Abstract
Antibiotic resistance threatens effective treatment of microbial infections globally. This situation has spurred the hunt for new antimicrobial compounds in both academia and the pharmaceutical industry. Here, we report how the widely used antitumor drug cisplatin may be repurposed as an effective antimicrobial against the nosocomial pathogen Pseudomonas aeruginosa. Cisplatin was found to effectively kill strains of P. aeruginosa. In such experiments, transcriptomic profiling showed upregulation of the recA gene, which is known to be important for DNA repair, implicating that cisplatin could interfere with DNA replication in P. aeruginosa. Cisplatin treatment significantly repressed the type III secretion system (T3SS), which is important for the secretion of exotoxins. Furthermore, cisplatin was also demonstrated to eradicate in vitro biofilms and in vivo biofilms in a murine keratitis model. This showed that cisplatin could be effectively used to eradicate biofilm infections which were otherwise difficult to be treated by conventional antibiotics. Although cisplatin is highly toxic for humans upon systemic exposure, a low toxicity was demonstrated with topical treatment. This indicated that higher-than-minimal inhibitory concentration (MIC) doses of cisplatin could be topically applied to treat persistent and recalcitrant P. aeruginosa infections.

Graphical Abstract
Introduction
Pseudomonas aeruginosa is a leading nosocomial pathogen which causes, among others, corneal, chronic otitis media, urinary tract (UTI) and respiratory tract infections [1]. P. aeruginosa is also the main cause of fatal infections in patients with cystic fibrosis (CF) [2] and cancer patients [3,4]. The success of P. aeruginosa as a leading pathogen is attributed to its ability to form resilient biofilms, resist antimicrobials and secrete virulence products.
Microbial cells resident in biofilms are encased by an extracellular matrix, which protects them from antimicrobial treatment and the host’s immune clearance [5]. Clinical P. aeruginosa isolates are mostly multidrug-resistant (MDR) strains [6], with robust ability to form biofilms [7]. P. aeruginosa also secretes virulence factors targeting important components of the immune system, such as the type III secretion systems (T3SS), which was shown to be associated with poor clinic outcomes in patients with lower respiratory infections [8] and ventilator-associated pneumonia [9]. Identifying antimicrobial compounds which actively target bacteria in the biofilm mode including virulence mechanisms that cripple immune defenses, may offer novel antimicrobial therapies against a variety of otherwise persistent P. aeruginosa infections.
Here, we screened our in-house collection of FDA-approved drugs and found that cisplatin was the most potent among several other Pt(II)-based compounds to kill P. aeruginosa. It was previously reported that cisplatin had antimicrobial effects on nosocomial pathogens, such as Escherichia coli, Klebsiella pneumonia and Staphylococcus aureus [10] and persister cells [11]. Transcriptomic analysis was employed to reveal the molecular mechanisms on how cisplatin inhibits the growth and production of virulence factors of P. aeruginosa. We also examined the effects of cisplatin treatment on in vitro P. aeruginosa biofilms and in a mouse model of corneal infection (keratitis). We showed that cisplatin is more effective than the clinically used antibiotic tobramycin in eradicating biofilms. Although cisplatin is highly toxic for intravenous applications, we showed that it has low toxicity when applied topically to wounds, as 25 mM (0.75 mg mL−1) did not have adverse effect on wound healing. This meant that higher doses (5–10 × MIC) of cisplatin could be safely used for topical applications. Given the low topical toxicity of cisplatin, it may be utilized as an attractive therapeutic agent for prevention and treatment of P. aeruginosa biofilm infections.
Materials and Methods
Bacterial strains and culture media
The bacterial strains used in this study are listed in Supporting Information File 1, Table S3. Luria–Bertani (LB) medium was used to maintain the bacterial strains. Growth assay and static biofilm cultivation were carried out at 37 °C in ABTGC (ABT minimal medium [12] supplemented with 0.4 g/L glucose and 0.4 g/L casamino acids). For marker selection in P. aeruginosa, 30 μg mL−1 gentamycin (Gm), 50 μg mL−1 tetracycline (Tc), 100 μg mL−1 streptomycin (Strep) or 200 μg mL−1 carbenicillin (Cb) were used, as appropriate.
Platinum complexes and solution preparation for MIC assay
Cisplatin, transplatin, cDPCP, oxaliplatin, K2PtCl4 and cis-PtCl2(CH3CN)2 were purchased from Sigma-Aldrich were used as received. Platinum complexes cis-PtCl2(Py)2 [13,14] and cis-PtCl2(PPh3)2 [15] were prepared according to the reported procedure. For minimal inhibitory concentration (MIC) assays, cisplatin, cDPCP, oxaliplatin, K2PtCl4 and cis-PtCl2(CH3CN)2 were dissolved in saline solution (0.85% w/v) at 2.5 mM concentration, while transplatin, cis-PtCl2(Py)2 and cis-PtCl2(PPh3)2 were dissolved in DMF (v/v) at 2.5 mM concentration.
Determination of minimal inhibitory concentration (MIC)
The MIC assays were performed using a microtiter broth dilution method as previously described (≈1 × 105 cells) in the NACLAR guidelines [16]. Overnight cultures of bacterial strains were diluted in ABTGC medium. Cisplatin and other Pt-containing compounds were diluted from a stock solution with ABTGC medium at a concentration 10 times higher than the required range. 10 μL of each diluted solution of Pt compounds were added to each corresponding well of a 96-well microtiter plate (polypropylene, Costar) and 90 μL of diluted bacterial culture in ABTGC medium were added before serial dilutions. The plate was incubated at 37 °C for 16–18 h. MIC was taken as the lowest concentration where no visual growth (based on OD600) of bacteria was detected. The experiments were performed in triplicate and representative results were shown.
RNA preparation
Bacterial cells were collected using the method described previously [17] with some modifications. Generally, PAO1 cells were cultivated either with (1.5 μM) or without cisplatin. The cells were harvested at the early-stationary phase (after approximately 8 h cultivation). Total RNA was extracted with an RNeasy Protect Bacteria Mini Kit with on-column DNase digestion (Qiagen). A Turbo DNA-free vigorous protocol was used for a second round of DNase treatment (Ambion). The 16S, 23S and 5S rRNA was removed using the Ribo-Zero Magnetic Kit (Bacteria) (Epicentre).
RNA sequencing and data analysis
Gene expression analysis was conducted via Illumina RNA sequencing (RNA-Seq technology). RNA-Seq was conducted for two biological replicates of each sample. The rRNA-depleted RNA was fragmented to 150–200 bp fragments, then first and second strand cDNA were synthesized with a cDNA-synthesis kit (ThermoScientific), followed by end repair and adapter ligation. After 12 cycles of PCR enrichment, the quality of the libraries was assessed using the Bioanalyzer (Agilent Technologies, USA). The libraries were sequenced using the Illumina HiSeq 2500 platform with a paired-end protocol and read lengths of 100 nt.
The sequencing data was analyzed as described previously [12]. Sequence reads were mapped onto PAO1 reference genome using the CLC genomics Workbench 8.0 (CLC Bio-Qiagen, Aarhus, Denmark). The differentially expressed genes were identified by performing a negative binomial test using the DESeq [18] package of R/Bioconductor [19], using the cut off of fold-change larger than 2 and a BH (Benjamini-Hochberg) adjusted P-value smaller than 0.05. The raw sequence reads were normalized by size factors, then Log2(N + 1) transformed. Hierarchical clustering analysis was performed using the transformed reads, and a heat-map was drawn for the differentially expressed genes between the cisplatin treated cells and control cells, using the heatmap.2 [19] package of R. Function enrichment analysis was conducted based on PseudoCAP Function Class (http://www.pseudomonas.com), and a dot plots figure was generated using the ggplot2 [20] package of R.
The RNA-Seq datasets are available at the NCBI Sequence Read Archives: SRS1038085 and SRS1038089.
qRT-PCR analysis
Total RNA was extracted using RNeasy Mini Kit (Qiagen) with on-column DNase digestion. The integrity, purity and concentration of the RNA were determined by NanoDrop spectrophotometry and Agilent 2200 TapeStation system.
Quantitative reverse transcriptase PCR (qRT-PCR) was performed using a two-step method. First-strand cDNA was synthesized from total RNA using SuperScript III First-Strand Synthesis SuperMix kit (Cat. No. 18080-400, Invitrogen). The cDNA was used as a template for qRT-PCR with a SYBR Select Master Mix kit (Cat. No. 4472953, Applied Biosystems by Life Technologies) on an Applied Biosystems StepOnePlus Real-Time PCR System with the specific primers (see Supporting Information File 1, Table S4). The three genes of GAPDH, gyrB, rpoD were used as endogenous control. Melting curve analyses were employed to verify the specific single-product amplification.
P. aeruginosa killing assay
The OD600 of overnight cultures of PAO1, ΔrecA and ΔrecA complementation strains were measured and adjusted to 0.3 in ABTGC medium with 0, 3.125, 6.25 and 12.5 µM cisplatin, respectively. The strains were incubated at 37 °C with shaking at 200 rpm for 4 h. The cultures were then harvested, serially diluted and plated on LB agar plates for incubation at 37 °C overnight. Colonies on the plate were enumerated and colony forming units (CFU) mL−1 were tabulated as follows: CFU mL−1 = average number of colonies X dilution factor X volume used to spread on LB agar plate. Experiments were conducted in triplicate, and the results are shown as the mean ± s.d.
P. aeruginosa biofilm killing assay by cisplatin and tobramycin
Biofilms were grown in 24-well plates (Nunc, Denmark) at 37 °C, as previously described [21]. Biofilms were washed 3 times with 0.9% NaCl and treated with ABTGC medium with 0, 3.125, 6.25 and 12.5 μM cisplatin, respectively. For tobramycin treatment, biofilms were treated with ABTGC medium with 5.3, 10.6 and 21.2 μM tobramycin, respectively. The treated biofilms were incubated at 37 °C for 4 h. The biofilms were harvested by scraping with a cell scraper, homogenized in 1 mL 0.9% NaCl, serially diluted and plated on LB agar plates for incubation at 37 °C overnight. Colonies on the plate were counted and CFU mL−1 was tabulated. Experiments were conducted in triplicate, and results are shown as the mean ± s.d.
RAW264.7 macrophage cytotoxicity assay
The murine macrophages (RAW264.7) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies), supplemented with 10% fetal bovine serum (FBS) (Gibco), in 75 cm2 cell culture flasks at a density of 1.0 × 106 cells mL−1 at 37 °C, 5% CO2 for 72 h. The 5.0 × 106 macrophages per well were seeded in 24-well plate (Nunc, Denmark) and grown at 37 °C, 5% CO2 overnight. As previously described [12], macrophages were washed once with PBS and treated with PAO1 in DMEM medium with 0, 3.125, 6.25 and 12.5 µM cisplatin at a multiplicity of infection of 100 bacteria cells: 1 macrophage. As control, the macrophages were treated with the T3SS deficient ΔpscJ mutant in DMEM medium. The co-culture was incubated at 37 °C, 5% CO2 for 2 h. The extracellular bacterial cells in DMEM were removed and the infected macrophages were washed 3 times with PBS. Fresh DMEM was added to the infected macrophages and a further incubation of the macrophages at 37 °C, 5% CO2 for 4 h was ensued. A solution containing 20 µM propidium iodide (PI) was added to the macrophages to stain for dead macrophages killed by PAO1 treated with cisplatin or ΔpscJ. Live and dead macrophages were imaged by fluorescence microscopy (Zeiss, Germany) at 200× and tabulated under % of dead macrophages. Experiments were conducted in triplicate, and the results are shown as the mean ± s.d.
Rabbit corneal wound healing model
New Zealand White Rabbits (n = 4, weighing 2 to 3 kg) purchased from National University of Singapore, were used for this study. All animal experiments were conducted in compliance with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research, the guide for the Care and Use of laboratory animals (National Research Council) and under the supervision of Singhealth Experimental Medical Centre (SEMC).
Four rabbits were randomly grouped into two groups of two rabbits each, comprising of the PBS control treated group and 0.75 mg mL–1 cisplatin treated group. Intra peritoneal injection of 1 mL of ketamine (100 mg mL−1) and 0.5 mL of xylazil (20 mg mL–1) had been used to anesthetize the rabbits. Corneas were then anesthetized by topical administration of xylocaine 1%. A corneal wound was made by using a 5 mm trephine and mechanical removal of epithelial cells was carried out by sterile mini blade (BD-Beaver) leaving the basal lamina intact [22,23]. All the groups were treated by topical administration of the respective drug 3 times per day. Cornea wound was visualized by the aid of cobalt-blue filter equipped slit lamp biomicroscopy (New-generation Zoom clinical Slit Lamp, NS-2D, Righton), staining with Minims fluorescein sodium eye drops (Bausch and Lomb, 2% w/v) which is used in ophthalmology clinic for disclosure of wound on the ocular surface [24,25]. Residual wound area (pixel square) was measured during the wound healing process by Image-J 1.44o version. Mann-Whitney U Test was employed to determine if a difference in re-epithelialization existed among the different groups by using GraphPad Prism 6.02. A probability value of p ≤ 0.05 was considered statistically significant.
Murine model for corneal infection
The corneas of the C57BL/6 mice were scratched with a scraper to create wounds. 10 µL of PAO1 planktonic cells (≈1 × 106 cells) were dripped onto each cornea and incubated for 24 h on the mice to allow the biofilms to form on the scratched corneas as previously described [26,27]. About 10 µL of cisplatin (final concentration 25 µM) or 0.9% NaCl as control were dripped onto the cornea 3 times per day at 4 h interval on the second day. The mice were kept for 72 h and then sacrificed.
The corneas were harvested, and the biofilm was disrupted from the corneas by crushing with mortar and pestle, followed by vortexing with glass beads for 15 min. The homogenized biofilm cells were serially diluted, plated on LB agar plates and incubated at 37 °C overnight. The number of colonies was counted, and CFU ml−1 was tabulated. Experiments were performed in triplicate, and the results are shown as the mean ± s.d.
Results and Discussion
Cisplatin inhibits P. aeruginosa planktonic growth
During our screening of in-house collection of FDA-approved drugs against P. aeruginosa growth, we identified that cisplatin had a minimal inhibitory concentration (MIC) of 6.25 μM against the P. aeruginosa PAO1 lab strain (Figure 1). We further tested the growth inhibitory effect of cisplatin against another P. aeruginosa lab strain PA14 and a P. aeruginosa mucoid multiple-drug resistant (MDR) CF clinical isolate 57388A and found that cisplatin had equivalent MIC at 6.25 μM against these two strains (Supporting Information File 1, Table S3). We next evaluated several other Pt(II)-based compounds for their growth inhibitory effects on P. aeruginosa, but these tested compounds had higher MIC against P. aeruginosa as compared to cisplatin (Figure 1).
![[1860-5397-14-284-1]](/bjoc/content/figures/1860-5397-14-284-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures and MICs of Pt-based compounds against P. aeruginosa PAO1.
Figure 1: Structures and MICs of Pt-based compounds against P. aeruginosa PAO1.
Mode of action
The growth inhibitory effects of cisplatin on both eukaryotic cells and microbial cells are attributed to the interactions of Pt(II) in cisplatin with DNA [28-30]. To reveal the growth arresting mechanisms and overall impact of cisplatin on the physiology of P. aeruginosa, we performed an RNA-sequencing (RNA-seq) based transcriptomic analysis on P. aeruginosa PAO1 after cultivation in sub-lethal concentration (1.5 µM) of cisplatin for 8 hours and compared the transcriptome with control transcriptomes of bacteria present in cisplatin-free medium. Using a negative binomial test with a BH adjusted P-value cut-off of 0.05 and a fold-change cut-off of 2, we found that sub-MIC cisplatin treatment induced the expression of 315 genes (Supporting Information File 1, Table S1) while repressed the expression of 72 genes (Supporting Information File 1, Table S2) in P. aeruginosa. The heat-map and function enrichment of the genes that were differently expressed between cisplatin-treated and control P. aeruginosa samples were illustrated in Figure 2 and Figure 3, respectively.
![[1860-5397-14-284-2]](/bjoc/content/figures/1860-5397-14-284-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Transcriptomic analysis of control and cisplatin-treated PAO1 cultures. Heatmap comparing the transcriptomes of control and cisplatin-treated PAO1 cultures.
Figure 2: Transcriptomic analysis of control and cisplatin-treated PAO1 cultures. Heatmap comparing the trans...
![[1860-5397-14-284-3]](/bjoc/content/figures/1860-5397-14-284-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Function enrichment of differentially expressed genes from the transcriptomic analysis. A dot-lot figure was generated using ggplot2 the package of R. Red circle highlights the genes involved in DNA replication and repair; blue circle highlights the genes involved in pyocin synthesis; green circle highlights the genes involved in protein secretion (T3SS).
Figure 3: Function enrichment of differentially expressed genes from the transcriptomic analysis. A dot-lot f...
The cisplatin treatment triggered the expression of a large fraction of the LexA-controlled SOS regulon [31], including genes involved in DNA replication, recombination, modification and repair (dnaE2, imuB, imuA, dinG, recA, recN, recX) and genes involved in pyocin synthesis (PA0614-PA0648), whose expression were previously reported to be induced by ciprofloxacin [31] and hydrogen peroxide treatments [32]. In addition, cisplatin treatment induced the expression of a series of genes involved in energy metabolism, which corroborated with previous proteomics work showing that cisplatin could interfere with stress response and energy metabolism in E. coli [33].
To further validate the impact of cisplatin on DNA replication, we compared the cisplatin sensitivity of the P. aeruginosa wild-type PAO1 strain and its DNA recombination-deficient recA mutant and found that the rec recombination pathway was essential for the cisplatin resistance in P. aeruginosa (Figure 4). Together with the transcriptome profiling, this result confirmed that cisplatin was able to interact with the P. aeruginosa DNA, resulting in up-regulation of stress response genes. This mechanism was also similar to the mechanism of action by another DNA crosslinker, mitomycin C which kills bacterial persister cells [34].
![[1860-5397-14-284-4]](/bjoc/content/figures/1860-5397-14-284-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Cisplatin fast-kill assay against the P. aeruginosa PAO1, ΔrecA mutant and the ΔrecACOM strain. P. aeruginosa strains were treated by ABTGC medium with varies concentrations of cisplatin for 4 h. CFUs were determined for cisplatin-treated P. aeruginosa cultures. Means and s.d. from triplicate experiments are shown.
Figure 4: Cisplatin fast-kill assay against the P. aeruginosa PAO1, ΔrecA mutant and the ΔrecACOM strain. P. ...
Anti-T3SS effect of cisplatin
Our transcriptomic analysis also revealed the expression of a large number of the secretion related genes, including those of the type III secretion system (T3SS), which were downregulated in PAO1 by cisplatin exposure (Table S2), which was similar to that by ciprofloxacin exposure [31]. Our qRT-PCR analysis confirmed that the expression of two selected T3SS genes, exoS and pscG, were downregulated by cisplatin treatment compared to control (Figure 5a). The downregulation of T3SS by the LexA-controlled SOS response [35] could be attributed to the induced expression of ptrB, a repressor of T3SS by cisplatin treatment (Supporting Information File 1, Table S3).
![[1860-5397-14-284-5]](/bjoc/content/figures/1860-5397-14-284-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Cisplatin treatment represses T3SS associated virulence. (A) Cisplatin treatment downregulated the expression of T3SS gene revealed by qRT-PCR analysis. Means and s.d. from triplicate experiments are shown. Student’s t-test was performed for testing differences between groups. *P ≤ 0.05. (B) Cisplatin treatment reduced cytotoxicity of P. aeruginosa PAO1 against mouse macrophage cells. Means and s.d. from triplicate experiments are shown. Student’s t-test was performed for testing differences between groups. *P ≤ 0.05.
Figure 5: Cisplatin treatment represses T3SS associated virulence. (A) Cisplatin treatment downregulated the ...
The T3SS is one of the major virulence mechanisms employed by P. aeruginosa and other microbial pathogens to impair the host immune systems during infection [36,37]. T3SS activity of P. aeruginosa was correlated with acute cytotoxicity to host epithelial cells and immune cells such as macrophages and neutrophils [38]. As we demonstrated that cisplatin treatment was able to reduce the T3SS of P. aeruginosa, we further tested the ability of cisplatin in attenuating the acute cytotoxicity of P. aeruginosa to macrophages. Cisplatin treatment of P. aeruginosa in the P. aeruginosa-macrophage co-cultures caused significant less death of the mouse macrophages compared to control samples (Figure 5b), suggesting the effectiveness of cisplatin against P. aeruginosa infection.
Antibiofilm effect of cisplatin
P. aeruginosa is notorious for its biofilm formation capacity, which might lead to persistent or recalcitrant infections. SOS response and DNA recombination are required for development of P. aeruginosa biofilm resistance [39-41]. Given cisplatin treatment was able to interfere with DNA repair, we hypothesized that cisplatin treatment could eradicate P. aeruginosa biofilm cells. We compared the biofilm killing effects of cisplatin and tobramycin at various concentrations. The MIC of cisplatin and tobramycin against planktonic P. aeruginosa cells were 6.25 µM and 2.65 µM, respectively. However, tobramycin could not kill the biofilm cells at 2 × MIC due to its limitation in biofilm penetration [42], while cisplatin was able to kill substantial amount of biofilm cells with nearly 100 times reduction of P. aeruginosa biofilm cells (Figure 6). This result suggested that cisplatin might penetrate the biofilms better than the otherwise eDNA trapped tobramycin to kill the P. aeruginosa cells [42]. The 4 × MIC and 8 × MIC of tobramycin treatment showed dose-dependent increase of biofilm killing capacity (Figure 6). Interestingly, cisplatin had combinatory effects with tobramycin in killing the PAO1 biofilms, as combinatorial treatment of 2 × MIC of cisplatin with 4 × MIC or 8 × MIC tobramycin killed the biofilm cells at a higher rate compared to the mono-compound treatment (Figure 6). These results suggest that combination of cisplatin and other conventional antimicrobials could be a useful strategy for eradicating persistent biofilm-associated infections.
![[1860-5397-14-284-6]](/bjoc/content/figures/1860-5397-14-284-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: P. aeruginosa biofilm killing assay by cisplatin, tobramycin and their combinations. P. aeruginosa biofilms were treated by ABTGC medium with varies concentrations of cisplatin and or tobramycin for 4 h. CFUs were determined for cisplatin and or tobramycin-treated P. aeruginosa biofilms. Means and s.d. from triplicate experiments are shown. Student’s t-test was performed for testing differences between groups. *P ≤ 0.05.
Figure 6: P. aeruginosa biofilm killing assay by cisplatin, tobramycin and their combinations. P. aeruginosa ...
Cisplatin treatment attenuates P. aeruginosa infection
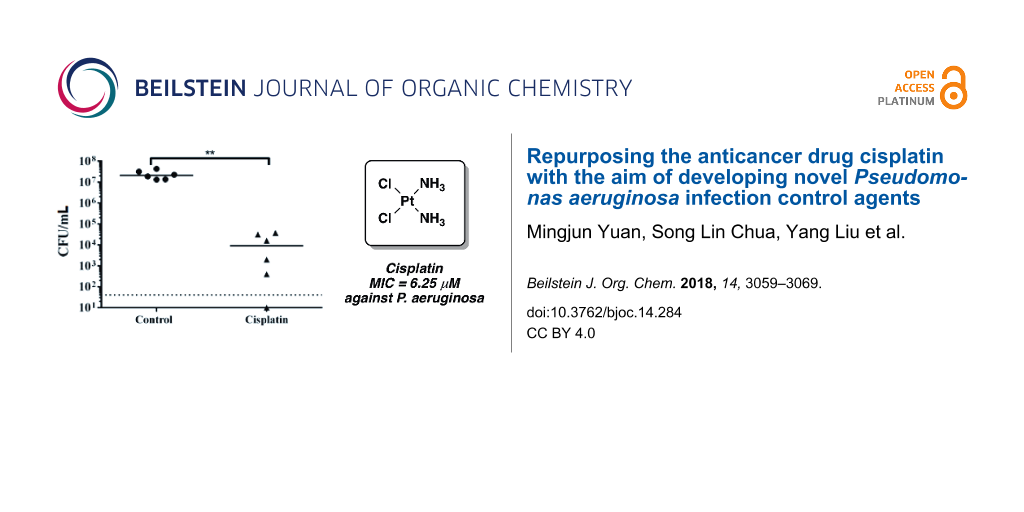
As cisplatin could reduce the synthesis of T3SS-mediated virulent products and kill biofilms of P. aeruginosa, we further tested if cisplatin treatment was able to eradicate in vivo P. aeruginosa infections using a mouse model of keratitis, where P. aeruginosa cells have biofilm-like morphology [26,27] and employ type III secretion during infections [43]. We firstly confirmed that cisplatin was not toxic and did not interfere with wound healing, with no observable inflammation or adverse effects, when applied topically on scratched corneas with no bacterial infection (Supporting Information File 1, Figure S1). We then allowed P. aeruginosa PAO1 to colonize and establish infection in the scratched corneas of mice for 24 h. 10 µL of 1 × MIC (6.25 µM) of cisplatin and control 0.9% NaCl were dripped at the site of P. aeruginosa infection 3 times (4 hour interval) on the second day. The mice were sacrificed on the third day and their corneas were harvested for CFU count. Cisplatin showed efficient killing capacity on P. aeruginosa cells from infected mouse corneas and there was a significant reduction in the bacterial loads from the cisplatin treated corneas as compared to the control corneas (Figure 7).
![[1860-5397-14-284-7]](/bjoc/content/figures/1860-5397-14-284-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Cisplatin treatment attenuates P. aeruginosa infections. CFU mL−1 of PAO1 obtained from corneas with and without cisplatin treatment. Dotted horizontal lines represent limit of detection. The mean and s.d. from six experiments were shown for in vivo biofilms. Student’s t-test was performed for testing differences between groups. *P < 0.01.
Figure 7: Cisplatin treatment attenuates P. aeruginosa infections. CFU mL−1 of PAO1 obtained from corneas wit...
Conclusion
Here, we have demonstrated how cisplatin displays antivirulence and antibiofilm effects against the opportunistic pathogen P. aeruginosa. Since biofilms are notoriously difficult to be cleared by conventional antibiotics, cisplatin possesses the additional advantage of killing biofilms. This makes cisplatin a more attractive antimicrobial for treating biofilm infections clinically. Even though cisplatin is known for its toxic side effects on cancer patients when administered intravenously, we showed indications that cisplatin could be applied topically to infection sites with low toxicity and minimal negative impact on wound repair. Transcriptomic analysis revealed that the working mechanism of cisplatin towards P. aeruginosa is rather unique and distinct from other conventional antibiotics, which may offer alternative therapeutic approaches towards persistent infections.
In recent years, metal-containing compounds have been identified as antimicrobial agents. Gallium was shown to disrupt the iron metabolism of P. aeruginosa and efficiently kill established biofilm [44]. In addition, the gold-containing drug, auranofin, was found to be a broad-spectrum bactericidal compound, that targets the thiol-redox homeostasis of a range of Gram-positive bacteria [45]. Further studies will be carried out to better understand the resistance mechanism and structural requirements of the Pt-based compounds as an alternative to the conventional antibiotics. Such compounds could also be used synergistically with specific enzymes that degrade the biofilm matrix [46] or biofilm-dispersal agents to boost the eradication of biofilms, to provide better treatment options for chronic and persistent infections.
Supporting Information
| Supporting Information File 1: Additional information. | ||
| Format: PDF | Size: 703.8 KB | Download |
Disclosure of Financial and Competing Interests
This research is supported by the National Research Foundation and Ministry of Education Singapore under its Research Centre of Excellence Programme and AcRF Tier 2 (MOE2016-T2-1-010) from Ministry of Education, Singapore. S.L. Chua is supported by the Lee Kong Chian School of Medicine (LKCMedicine) Postdoctoral Fellowship 2015. The authors declare no other conflict of interest. No writing assistance was utilized in the writing of the manuscript.
Ethical Conduct of Research
All animal experiments were conducted in compliance with the guide for the Care and Use of laboratory animals (National Research Council) under Nanyang Technological University Institutional Animal Care and Use Committee (IACUC) protocol number ARF SBS/NIE-A0192, which was approved by the Nanyang Technological University-IACUC.
References
-
Lyczak, J. B.; Cannon, C. L.; Pier, G. B. Microbes Infect. 2000, 2, 1051–1060. doi:10.1016/s1286-4579(00)01259-4
Return to citation in text: [1] -
Govan, J. R.; Deretic, V. Microbiol. Rev. 1996, 60, 539–574.
Return to citation in text: [1] -
Markou, P.; Apidianakis, Y. Front. Cell. Infect. Microbiol. 2014, 3, 115. doi:10.3389/fcimb.2013.00115
Return to citation in text: [1] -
Rolston, K. V. I.; Bodey, G. P. Cancer Invest. 1992, 10, 43–59. doi:10.3109/07357909209032787
Return to citation in text: [1] -
Yang, L.; Liu, Y.; Wu, H.; Song, Z.; Høiby, N.; Molin, S.; Givskov, M. FEMS Immunol. Med. Microbiol. 2012, 65, 146–157. doi:10.1111/j.1574-695x.2011.00858.x
Return to citation in text: [1] -
Zavascki, A. P.; Carvalhaes, C. G.; Picão, R. C.; Gales, A. C. Expert Rev. Anti-Infect. Ther. 2010, 8, 71–93. doi:10.1586/eri.09.108
Return to citation in text: [1] -
Sanchez, C. J., Jr.; Mende, K.; Beckius, M. L.; Akers, K. S.; Romano, D. R.; Wenke, J. C.; Murray, C. K. BMC Infect. Dis. 2013, 13, 47. doi:10.1186/1471-2334-13-47
Return to citation in text: [1] -
Roy-Burman, A.; Savel, R. H.; Racine, S.; Swanson, B. L.; Revadigar, N. S.; Fujimoto, J.; Sawa, T.; Frank, D. W.; Wiener-Kronish, J. P. J. Infect. Dis. 2001, 183, 1767–1774. doi:10.1086/320737
Return to citation in text: [1] -
Hauser, A. R.; Cobb, E.; Bodí, M.; Mariscal, D.; Vallés, J.; Engel, J. N.; Rello, J. Crit. Care Med. 2002, 30, 521–528. doi:10.1097/00003246-200203000-00005
Return to citation in text: [1] -
Joyce, K.; Saxena, S.; Williams, A.; Damurjian, C.; Auricchio, N.; Aluotto, S.; Tynan, H.; Demain, A. L. J. Antibiot. 2010, 63, 530–532. doi:10.1038/ja.2010.64
Return to citation in text: [1] -
Chowdhury, N.; Wood, T. L.; Martínez-Vázquez, M.; García-Contreras, R.; Wood, T. K. Biotechnol. Bioeng. 2016, 113, 1984–1992. doi:10.1002/bit.25963
Return to citation in text: [1] -
Chua, S. L.; Liu, Y.; Yam, J. K. H.; Chen, Y.; Vejborg, R. M.; Tan, B. G. C.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Nat. Commun. 2014, 5, 4462. doi:10.1038/ncomms5462
Return to citation in text: [1] [2] [3] -
Rajput, J.; Moss, J. R.; Hutton, A. T.; Hendricks, D. T.; Arendse, C. E.; Imrie, C. J. Organomet. Chem. 2004, 689, 1553–1568. doi:10.1016/j.jorganchem.2004.01.034
Return to citation in text: [1] -
Griffith, D.; Bergamo, A.; Pin, S.; Vadori, M.; Müller-Bunz, H.; Sava, G.; Marmion, C. J. Polyhedron 2007, 26, 4697–4706. doi:10.1016/j.poly.2007.03.011
Return to citation in text: [1] -
Ramos-Lima, F. J.; Quiroga, A. G.; Pérez, J. M.; Font-Bardía, M.; Solans, X.; Navarro-Ranninger, C. Eur. J. Inorg. Chem. 2003, 1591–1598. doi:10.1002/ejic.200390209
Return to citation in text: [1] -
Wiegand, I.; Hilpert, K.; Hancock, R. E. W. Nat. Protoc. 2008, 3, 163–175. doi:10.1038/nprot.2007.521
Return to citation in text: [1] -
Tan, S. Y.-Y.; Liu, Y.; Chua, S. L.; Vejborg, R. M.; Jakobsen, T. H.; Chew, S. C.; Li, Y.; Nielsen, T. E.; Tolker-Nielsen, T.; Yang, L.; Givskov, M. Antimicrob. Agents Chemother. 2014, 58, 6648–6659. doi:10.1128/aac.02620-13
Return to citation in text: [1] -
Anders, S.; Huber, W. Genome Biol. 2010, 11, R106. doi:10.1186/gb-2010-11-10-r106
Return to citation in text: [1] -
Gentleman, R. C.; Carey, V. J.; Bates, D. M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A. J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J. Y.; Zhang, J. Genome Biol. 2004, 5, R80. doi:10.1186/gb-2004-5-10-r80
Return to citation in text: [1] [2] -
Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company, Inc.: New York City, NY, U.S.A., 2009; p 216. doi:10.1007/978-0-387-98141-3
Return to citation in text: [1] -
Chua, S. L.; Hultqvist, L. D.; Yuan, M.; Rybtke, M.; Nielsen, T. E.; Givskov, M.; Tolker-Nielsen, T.; Yang, L. Nat. Protoc. 2015, 10, 1165–1180. doi:10.1038/nprot.2015.067
Return to citation in text: [1] -
Koh, J.-J.; Lin, S.; Aung, T. T.; Lim, F.; Zou, H.; Bai, Y.; Li, J.; Lin, H.; Pang, L. M.; Koh, W. L.; Salleh, S. M.; Lakshminarayanan, R.; Zhou, L.; Qiu, S.; Pervushin, K.; Verma, C.; Tan, D. T. H.; Cao, D.; Liu, S.; Beuerman, R. W. J. Med. Chem. 2015, 58, 739–752. doi:10.1021/jm501285x
Return to citation in text: [1] -
Aung, T. T.; Yam, J. K. H.; Lin, S.; Salleh, S. M.; Givskov, M.; Liu, S.; Lwin, N. C.; Yang, L.; Beuerman, R. W. Antimicrob. Agents Chemother. 2016, 60, 24–35. doi:10.1128/aac.01509-15
Return to citation in text: [1] -
Reidy, J. J.; Zarzour, J.; Thompson, H. W.; Beuerman, R. W. Br. J. Ophthalmol. 1994, 78, 377–380. doi:10.1136/bjo.78.5.377
Return to citation in text: [1] -
Brazzell, R. K. S. M.; Aquavella, J. V.; Beuerman, R. W.; Baird, L. Invest. Ophthalmol. Visual Sci. 1991, 32, 336–340.
Return to citation in text: [1] -
Saraswathi, P.; Beuerman, R. W. The ocular surface; 2015.
Return to citation in text: [1] [2] -
Yam, J. K. H.; Aung, T. T.; Chua, S. L.; Cheng, Y.; Kohli, G. S.; Zhou, J.; Constancias, F.; Liu, Y.; Cai, Z.; Salido, M. M. S.; Drautz-Moses, D. I.; Rice, S. A.; Schuster, S. C.; Boo, Z. Z.; Wu, B.; Kjelleberg, S.; Tolker-Nielsen, T.; Beuerman, R. W.; Givskov, M.; Yang, L. Environ. Microbiol. 2018, in press.
Return to citation in text: [1] [2] -
Fichtinger-Schepman, A. M.; van Oosterom, A. T.; Lohman, P. H.; Berends, F. Cancer Res. 1987, 47, 3000–3004.
Return to citation in text: [1] -
Onoa, G. B.; Cervantes, G.; Moreno, V.; Prieto, M. J. Nucleic Acids Res. 1998, 26, 1473–1480. doi:10.1093/nar/26.6.1473
Return to citation in text: [1] -
Siddik, Z. H. Oncogene 2003, 22, 7265–7279. doi:10.1038/sj.onc.1206933
Return to citation in text: [1] -
Cirz, R. T.; O'Neill, B. M.; Hammond, J. A.; Head, S. R.; Romesberg, F. E. J. Bacteriol. 2006, 188, 7101–7110. doi:10.1128/jb.00807-06
Return to citation in text: [1] [2] [3] -
Chang, W.; Small, D. A.; Toghrol, F.; Bentley, W. E. BMC Genomics 2005, 6, 115. doi:10.1186/1471-2164-6-115
Return to citation in text: [1] -
Stefanopoulou, M.; Kokoschka, M.; Sheldrick, W. S.; Wolters, D. A. Proteomics 2011, 11, 4174–4188. doi:10.1002/pmic.201100203
Return to citation in text: [1] -
Kwan, B. W.; Chowdhury, N.; Wood, T. K. Environ. Microbiol. 2015, 17, 4406–4414. doi:10.1111/1462-2920.12873
Return to citation in text: [1] -
Wu, W.; Jin, S. J. Bacteriol. 2005, 187, 6058–6068. doi:10.1128/jb.187.17.6058-6068.2005
Return to citation in text: [1] -
Hauser, A. R. Nat. Rev. Microbiol. 2009, 7, 654–665. doi:10.1038/nrmicro2199
Return to citation in text: [1] -
Coburn, B.; Sekirov, I.; Finlay, B. B. Clin. Microbiol. Rev. 2007, 20, 535–549. doi:10.1128/cmr.00013-07
Return to citation in text: [1] -
Finck-Barbançon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J. P.; Fleiszig, S. M. J.; Wu, C.; Mende-Mueller, L.; Frank, D. W. Mol. Microbiol. 1997, 25, 547–557. doi:10.1046/j.1365-2958.1997.4891851.x
Return to citation in text: [1] -
Poole, K. Trends Microbiol. 2012, 20, 227–234. doi:10.1016/j.tim.2012.02.004
Return to citation in text: [1] -
Stewart, P. S.; Franklin, M. J.; Williamson, K. S.; Folsom, J. P.; Boegli, L.; James, G. A. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. doi:10.1128/aac.00433-15
Return to citation in text: [1] -
Boles, B. R.; Thoendel, M.; Singh, P. K. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16630–16635. doi:10.1073/pnas.0407460101
Return to citation in text: [1] -
Chiang, W.-C.; Nilsson, M.; Jensen, P. Ø.; Høiby, N.; Nielsen, T. E.; Givskov, M.; Tolker-Nielsen, T. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. doi:10.1128/aac.00001-13
Return to citation in text: [1] [2] -
Zolfaghar, I.; Evans, D. J.; Ronaghi, R.; Fleiszig, S. M. J. Infect. Immun. 2006, 74, 3880–3889. doi:10.1128/iai.01891-05
Return to citation in text: [1] -
Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B. E.; Singh, P. K. J. Clin. Invest. 2007, 117, 877–888. doi:10.1172/jci30783
Return to citation in text: [1] -
Harbut, M. B.; Vilchèze, C.; Luo, X.; Hensler, M. E.; Guo, H.; Yang, B.; Chatterjee, A. K.; Nizet, V.; Jacobs, W. R., Jr.; Schultz, P. G.; Wang, F. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4453–4458. doi:10.1073/pnas.1504022112
Return to citation in text: [1] -
Yu, S.; Su, T.; Wu, H.; Liu, S.; Wang, D.; Zhao, T.; Jin, Z.; Du, W.; Zhu, M.-J.; Chua, S. L.; Yang, L.; Zhu, D.; Gu, L.; Ma, L. Z. Cell Res. 2015, 25, 1352–1367. doi:10.1038/cr.2015.129
Return to citation in text: [1]
| 31. | Cirz, R. T.; O'Neill, B. M.; Hammond, J. A.; Head, S. R.; Romesberg, F. E. J. Bacteriol. 2006, 188, 7101–7110. doi:10.1128/jb.00807-06 |
| 32. | Chang, W.; Small, D. A.; Toghrol, F.; Bentley, W. E. BMC Genomics 2005, 6, 115. doi:10.1186/1471-2164-6-115 |
| 33. | Stefanopoulou, M.; Kokoschka, M.; Sheldrick, W. S.; Wolters, D. A. Proteomics 2011, 11, 4174–4188. doi:10.1002/pmic.201100203 |
| 1. | Lyczak, J. B.; Cannon, C. L.; Pier, G. B. Microbes Infect. 2000, 2, 1051–1060. doi:10.1016/s1286-4579(00)01259-4 |
| 6. | Zavascki, A. P.; Carvalhaes, C. G.; Picão, R. C.; Gales, A. C. Expert Rev. Anti-Infect. Ther. 2010, 8, 71–93. doi:10.1586/eri.09.108 |
| 17. | Tan, S. Y.-Y.; Liu, Y.; Chua, S. L.; Vejborg, R. M.; Jakobsen, T. H.; Chew, S. C.; Li, Y.; Nielsen, T. E.; Tolker-Nielsen, T.; Yang, L.; Givskov, M. Antimicrob. Agents Chemother. 2014, 58, 6648–6659. doi:10.1128/aac.02620-13 |
| 42. | Chiang, W.-C.; Nilsson, M.; Jensen, P. Ø.; Høiby, N.; Nielsen, T. E.; Givskov, M.; Tolker-Nielsen, T. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. doi:10.1128/aac.00001-13 |
| 5. | Yang, L.; Liu, Y.; Wu, H.; Song, Z.; Høiby, N.; Molin, S.; Givskov, M. FEMS Immunol. Med. Microbiol. 2012, 65, 146–157. doi:10.1111/j.1574-695x.2011.00858.x |
| 12. | Chua, S. L.; Liu, Y.; Yam, J. K. H.; Chen, Y.; Vejborg, R. M.; Tan, B. G. C.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Nat. Commun. 2014, 5, 4462. doi:10.1038/ncomms5462 |
| 42. | Chiang, W.-C.; Nilsson, M.; Jensen, P. Ø.; Høiby, N.; Nielsen, T. E.; Givskov, M.; Tolker-Nielsen, T. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. doi:10.1128/aac.00001-13 |
| 3. | Markou, P.; Apidianakis, Y. Front. Cell. Infect. Microbiol. 2014, 3, 115. doi:10.3389/fcimb.2013.00115 |
| 4. | Rolston, K. V. I.; Bodey, G. P. Cancer Invest. 1992, 10, 43–59. doi:10.3109/07357909209032787 |
| 15. | Ramos-Lima, F. J.; Quiroga, A. G.; Pérez, J. M.; Font-Bardía, M.; Solans, X.; Navarro-Ranninger, C. Eur. J. Inorg. Chem. 2003, 1591–1598. doi:10.1002/ejic.200390209 |
| 38. | Finck-Barbançon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J. P.; Fleiszig, S. M. J.; Wu, C.; Mende-Mueller, L.; Frank, D. W. Mol. Microbiol. 1997, 25, 547–557. doi:10.1046/j.1365-2958.1997.4891851.x |
| 16. | Wiegand, I.; Hilpert, K.; Hancock, R. E. W. Nat. Protoc. 2008, 3, 163–175. doi:10.1038/nprot.2007.521 |
| 39. | Poole, K. Trends Microbiol. 2012, 20, 227–234. doi:10.1016/j.tim.2012.02.004 |
| 40. | Stewart, P. S.; Franklin, M. J.; Williamson, K. S.; Folsom, J. P.; Boegli, L.; James, G. A. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. doi:10.1128/aac.00433-15 |
| 41. | Boles, B. R.; Thoendel, M.; Singh, P. K. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16630–16635. doi:10.1073/pnas.0407460101 |
| 10. | Joyce, K.; Saxena, S.; Williams, A.; Damurjian, C.; Auricchio, N.; Aluotto, S.; Tynan, H.; Demain, A. L. J. Antibiot. 2010, 63, 530–532. doi:10.1038/ja.2010.64 |
| 12. | Chua, S. L.; Liu, Y.; Yam, J. K. H.; Chen, Y.; Vejborg, R. M.; Tan, B. G. C.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Nat. Commun. 2014, 5, 4462. doi:10.1038/ncomms5462 |
| 35. | Wu, W.; Jin, S. J. Bacteriol. 2005, 187, 6058–6068. doi:10.1128/jb.187.17.6058-6068.2005 |
| 9. | Hauser, A. R.; Cobb, E.; Bodí, M.; Mariscal, D.; Vallés, J.; Engel, J. N.; Rello, J. Crit. Care Med. 2002, 30, 521–528. doi:10.1097/00003246-200203000-00005 |
| 13. | Rajput, J.; Moss, J. R.; Hutton, A. T.; Hendricks, D. T.; Arendse, C. E.; Imrie, C. J. Organomet. Chem. 2004, 689, 1553–1568. doi:10.1016/j.jorganchem.2004.01.034 |
| 14. | Griffith, D.; Bergamo, A.; Pin, S.; Vadori, M.; Müller-Bunz, H.; Sava, G.; Marmion, C. J. Polyhedron 2007, 26, 4697–4706. doi:10.1016/j.poly.2007.03.011 |
| 36. | Hauser, A. R. Nat. Rev. Microbiol. 2009, 7, 654–665. doi:10.1038/nrmicro2199 |
| 37. | Coburn, B.; Sekirov, I.; Finlay, B. B. Clin. Microbiol. Rev. 2007, 20, 535–549. doi:10.1128/cmr.00013-07 |
| 8. | Roy-Burman, A.; Savel, R. H.; Racine, S.; Swanson, B. L.; Revadigar, N. S.; Fujimoto, J.; Sawa, T.; Frank, D. W.; Wiener-Kronish, J. P. J. Infect. Dis. 2001, 183, 1767–1774. doi:10.1086/320737 |
| 34. | Kwan, B. W.; Chowdhury, N.; Wood, T. K. Environ. Microbiol. 2015, 17, 4406–4414. doi:10.1111/1462-2920.12873 |
| 7. | Sanchez, C. J., Jr.; Mende, K.; Beckius, M. L.; Akers, K. S.; Romano, D. R.; Wenke, J. C.; Murray, C. K. BMC Infect. Dis. 2013, 13, 47. doi:10.1186/1471-2334-13-47 |
| 11. | Chowdhury, N.; Wood, T. L.; Martínez-Vázquez, M.; García-Contreras, R.; Wood, T. K. Biotechnol. Bioeng. 2016, 113, 1984–1992. doi:10.1002/bit.25963 |
| 31. | Cirz, R. T.; O'Neill, B. M.; Hammond, J. A.; Head, S. R.; Romesberg, F. E. J. Bacteriol. 2006, 188, 7101–7110. doi:10.1128/jb.00807-06 |
| 19. | Gentleman, R. C.; Carey, V. J.; Bates, D. M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A. J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J. Y.; Zhang, J. Genome Biol. 2004, 5, R80. doi:10.1186/gb-2004-5-10-r80 |
| 18. | Anders, S.; Huber, W. Genome Biol. 2010, 11, R106. doi:10.1186/gb-2010-11-10-r106 |
| 26. | Saraswathi, P.; Beuerman, R. W. The ocular surface; 2015. |
| 27. | Yam, J. K. H.; Aung, T. T.; Chua, S. L.; Cheng, Y.; Kohli, G. S.; Zhou, J.; Constancias, F.; Liu, Y.; Cai, Z.; Salido, M. M. S.; Drautz-Moses, D. I.; Rice, S. A.; Schuster, S. C.; Boo, Z. Z.; Wu, B.; Kjelleberg, S.; Tolker-Nielsen, T.; Beuerman, R. W.; Givskov, M.; Yang, L. Environ. Microbiol. 2018, in press. |
| 19. | Gentleman, R. C.; Carey, V. J.; Bates, D. M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A. J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J. Y.; Zhang, J. Genome Biol. 2004, 5, R80. doi:10.1186/gb-2004-5-10-r80 |
| 43. | Zolfaghar, I.; Evans, D. J.; Ronaghi, R.; Fleiszig, S. M. J. Infect. Immun. 2006, 74, 3880–3889. doi:10.1128/iai.01891-05 |
| 44. | Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B. E.; Singh, P. K. J. Clin. Invest. 2007, 117, 877–888. doi:10.1172/jci30783 |
| 28. | Fichtinger-Schepman, A. M.; van Oosterom, A. T.; Lohman, P. H.; Berends, F. Cancer Res. 1987, 47, 3000–3004. |
| 29. | Onoa, G. B.; Cervantes, G.; Moreno, V.; Prieto, M. J. Nucleic Acids Res. 1998, 26, 1473–1480. doi:10.1093/nar/26.6.1473 |
| 30. | Siddik, Z. H. Oncogene 2003, 22, 7265–7279. doi:10.1038/sj.onc.1206933 |
| 31. | Cirz, R. T.; O'Neill, B. M.; Hammond, J. A.; Head, S. R.; Romesberg, F. E. J. Bacteriol. 2006, 188, 7101–7110. doi:10.1128/jb.00807-06 |
| 24. | Reidy, J. J.; Zarzour, J.; Thompson, H. W.; Beuerman, R. W. Br. J. Ophthalmol. 1994, 78, 377–380. doi:10.1136/bjo.78.5.377 |
| 25. | Brazzell, R. K. S. M.; Aquavella, J. V.; Beuerman, R. W.; Baird, L. Invest. Ophthalmol. Visual Sci. 1991, 32, 336–340. |
| 26. | Saraswathi, P.; Beuerman, R. W. The ocular surface; 2015. |
| 27. | Yam, J. K. H.; Aung, T. T.; Chua, S. L.; Cheng, Y.; Kohli, G. S.; Zhou, J.; Constancias, F.; Liu, Y.; Cai, Z.; Salido, M. M. S.; Drautz-Moses, D. I.; Rice, S. A.; Schuster, S. C.; Boo, Z. Z.; Wu, B.; Kjelleberg, S.; Tolker-Nielsen, T.; Beuerman, R. W.; Givskov, M.; Yang, L. Environ. Microbiol. 2018, in press. |
| 12. | Chua, S. L.; Liu, Y.; Yam, J. K. H.; Chen, Y.; Vejborg, R. M.; Tan, B. G. C.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Nat. Commun. 2014, 5, 4462. doi:10.1038/ncomms5462 |
| 22. | Koh, J.-J.; Lin, S.; Aung, T. T.; Lim, F.; Zou, H.; Bai, Y.; Li, J.; Lin, H.; Pang, L. M.; Koh, W. L.; Salleh, S. M.; Lakshminarayanan, R.; Zhou, L.; Qiu, S.; Pervushin, K.; Verma, C.; Tan, D. T. H.; Cao, D.; Liu, S.; Beuerman, R. W. J. Med. Chem. 2015, 58, 739–752. doi:10.1021/jm501285x |
| 23. | Aung, T. T.; Yam, J. K. H.; Lin, S.; Salleh, S. M.; Givskov, M.; Liu, S.; Lwin, N. C.; Yang, L.; Beuerman, R. W. Antimicrob. Agents Chemother. 2016, 60, 24–35. doi:10.1128/aac.01509-15 |
| 20. | Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company, Inc.: New York City, NY, U.S.A., 2009; p 216. doi:10.1007/978-0-387-98141-3 |
| 45. | Harbut, M. B.; Vilchèze, C.; Luo, X.; Hensler, M. E.; Guo, H.; Yang, B.; Chatterjee, A. K.; Nizet, V.; Jacobs, W. R., Jr.; Schultz, P. G.; Wang, F. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4453–4458. doi:10.1073/pnas.1504022112 |
| 21. | Chua, S. L.; Hultqvist, L. D.; Yuan, M.; Rybtke, M.; Nielsen, T. E.; Givskov, M.; Tolker-Nielsen, T.; Yang, L. Nat. Protoc. 2015, 10, 1165–1180. doi:10.1038/nprot.2015.067 |
| 46. | Yu, S.; Su, T.; Wu, H.; Liu, S.; Wang, D.; Zhao, T.; Jin, Z.; Du, W.; Zhu, M.-J.; Chua, S. L.; Yang, L.; Zhu, D.; Gu, L.; Ma, L. Z. Cell Res. 2015, 25, 1352–1367. doi:10.1038/cr.2015.129 |
© 2018 Yuan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)