sample that decayed during an attempted recrystallization from methanol. A possible reason for this instability at a temperature far below the boiling point became clear on addition of some solid potassium tert-butoxide (KOt-Bu) to an NMR tube that contained purified 7 in DMSO at rt: After the immediate appearance of a yellow tint, the next 1H NMR spectrum revealed that 7 was almost completely transformed into the acrylonitrile derivative 1 (bottom line of Scheme 2). This suggested that the nucleofugal carbanion unit of 2K had escaped from the potassium alkoxide 10 with formation of 2K (yellow tint) and t-BuCH=O, whereupon 2K was trapped by proton sources such as DMSO or unconsumed 7 to produce 1. As a consequence, the general procedure (GP) of adding carbonyl compounds to deprotonated 1 commends to use an acidic work-up and a strict exclusion of bases that are stronger than diluted aqueous NaHCO3.
![[1860-5397-14-281-i2]](/bjoc/content/inline/1860-5397-14-281-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of β-shielded α-lithioacrylonitrile 2Li and its reaction with aldehydes 4–6 to adducts 7–9. Cleavage of the potassium alkoxide 10 (bottom line) gave 2K and then 1.
Scheme 2: Preparation of β-shielded α-lithioacrylonitrile 2Li and its reaction with aldehydes 4–6 to adducts 7...
Benzaldehyde (5) and cyclopropanecarboxaldehyde (6) added also readily to 2Li with production of 8 and 9, respectively. As already reported above for 7, adducts 8 and 9 showed also four CH3 NMR signals on account of the stereogenic carbon centers C–OH. The NMR AB-type interproton 3J splittings of the H–C–O–H moieties in 8 and 9 disclosed that intermolecular scrambling of the hydroxy protons was retarded through steric congestion.
The adduct 13 (Scheme 3) of adamantan-2-one (11) was formed (like the above aldehyde adducts) through addition at C-α (rather than at nitrogen), as shown by the δ values of the tetramethylindane part which were consistent with those of 7–9. Complete NMR assignments for the 2-hydroxyadamantan-2-yl part of 13 were achieved with the two-dimensional NOESY and HSQC techniques at rt. Except for the OH signal, all other resonances were changed only insignificantly on cooling to −60 °C. Cleavage of 13 via the lithium alkoxide 12 (top line of Scheme 3) was fast on the laboratory time scale at rt in the solvents THF, Et2O, t-BuOMe, or toluene. As mentioned in the Introduction, the successful transfer of 2Li from 11 to 4 depends on the exclusion of proton sources (including the alcohol 13) that would protonate 2Li with formation of 1. Thus, the slow addition of 13 to a well-stirred solution of methyllithium (MeLi, 2 equiv) liberated gaseous CH4 (1 equiv) so that 13 was completely consumed before the electrophile t-BuCH=O (4; 4 equiv) was introduced and furnished adduct 7 but no trace of 1. The worst case (with 1 as a preponderant product) may be encountered when the deprotonating base is added to the alcohol 13 either too slowly or in a less than stoichiometric amount. For instance (bottom line of Scheme 3), the heterogeneous, slow deprotonation of 13 in THF by the insoluble base potassium hydride (KH) afforded 11 and 1 as the only products via cleavage of the potassium alkoxide 14a and protonation of the emerging 2K by residual 13, so that the final introduction of pivalaldehyde (4) produced no adduct 7. However, the same result was also found when 13 was added to a homogeneous solution of PhCH2K in THF; this suggested that 13 had transferred its proton to the emerging, thermally stable [4] cleavage product 2K faster than to the residual PhCH2K (perhaps a problem of slow mixing). On the other hand, deprotonation of 13 by the Grignard reagent MeMgBr in THF was complete (quantitative CH4 evolution) within 20 min at 0 °C; the subsequent cleavage reaction of the generated magnesium alkoxide 14b was very slow at rt, creating 2MgBr (Scheme 3) in the presence of t-BuCH=O (4) and therefrom 7 together with only a small amount of 1. This almost clean production of 7 agrees with the versatile [5] reactions of H2C=C(CN)–MgBr with many electrophiles. In most of the above trapping experiments, a slow disproportionation of t-BuCH=O was observed to generate t-BuCH2OH and t-BuCO2Li as side-products.
![[1860-5397-14-281-i3]](/bjoc/content/inline/1860-5397-14-281-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reversible formation of the adduct 13 of adamantan-2-one (11).
Scheme 3: Reversible formation of the adduct 13 of adamantan-2-one (11).
The adduct 18 (Scheme 4) of fluoren-9-one (15) had again a triangular ligand-binding system at C-α, as shown by the completely assigned NMR resonances. As the only unexpected contrast to 7–9 and 13, the 3-CH3 protons were observed to absorb at a higher δ value (1.98 ppm) than that of the 1-CH3 protons, which amounts to an increase of 0.34 ppm relative to 3-CH3 of 13. We ascribed this to a ring-current effect by the 9´-hydroxyfluoren-9´-yl substituent that can be expected to prefer a perpendicular conformation relative to the indane rings with the 9´-OH group pointing toward 3-CH3. As a peculiar line-width effect, the NMR signals of 3-CH3 (1H and 13C) and of 4-H were significantly broadened at rt but had the usual narrow line widths at −45 °C. Formation of the primary lithium alkoxide 16 (Scheme 4) was shown to be readily reversible as follows. Without protolysis and work-up of the green-colored solution, 16 was kept in THF at rt for 30 min and then treated with t-BuCH=O (4; 1.5 equiv); after a further period of 2 hours at rt, the GP work-up furnished the adduct 7 of t-BuCH=O along with the co-product fluoren-9-one (15), some fluoren-9-ol [6], and a small amount of 1, but no 18 (would derive from residual 16). Thus, 16 had released the nucleofugal carbanion unit of 2Li which was trapped by 4 to give the alkoxide 17 of 7.
![[1860-5397-14-281-i4]](/bjoc/content/inline/1860-5397-14-281-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation and cleavage of the adduct 18 of fluoren-9-one (15).
Scheme 4: Preparation and cleavage of the adduct 18 of fluoren-9-one (15).
In view of the apparently modest spatial demand of the α-cyano substituent in the adducts of 2Li, it seemed surprising that the following ketones generated no (or no kinetically stable) C=O adducts at rt: t-Bu2C=O, pivalophenone (t-Bu–C(O)–Ph), benzophenone (Ph2C=O) [7], bis(1-methylcyclopropyl)ketone [8], and dicyclopropyl ketone (19). Instead of an adduct of 19 to 2Li, the carboxyalcohol 21 (a known [9] product of 19 with LDA) in Scheme 5 and the acrylonitrile derivative 1 were obtained from 19; this run was conducted in the absence of LDA with a sample of 2Li that had been prepared in Et2O through an Sn/Li interchange reaction [4] of 3 with n-BuLi. Therefore, 2Li should have deprotonated 19 with formation of 20 that was trapped by 19 to give 21. But why was 21 not formed from 19 with the α-phenylalkenyllithium 22 (related to 2Li) that produced [10] the “normal” carbonyl adduct 23 of 19? This “normal” C=O addition reaction was obviously faster than the deprotonation of 19 and also apparently irreversible under the reaction conditions, whereas the unobserved C=O addition of 19 to 2Li might be possible yet quickly reversible with a terminating proton transfer from 19 to 2Li as shown in Scheme 5. Alternatively, carbonyl additions to 2Li might be generally retarded (relative to protonations) on account of the clustered [4] ground state structure of 2Li in solution.
![[1860-5397-14-281-i5]](/bjoc/content/inline/1860-5397-14-281-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proton transfer from dicyclopropyl ketone (19) to 2Li.
Scheme 5: Proton transfer from dicyclopropyl ketone (19) to 2Li.
Electrophilic cations (Li+ or K+) are not necessary (albeit perhaps helpful) for the addition of the carbanion unit of 2Li or 2K to carbonyl compounds: Generated through desilylation of 24 (Scheme 6) by tetrabutylammonium fluoride (Bu4N+F−; ≤0.05 equiv), the ion pair 25 was trapped by pivalaldehyde (4) with formation of 7 along with a comparable amount of the alkene 1. However, the primary alkoxide product, as formed by 25 and 4, was supposedly blocked by FSiMe3 and hence unsuitable for an analysis of the retro-addition reaction (see Supporting Information File 1).
![[1860-5397-14-281-i6]](/bjoc/content/inline/1860-5397-14-281-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Metal-free release of the carbanion unit in 25 and its seizure by t-BuCH=O (→ 7); Bu = n-butyl.
Scheme 6: Metal-free release of the carbanion unit in 25 and its seizure by t-BuCH=O (→ 7); Bu = n-butyl.
Conclusion
(i) The nucleofugal carbanion (α-deprotonated 1) can escape with surprising ease from alkoxides, perhaps with some assistance by a metal cation if present. Conversely, metal cation assistance was not necessary for the rapid carbanion release from the α-silyl compound 24 in the presence of Bu4N+F− in catalytic amounts.
(ii) Most of the above alkoxide fission reactions were conducted in the presence of an electrophile for trapping the released nucleofugal carbanion; this provided a first evidence for the retro-addition process with an alkenylmetal intermediate. These fissions were slow in case of the magnesium alkoxide but rapid for the lithium or potassium alkoxides at ambient temperatures.
(iii) The alternative trapping [3] of the carbonyl component by means of a nucleophile was not tried here but might serve to accumulate the organometallic equilibrium component for the purpose of spectroscopic characterization or X-ray diffraction analyses.
(iv) Fission of the lithium alkoxide of the adduct 13 of adamantan-2-one appeared to be comparably fast on the laboratory time scale in the solvents THF, Et2O, t-BuOMe, or toluene. A similarly weak solvent dependence had been observed [4] for the heterolytic cis/trans stereoinversion of 2Li.
(v) For more profound mechanistic investigations, one should be aware of the established [4] clustered ground state of 2Li and the possibility that clustered species might be the reactive components even in THF solutions, whereas the retro-addition processes may perhaps involve monomeric nucleofuges.
Experimental
General remarks. Stoichiometric formation of alkoxides through deprotonation of the alcohols was conveniently performed with methyllithium or methylmagnesium bromide and gas-volumetric control of the liberated methane (ca. 25 mL/mmol at 22 °C). All 1H and 13C NMR shifts δ were referenced with internal Me4Si. NMR abbreviations were as follows: d = doublet, m = multiplet, q = quartet, quat = quaternary, s = singlet, t = triplet.
General procedure (GP) for carbonyl addition to α-lithio-α-(1,1,3,3-tetramethylindan-2-ylidene)acetonitrile (2Li). A dry Schlenk flask was charged with a magnetic stirring bar, anhydrous Et2O or toluene (2 mL), and N,N-diisopropylamine (0.161 mL, 1.14 mmol). This solution was stirred at –30 °C under argon gas cover during the addition of n-BuLi (1.05 mmol) in hexane (0.47 mL). The created LDA (up to 1.05 mmol) solution was treated with the solid nitrile 1 (200 mg, 0.95 mmol) and stirred further at rt till 1 was completely dissolved, whereupon the carbonyl compound (1.24 mmol, neat or in a solvent) was added to the yellow solution of 2Li and stirred for another 30 min. The mixture was poured into aqueous HCl (2 M, 10 mmol) and extracted with Et2O (3 × 20 mL). (Note that the product might decay in a non-acidic milieu.) The combined Et2O extracts were shaken first with water (1 × 20 mL), then with diluted aqueous NaHCO3, and again distilled water (2 × 20 mL), then dried over Na2SO4, filtered, and concentrated.
3´-Hydroxy-4´,4´-dimethyl-2´-(1,1,3,3-tetramethylindan-2-ylidene)pentanenitrile (7). The GP protocol was used to treat LDA (1.56 mmol) in toluene with the β-shielded acrylonitrile 1 (300 mg, 1.42 mmol), followed by pivalaldehyde (4, 0.200 mL, 1.84 mmol). After work-up, the crude material (401 mg) distilled at 120–130 °C (bath temp.)/0.03 mbar to give pure 7 (132 mg, 29%) with mp 108–112 °C; 7 would decay on recrystallization. 1H NMR (CDCl3, 400 MHz) δ 1.16 (s, 9H, t-Bu), 1.59 and 1.61 (2 s, 2 × 3H, 2 × 3-CH3), 1.69 and 1.71 (2 s, 2 × 3H, 2 × 1-CH3), 1.97 (broad s, 1H, OH), 4.71 (s, 1H, H–CO), 7.14 and 7.17 (2 m, 2 × 1H, 4-/7-H), 7.27 (m, 2H, 5-/6-H) ppm, assigned through comparison with 8; 13C NMR (CDCl3, 100.6 MHz) δ 27.0 (q, Me3C), 28.4 and 28.7 (2 q, 2 × 1-CH3), 30.9 and 32.4 (2 q, 2 × 3-CH3), 35.7 (quat, Me3C), 49.42 (quat, C-3), 49.57 (quat, C-1), 74.6 (d, HCO), 110.9 (quat, C-α), 118.3 (quat, C≡N), 122.1 (d, C-4), 122.4 (d, C-7), 127.7 and 127.9 (2 d, C-5/-6), 147.6 (quat, C-7a), 148.0 (quat, C-3a), 180.6 ppm (quat, C-2); IR (KBr): 3475 (O–H), 2927, 2869, 2214 (s, C≡N), 1610, 1487, 1464, 1368, 1274, 1192, 1060, 1013, 762 cm−1; anal. calcd for C20H27NO (297.4): C, 80.76; H, 9.15; N, 4.71; found: C, 81.15; H, 9.42; N, 4.76.
3´-Hydroxy-3´-phenyl-2´-(1,1,3,3-tetramethylindan-2-ylidene)propionitrile (8). A solution of LDA (1.14 mmol) in dry Et2O was prepared according to the GP and treated with the acrylonitrile derivative 1 (200 mg, 0.95 mmol). The yellow solution (2Li, 0.95 mmol) was decolorized on the addition of benzaldehyde (5, 0.126 mL, 1.24 mmol). Warm-up (GP) afforded a colorless powder (262 mg, 87%); this almost pure material was recrystallized from toluene (5 mL) to furnish a powder (108 mg, 36%) with mp 190–192 °C; 8 was insoluble in CCl4. 1H NMR (CDCl3, 400 MHz) δ 1.58 (s, 3H, 1 × 3-CH3), 1.71 and 1.72 (2 s, 2 × 3H, 2 × 1-CH3), 1.75 (s, 3H, 1 × 3-CH3), 2.33 (d, 3J = 6.7 Hz, 1H, OH), 6.15 (d, 3J = 6.7 Hz, 1H, H–CO), 7.17 and 7.20 (2 m, 2 × 1H, 4-/7-H), 7.30 (m, 2H, 5-/6-H), 7.35 (t, 3J = 7.0 Hz, 1H, para-H), 7.42 (tm, 3J = 7.2 Hz, 2H, 2 × meta-H), 7.51 (dm, 3J = 7.4 Hz, 2H, 2 × ortho-H) ppm, assigned through comparison with 7; 13C NMR (CDCl3, 100.6 MHz) δ 28.8 and 29.0 (2 q, 2 × 1-CH3), 30.6 and 31.5 (2 q, 2 × 3-CH3), 49.3 (quat, C-3), 49.5 (quat, C-1), 69.6 (d, HCO), 112.4 (quat, C-α), 116.9 (quat, C≡N), 122.1 (d, C-4), 122.5 (d, C-7), 126.1 (d, 2 × C-ortho), 127.9 and 128.0 (2 d, C-5/-6), 128.4 (d, C-para), 128.8 (d, 2 × C-meta), 140.0 (quat, C-ipso), 147.6 (quat, C-7a), 147.8 (quat, C-3a), 179.5 (quat, C-2) ppm, assigned through comparison with 7 and benzyl alcohol; IR (KBr): 3404 (O–H), 2971, 2228 (C≡N), 1487, 1458, 1368, 1052, 751, 716 cm−1; anal. calcd for C22H23NO (317.4): C, 83.24; H, 7.30; N, 4.41; found: C, 83.39; H, 7.33; N, 4.48.
3´-Cyclopropyl-3´-hydroxy-2´-(1,1,3,3-tetramethylindan-2-ylidene)propionitrile (9). Using the GP protocol, the acrylonitrile derivative 1 (200 mg, 0.95 mmol) was deprotonated with LDA (1.05 mmol) in Et2O to generate 2Li which was treated with cyclopropanecarboxaldehyde (6, 0.092 mL, 1.24 mmol). The crude product (259 mg) was crystallized from ethanol (2 mL) to give colorless 9 (126 mg, 47%) with mp 146–149 °C. 1H NMR (CDCl3, 400 MHz) δ 0.39 (m, 1H of cyclopropyl-CH2 cis to HCO), 0.65 (m, 2 × trans-H), 0.76 (m, 1 cis-H), 1.41 (m, 1H, tert-CH of cyclopropyl), 1.49 and 1.57 (2 s, 2 × 3H, 2 × 3-CH3), 1.69 and 1.71 (2 s, 2 × 3H, 2 × 1-CH3), 1.98 (d, 3J = 4.6 Hz, 1H, exchangeable with D2O, OH), 4.35 (dd, 3J = 7.7 and 4.6 Hz, 1H, H–CO), 7.14 and 7.18 (2 m, 2 × 1H, 4-/7-H), 7.27 (m, 2H, 5-/6-H) ppm, assigned through comparison with 7; 13C NMR (CDCl3, 100.6 MHz) δ 2.8 and 3.8 (2 t, 2 diastereotopic cylopropyl-CH2), 16.4 (d, tert-C of cyclopropyl), 28.83 and 28.88 (2 q, 2 × 1-CH3), 30.3 and 32.0 (2 q, 2 × 3-CH3), 49.11 and 49.16 (2 quat, C-3 and C-1), 71.2 (d, HCO), 112.3 (quat, C-α), 117.4 (quat, C≡N), 122.1 (d, C-4), 122.5 (d, C-7), 127.76 and 127.87 (2 d, C-5/-6), 147.71 (quat, C-7a), 147.92 (quat, C-3a), 178.6 (quat, C-2) ppm, assigned as above; IR (KBr): 3471 (O–H), 2993, 2961, 2928, 2214 (s, C≡N), 1629, 1487, 1461, 1039, 759 cm−1; anal. calcd for C19H23NO (281.4): C, 81.10; H, 8.24; N, 4.98; found: C, 80.92; H, 8.11; N, 4.97.
α-(2´-Hydroxyadamantan-2´-yl)-α-(1,1,3,3-tetramethylindan-2-ylidene)acetonitrile (13). The technique of the GP was used to generate 2Li in Et2O from LDA (1.05 mmol) and acrylonitrile derivative 1 (200 mg, 0.95 mmol). After continued stirring till 1 was completely dissolved, a solution of adamantan-2-one (11, 186 mg, 1.24 mmol) in dry Et2O (4 mL) was added slowly (1 drop per s) at rt, whereupon the mixture was stirred for 30 min and then worked up (GP). The crude material (321 mg after drying in presence of solid KOH) was recrystallized from CCl4 (5 mL) to give colorless platelets 13 (141 mg, 41%) with mp 179–180.5 °C. 1H NMR (CDCl3, 400 MHz, 25 °C) δ 1.64 (s, 6H, 2 × 3-CH3), 1.68 (dm. 2J = 12.5 Hz, 2H, 1 × 4´-H and 1 × 9´-H), 1.74 (broadened t, 3J ≈ 2 Hz, 2H, 2 × enantiotopic 6´-H), 1.79 (s, 6H, 2 × 1-CH3), 1.83 (m, 3J ≈ 2.5 Hz, 1H, 5´-H), 1.85 (dm, 2J obscured, 2H, 1 × 8´-H and 1 × 10´-H), 1.86 (s, 1H, OH, δ = 2.30 ppm at –60 °C), 1.89 (m, 3J = 2.8 Hz, 1H, 7´-H), 1.96 (broadened d, 2J = 13 Hz, 2H, 1 × 8´-H and 1 × 10´-H), 2.33 (broadened d, 2J = 12.5 Hz, 2H, 1 × 4´-H and 1 × 9´-H), 2.70 (broad, 2H, 1´-/3´-H), 7.12 (m, 1H, 4-H), 7.17 (m, 1H, 7-H), 7.26 (m, 2H, 6-/5-H) ppm, assigned through the NOESY correlations 4-H ↔ 3-CH3 ↔ 1´-/3´-H ↔ all four 4´-/9´-H ↔ 5´-H ↔ 2 × 6´-H ↔ 7´-H ↔ 8´-/10´-H ↔ 1´-/3´-H ↔ OH, and 7-H ↔ 1-CH3 (this without any cross-peaks to the adamantane protons); 13C NMR (CDCl3, 100.6 MHz) δ 25.9 (d, CH-5´), 26.5 (d, CH-7´), 28.8 (q, 2 × 1-CH3), 30.0 (q, 2 × 3-CH3), 32.9 (t, CH2-4´-/9´), 35.6 (t, CH2-8´-/10´), 37.0 (t, CH2-6´), 37.4 (d, CH-1´-/3´), 50.5 (quat, C-3), 51.5 (quat, C-1), 77.1 (quat, C-2´), 118.2 and 119.4 (2 × quat, C-α or C≡N), 121.9 (d, C-4), 122.5 (d, C-7), 127.45 and 127.57 (2 d, C-5/-6), 147.4 (quat, C-7a), 149.2 (quat, C-3a), 182.6 (quat, C-2) ppm, assigned through DEPT, HSQC, and comparison with 7–9, no other significant changes at −60 °C; IR (KBr): 3471 (sharp, O–H), 2855, 2201 (sharp, C≡N), 1602 (w), 1488, 1453, 1353, 1020, 760 (s) cm−1; anal. calcd for C25H31NO (361.53): C, 83.06; H, 8.64; N, 3.87; found: C, 82.90; H, 8.50; N, 3.61.
α-(9´-Hydroxyfluoren-9´-yl)-α-(1,1,3,3-tetramethylindan-2-ylidene)acetonitrile (18). Using the GP protocol, the solid acrylonitrile derivative 1 (200 mg, 0.95 mmol) was added to a solution of LDA (1.05 mmol) in dry Et2O, followed by fluoren-9-one (15, 223 mg, 1.24 mmol) which changed the color of the mixture from yellow to dark green. The crude material (406 mg) obtained after work-up (GP) was recrystallized from toluene (9 mL) to afford colorless 18 (147 mg, 40%) with mp 243–245 °C. 1H NMR (CDCl3, 400 MHz, 25 °C) δ 1.66 (sharp s, 6H, 2 × 1-CH3), 1.98 (broadened s, 6H, 2 × 3-CH3), 2.35 (s, 1H, OH), 7.15 (dm, 1H, 7-H), 7.22 (broadened d, 1H, 4-H, narrowed at −45 °C), 7.25 (td, 1H, 5-H), 7.28 (td, 1H, 6-H), 7.33 (td, 3J = 7.5 Hz, 2H, 2´-/7´-H), 7.42 (td, 3J = 7.0 Hz, 2H, 3´-/6´-H), 7.48 (d, 3J = 7.6 Hz, 2H, 1´-/8´-H), 7.69 (d, 3J = 7.5 Hz, 2H, 4´-/5´-H) ppm, assigned through comparison with 13 and the NOESY correlations 3-CH3 ↔ 1´-/8´-H ↔ 1-CH3 (weaker) ↔ 7-H ↔ 6-H, and 1´-/8´-H ↔ 2´-/7´-H ↔ 3´-/6´-H ↔ 4´-/5´-H; 1H NMR (CDCl3, 400 MHz, −45 °C) δ 1.65, 1.98, 2.39, 7.20, 7.27, 7.30, 7.33, 7.37, 7.46, 7.49, 7.69 ppm; 13C NMR (CDCl3, 100.6 MHz) δ 29.1 (qq, 1J = 128 Hz, 3J = 4.5 Hz, 2 × 1-CH3), 31.7 (broadened qq, sharp at −45 °C, 1J = 128 Hz, 3J = ca. 4.5 Hz, 2 × 3-CH3), 50.1 (unresolved m, C-3), 51.0 (unresolved m, C-1), 85.7 (m, C-9´), 111.1 (very weak d, stronger at −45 °C, 3J = 8.5 Hz to OH, C-α), 117.9 (sharp s, C≡N), 120.6 (dd, 1J = 160 Hz, 3J = 8 Hz, CH-4´/-5´), 122.3 and 122.4 (2 dd, 1J = 159 Hz, 3J = 8 Hz, CH-7/-4), 124.1 (dd, 1J = 160 Hz, 3J = 8 Hz, CH-1´/-8´), 127.2 and 127.5 (2 dd, 1J = 160 Hz, 3J = 7.5 Hz, CH-5/-6), 128.8 (dd, 1J = 162 Hz, 3J = 7.3 Hz, CH-2´/-7´), 130.1 (dd, 1J = 159 Hz, 3J = 7.2 Hz, CH-3´/-6´), 139.9 (broadened t, 3J = 7 Hz, C-4a´/4b´), 147.4 (m, C-7a), 147.9 (t, 3J = 7.2 Hz, C-8a´/9a´), 149.6 (unresolved m, C-3a), 179.4 (m, C-2) ppm, assigned through comparison with 7, 8, 9, and fluoren-9-one → HETCOR → COLOCS(7 Hz); COLOCS cross-peaks of 3J: 1-CH3 → C-2 and C-7a, 3-CH3 → C-2 and C-3a, OH → C-α, 4-H → C-6, 5-H → C-7, 6-H → C-4, 7-H → C-5, 1´-H → C-3´ and C-4a´, 2´-H → C-4´ and C-9a´, 3´-H → C-1´ and C-4a´, 4´-H → C-2´ and C-9a´; IR (KBr): 3525 (w). 3417 (s), 3022, 2959, 2925, 2215 (sharp, C≡N), 1607, 1488, 1451, 1366, 1207, 771, 761, 754, 735 cm−1; anal. calcd for C28H25NO (391.5): C, 85.90; H, 6.44; N, 3.58; found: C, 86.13; H, 6.04; N, 3.60.
Cyclopropyl 1-[α,α-(dicyclopropyl)hydroxymethyl]cyclopropyl ketone (21) [9]. 1H NMR (CDCl3, 400 MHz) δ 0.27 (m, 4H), 0.49 (m, 4H), 0.80 (m, 2H), 0.86 (m, 4H), 0.98 (m, 2H), 1.27 (m, 2H), 1.46 (m, 1 tert-H), 4.3 (broadened s, 1H, OH) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 0.28 (2 × CH2), 0.39 (2 × CH2), 11.1 (2 × CH2), 11.3 (2 × CH2), 15.7 (1 × CH), 17.6 (2 × CH), 39.8 (1 × quat C), 70.7 (1 × quat, C–O), 212.9 (1 × quat, C=O) ppm.
Supporting Information
| Supporting Information File 1: Ion-pair intermediate through desilylation with Bu4N+ F−. | ||
| Format: PDF | Size: 148.7 KB | Download |
References
-
Kaiser, E. M.; Hauser, C. R. J. Org. Chem. 1968, 33, 3402–3404. doi:10.1021/jo01273a009
and cited references.
Return to citation in text: [1] [2] -
Jones, P.; Knochel, P. J. Org. Chem. 1999, 64, 186–195. doi:10.1021/jo981623m
and quoted literature.
Return to citation in text: [1] [2] -
Knorr, R.; Böhrer, G.; Schubert, B.; Böhrer, P. Chem. – Eur. J. 2012, 18, 7506–7515. doi:10.1002/chem.201102867
Return to citation in text: [1] [2] [3] [4] -
Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Thibonnet, J.; Anh Vu, V.; Bérillon, L.; Knochel, P. Tetrahedron 2002, 58, 4787–4799. doi:10.1016/s0040-4020(02)00439-8
Return to citation in text: [1] -
Wang, J.; Wan, W.; Jiang, H.; Gao, Y.; Jiang, X.; Lin, H.; Zhao, W.; Hao, J. Org. Lett. 2010, 12, 3874–3877. doi:10.1021/ol101553a
compound 6 in the Supporting Infornation therein.
Return to citation in text: [1] -
Melamed, U.; Feit, B. A. J. Chem. Soc., Perkin Trans. 1 1978, 1232–1236. doi:10.1039/p19780001232
They reported that Ph2C=C(Li)–CN added readily to Ph2C=O (compound 14 therein).
Return to citation in text: [1] -
Böhrer, G.; Knorr, R.; Böhrer, P. Chem. Ber. 1990, 123, 2161–2166. doi:10.1002/cber.19901231112
compound 12 therein.
Return to citation in text: [1] -
Perevalova, E. G.; Belesov, I. G.; Struchkov, Y. T.; Leschova, I. F.; Kalyuzhnaya, Y. S.; Voyevodskaya, T. I.; Slovokhotov, Y. L.; Granberg, K. I. J. Organomet. Chem. 1985, 286, 129–143. doi:10.1016/0022-328x(85)87242-9
on pp 133 and 134 therein.
Return to citation in text: [1] [2] -
Knorr, R.; Menke, T.; Behringer, C.; Ferchland, K.; Mehlstäubl, J.; Lattke, E. Organometallics 2013, 32, 4070–4081. doi:10.1021/om4000852
compound 27b therein.
Return to citation in text: [1]
| 3. | Knorr, R.; Böhrer, G.; Schubert, B.; Böhrer, P. Chem. – Eur. J. 2012, 18, 7506–7515. doi:10.1002/chem.201102867 |
| 10. |
Knorr, R.; Menke, T.; Behringer, C.; Ferchland, K.; Mehlstäubl, J.; Lattke, E. Organometallics 2013, 32, 4070–4081. doi:10.1021/om4000852
compound 27b therein. |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 1. |
Kaiser, E. M.; Hauser, C. R. J. Org. Chem. 1968, 33, 3402–3404. doi:10.1021/jo01273a009
and cited references. |
| 2. |
Jones, P.; Knochel, P. J. Org. Chem. 1999, 64, 186–195. doi:10.1021/jo981623m
and quoted literature. |
| 3. | Knorr, R.; Böhrer, G.; Schubert, B.; Böhrer, P. Chem. – Eur. J. 2012, 18, 7506–7515. doi:10.1002/chem.201102867 |
| 3. | Knorr, R.; Böhrer, G.; Schubert, B.; Böhrer, P. Chem. – Eur. J. 2012, 18, 7506–7515. doi:10.1002/chem.201102867 |
| 9. |
Perevalova, E. G.; Belesov, I. G.; Struchkov, Y. T.; Leschova, I. F.; Kalyuzhnaya, Y. S.; Voyevodskaya, T. I.; Slovokhotov, Y. L.; Granberg, K. I. J. Organomet. Chem. 1985, 286, 129–143. doi:10.1016/0022-328x(85)87242-9
on pp 133 and 134 therein. |
| 3. | Knorr, R.; Böhrer, G.; Schubert, B.; Böhrer, P. Chem. – Eur. J. 2012, 18, 7506–7515. doi:10.1002/chem.201102867 |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 2. |
Jones, P.; Knochel, P. J. Org. Chem. 1999, 64, 186–195. doi:10.1021/jo981623m
and quoted literature. |
| 7. |
Melamed, U.; Feit, B. A. J. Chem. Soc., Perkin Trans. 1 1978, 1232–1236. doi:10.1039/p19780001232
They reported that Ph2C=C(Li)–CN added readily to Ph2C=O (compound 14 therein). |
| 1. |
Kaiser, E. M.; Hauser, C. R. J. Org. Chem. 1968, 33, 3402–3404. doi:10.1021/jo01273a009
and cited references. |
| 8. |
Böhrer, G.; Knorr, R.; Böhrer, P. Chem. Ber. 1990, 123, 2161–2166. doi:10.1002/cber.19901231112
compound 12 therein. |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 5. | Thibonnet, J.; Anh Vu, V.; Bérillon, L.; Knochel, P. Tetrahedron 2002, 58, 4787–4799. doi:10.1016/s0040-4020(02)00439-8 |
| 9. |
Perevalova, E. G.; Belesov, I. G.; Struchkov, Y. T.; Leschova, I. F.; Kalyuzhnaya, Y. S.; Voyevodskaya, T. I.; Slovokhotov, Y. L.; Granberg, K. I. J. Organomet. Chem. 1985, 286, 129–143. doi:10.1016/0022-328x(85)87242-9
on pp 133 and 134 therein. |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 6. |
Wang, J.; Wan, W.; Jiang, H.; Gao, Y.; Jiang, X.; Lin, H.; Zhao, W.; Hao, J. Org. Lett. 2010, 12, 3874–3877. doi:10.1021/ol101553a
compound 6 in the Supporting Infornation therein. |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
| 4. | Knorr, R.; Schmidt, B.; Mehlstäubl, J.; von Roman, T. J. Organomet. Chem. 2018, 871, 185–196. doi:10.1016/j.jorganchem.2018.07.012 |
© 2018 Knorr and Schmidt; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)