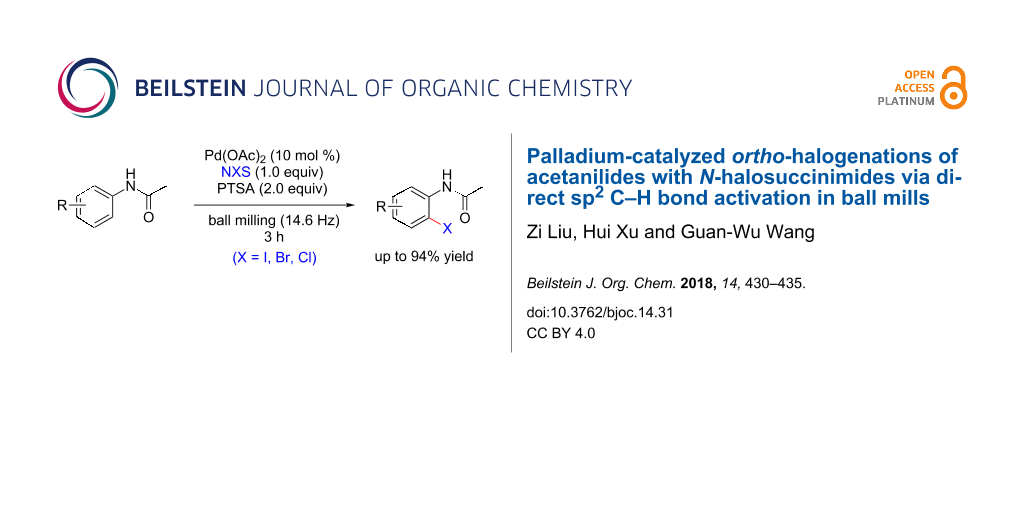
Abstract
A solvent-free palladium-catalyzed ortho-iodination of acetanilides using N-iodosuccinimide as the iodine source has been developed under ball-milling conditions. This present method avoids the use of hazardous organic solvents, high reaction temperature, and long reaction time and provides a highly efficient methodology to realize the regioselective functionalization of acetanilides in yields up to 94% in a ball mill. Furthermore, the current methodology can be extended to the synthesis of ortho-brominated and ortho-chlorinated products in good yields by using the corresponding N-halosuccinimides.

Graphical Abstract
Introduction
Aryl halides have been widely utilized in organic syntheses, which give access to a range of complex natural products [1,2]. However, traditional halogenations of aromatic compounds by direct electrophilic halogenation [3] and Sandmeyer reaction [4] have several drawbacks such as low regioselectivities, complicated reaction procedures and even a risk of danger. Thus, it is necessary to discover new approaches to the regioselective construction of C–X bonds. With the development of transition-metal-catalyzed cross-coupling reactions, a series of halogenations at the ortho-position of the directing groups have been disclosed [5-18]. Nevertheless, from the viewpoint of green chemistry, the reduction or even elimination of organic solvents, shorter reaction times, simplification of work-up procedures and improvement of product yields are highly demanding. In recent years, the application of mechanochemical techniques in organic syntheses has attracted increasing attention [19-28].
A few mechanochemical ortho-C–H bond activation reactions under the catalysis of rhodium and palladium salts have been reported [29-38]. Hernández and Bolm reported the rhodium-catalyzed bromination and iodination of 2-phenylpyridine using N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS), respectively, as the halogen source [30]. However, the mechanochemical ortho-halogenation using the cheaper palladium catalysts has not been reported yet. In continuing our interest in mechanochemistry [21,22,39-41] and C–H activation reactions [42-44], we have independently investigated the solvent-free ortho-iodination of acetanilides under ball-milling conditions [45]. In addition, the current reaction can be extended to ortho-bromination and ortho-chlorination by using the corresponding N-halosuccinimides. Herein, we report these regioselective ortho-halogenations in detail.
Results and Discussion
To begin our study, N-(p-tolyl)acetamide (1a) was chosen as the model substrate to react with NIS using Pd(OAc)2 as the catalyst to optimize reaction parameters such as additive, reaction time and reagent ratio. The reaction of 1a (0.4 mmol) with NIS (0.4 mmol) was initially performed under the catalysis of Pd(OAc)2 (10 mol %) in a Spex SamplePrep 8000 mixer mill at a frequency of 875 cycles per minute at room temperature for 3 h. Unfortunately, the desired iodinated product was not detected (Table 1, entry 1). Then, various acids were examined because the addition of acids into the reaction system could promote the C–H bond halogenation according to the previous literature [46]. As desired, compound 2a was isolated in 87% yield when p-toluenesulfonic acid (PTSA) was employed (Table 1, entry 2). A control experiment was conducted for the reaction of 1a with NIS in the absence of Pd(OAc)2, yet still with PTSA as the promoter, and no iodinated product was furnished (Table 1, entry 3). The use of D-camphorsulfonic acid (D-CSA) or mesitylenesulfonic acid dihydrate provided inferior results than that obtained in the presence of PTSA (Table 1, entries 4 and 5 vs entry 2). Furthermore, no desired product was obtained when pyridine-2-sulfonic acid, 2-nitrobenzoic acid, 2-aminoethanesulfonic acid or tungstophosphoric acid hydrate (HPA) was used in the reaction (Table 1, entries 6–9). Thus, the combination of Pd(OAc)2 with PTSA was essential for the reaction to take place effectively. Subsequently, the ratio of substrates was investigated, and the results demonstrated that the amount of both NIS and PTSA affected the product yield. Decreasing or increasing the amount of PTSA was not beneficial to the reaction (Table 1, entries 10 and 11). When the amount of NIS was increased from 1.0 equiv to 1.5 equiv and 2.0 equiv, the yield of the iodinated product did not further go up (Table 1, entries 12 and 13). The iodination was slightly less efficient for a shorter time of 2 h (Table 1, entry 14), and prolongation of the reaction time from 3 h to 4 h did not lead to a superior result (Table 1, entry 15).
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-31-i5.svg?max-width=637&scale=1.18182)
|
||||
|---|---|---|---|---|
| entry | ratio of reagentsb | additive | time (h) | yieldc (%) |
| 1 | 1:0.1:1:0 | – | 3 h | N.R. |
| 2 | 1:0.1:1:2 | PTSA | 3 h | 87 |
| 3 | 1:0:1:2 | PTSA | 3 h | N.R. |
| 4 | 1:0.1:1:2 | D-CSA | 3 h | 62 |
| 5 | 1:0.1:1:2 | mesitylenesulfonic acid dihydrate | 3 h | 56 |
| 6 | 1:0.1:1:2 | pyridine-2-sulfonic acid | 3 h | N.R. |
| 7 | 1:0.1:1:2 | 2-nitrobenzoic acid | 3 h | N.R. |
| 8 | 1:0.1:1:2 | 2-aminoethanesulfonic acid | 3 h | N.R. |
| 9 | 1:0.1:1:2 | HPA | 3 h | N.R. |
| 10 | 1:0.1:1:1.5 | PTSA | 3 h | 81 |
| 11 | 1:0.1:1:2.5 | PTSA | 3 h | 86 |
| 12 | 1:0.1:1.5:2 | PTSA | 3 h | 88 |
| 13 | 1:0.1:2:2 | PTSA | 3 h | 86 |
| 14 | 1:0.1:1:2 | PTSA | 2 h | 80 |
| 15 | 1:0.1:1:2 | PTSA | 4 h | 87 |
aUnless otherwise specified, all the reactions were carried out in a Spex SamplePrep 8000 mixer mill using 1a (0.4 mmol). bThe reagent ratio referred to 1a:Pd(OAc)2:NIS:additive. cIsolated yield. N.R. = no reaction.
To demonstrate the generality of this protocol, the regioselective iodination of a series of acetanilides was then examined in the presence of Pd(OAc)2 and PTSA under the ball-milling conditions (Table 2). Gratifyingly, the ortho-iodinated acetanilides were obtained in moderate to good isolated yields. Both p-Me and m-Me-substituted acetanilides provided products 2a and 2b in excellent yields of 87% and 80%, respectively (Table 2, entries 1 and 2). As expected, 3,4-dimethylacetanilide underwent iodination successfully at the less sterically hindered ortho-position and gave product 2c in 85% yield (Table 2, entry 3). The unsubstituted acetanilide provided the desired product 2d in 77% yield (Table 2, entry 4). It is worth mentioning that the presence of a potentially reactive group, such as fluoro, chloro, and bromo substituents in the acetanilides was tolerable, and products 2e–i were isolated in 51–94% yields (Table 2, entries 5–9), highlighting the functional group compatibility of the current protocol. The presence of an acetyl group at the para-position of the phenyl ring of acetanilide 1j decreased the yield of the corresponding product 2j to 11% (Table 2, entry 10). Unfortunately, substrates bearing a strong electron-donating methoxy group and a strong electron-withdrawing nitro group could not afford any desired products, and the reason is not quite clear right now.
Table 2: Substrate scope.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-31-i6.svg?max-width=637&scale=1.0)
|
|||
| entry | substrate 1 | product 2b | yieldc (%) |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-31-i7.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-31-i8.svg?max-width=637&scale=1.0)
2a |
87 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-31-i9.svg?max-width=637&scale=1.0)
1b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-31-i10.svg?max-width=637&scale=1.0)
2b |
80 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-31-i11.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-31-i12.svg?max-width=637&scale=1.0)
2c |
85 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-31-i13.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-31-i14.svg?max-width=637&scale=1.0)
2d |
77 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-31-i15.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-31-i16.svg?max-width=637&scale=1.0)
2e |
94 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-31-i17.svg?max-width=637&scale=1.0)
1f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-31-i18.svg?max-width=637&scale=1.0)
2f |
71 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-31-i19.svg?max-width=637&scale=1.0)
1g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-31-i20.svg?max-width=637&scale=1.0)
2g |
74 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-31-i21.svg?max-width=637&scale=1.0)
1h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-31-i22.svg?max-width=637&scale=1.0)
2h |
51 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-31-i23.svg?max-width=637&scale=1.0)
1i |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-31-i24.svg?max-width=637&scale=1.0)
2i |
70 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-31-i25.svg?max-width=637&scale=1.0)
1j |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-31-i26.svg?max-width=637&scale=1.0)
2j |
11 |
aAll the reactions were carried out in a Spex SamplePrep 8000 mixer mill using 1 (0.4 mmol), NIS (0.4 mmol), Pd(OAc)2 (10 mol %) and PTSA (0.8 mmol) for 3 h. bProperly characterized by 1H NMR, 13C NMR, and HRMS spectral data. cIsolated yield.
In an aim to investigate the influence of the milling frequency, the model reaction of 1a with NIS was conducted by employing different types of mixer mills with different milling frequencies. Ortho-iodized acetanilide 2a was furnished in 90% yield after milling for 2 h by using a Retsch MM 200 mixer mill (30 Hz, Scheme 1a). At a milling frequency of 50 Hz in a Spex SamplePrep 5100 mixer mill, the iodination was accomplished within 1.5 h to afford the corresponding product 2a in 92% yield (Scheme 1b). According to the above experimental results, it could be concluded that the higher milling frequency had a beneficial effect on the reaction efficiency in terms of product yield and reaction time.
![[1860-5397-14-31-i1]](/bjoc/content/inline/1860-5397-14-31-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The influence of the milling frequency on the reaction of 1a with NIS.
Scheme 1: The influence of the milling frequency on the reaction of 1a with NIS.
To illustrate the superiority of the ball-milling technique, the reaction was also investigated in an organic solvent. The reaction of 1a with NIS conducted in toluene at room temperature for 3 h provided the desired product 2a in only 49% yield, which was inferior to those obtained by our mechanochemical approaches (Scheme 2).
![[1860-5397-14-31-i2]](/bjoc/content/inline/1860-5397-14-31-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Palladium-catalyzed ortho-iodination of 1a in toluene.
Scheme 2: Palladium-catalyzed ortho-iodination of 1a in toluene.
The plausible mechanism is proposed and depicted in Scheme 3. The addition of PTSA was essential for the present reaction. It is believed the more active Pd(OTs)2 is formed in situ from Pd(OAc)2 and TsOH [46,47]. The formed Pd(OTs)2 inserts into the ortho C–H bond of the anilides after coordination to the oxygen atom of the amide moiety, affording the species A. Oxidative addition of the species A with NIS generates the Pd(IV) complex B. Finally, the iodinated product is provided by reductive elimination along with regeneration of Pd(OTs)2 in the presence of TsOH.
![[1860-5397-14-31-i3]](/bjoc/content/inline/1860-5397-14-31-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
It was intriguing to find that N-bromosuccinimide (NBS) and N-chlorosuccinimide (NCS) could also be used as reaction partners to react with the representative acetanilide 1a under identical ball-milling conditions. The corresponding ortho-brominated and ortho-chlorinated products 3a and 4a were obtained in 73% and 77% yields, respectively (Scheme 4).
![[1860-5397-14-31-i4]](/bjoc/content/inline/1860-5397-14-31-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Palladium-catalyzed ortho-bromination and chlorination of 1a in a ball mill.
Scheme 4: Palladium-catalyzed ortho-bromination and chlorination of 1a in a ball mill.
Conclusion
In summary, we have developed a solvent-free and efficient protocol to synthesize ortho-iodinated acetanilide derivatives with Pd(OAc)2 as the catalyst and N-iodosuccinimide as the halogen source under mechanical milling conditions. This protocol shows its advantages in terms of high regioselectivity, simple operation and environmentally friendliness. In addition, the present protocol can be extended to the synthesis of ortho-brominated and chlorinated acetanilides delivering good yields by using the corresponding N-halosuccinimides.
Supporting Information
| Supporting Information File 1: Experimental, analytical data and NMR spectra of 2a–j, 3a and 4a. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Evans, D. A.; Katz, J. L.; Peterson, G. S.; Hintermann, T. J. Am. Chem. Soc. 2001, 123, 12411–12413. doi:10.1021/ja011943e
Return to citation in text: [1] -
Pelletier, J. C.; Youssefyeh, R. D.; Campbell, H. F. Substituted Saturated and Unsaturated Indole Quinoline and Benzazepine Carboxamides and Their Use as Pharmacological Agents. U.S. Patent 4920219 A, April 24, 1990.
Return to citation in text: [1] -
de la Mare, P. B. D., Ed. Electrophilic Halogenation; Cambridge University Press: New York, 1976.
Return to citation in text: [1] -
Hodgson, H. H. Chem. Rev. 1947, 40, 251–277. doi:10.1021/cr60126a003
Return to citation in text: [1] -
Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Org. Lett. 2006, 8, 2523–2526. doi:10.1021/ol060747f
Return to citation in text: [1] -
Bedford, R. B.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Angew. Chem., Int. Ed. 2011, 50, 5524–5527. doi:10.1002/anie.201101606
Return to citation in text: [1] -
Dubost, E.; Fossey, C.; Cailly, T.; Rault, S.; Fabis, F. J. Org. Chem. 2011, 76, 6414–6420. doi:10.1021/jo200853j
Return to citation in text: [1] -
Schröder, N.; Wencel-Delord, J.; Glorius, F. J. Am. Chem. Soc. 2012, 134, 8298–8301. doi:10.1021/ja302631j
Return to citation in text: [1] -
John, A.; Nicholas, K. M. J. Org. Chem. 2012, 77, 5600–5605. doi:10.1021/jo300713h
Return to citation in text: [1] -
Ma, X.-T.; Tian, S.-K. Adv. Synth. Catal. 2013, 355, 337–340. doi:10.1002/adsc.201200902
Return to citation in text: [1] -
Zhao, X.; Dimitrijević, E.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 3466–3467. doi:10.1021/ja900200g
Return to citation in text: [1] -
Wang, W.; Pan, C.; Chen, F.; Cheng, J. Chem. Commun. 2011, 47, 3978–3980. doi:10.1039/c0cc05557c
Return to citation in text: [1] -
Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215–5219. doi:10.1002/anie.200705613
Return to citation in text: [1] -
Mei, T.-S.; Wang, D.-H.; Yu, J.-Q. Org. Lett. 2010, 12, 3140–3143. doi:10.1021/ol1010483
Return to citation in text: [1] -
Li, J.-J.; Mei, T.-S.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 6452–6455. doi:10.1002/anie.200802187
Return to citation in text: [1] -
Wan, X.; Ma, Z.; Li, B.; Zhang, K.; Cao, S.; Zhang, S.; Shi, Z. J. Am. Chem. Soc. 2006, 128, 7416–7417. doi:10.1021/ja060232j
Return to citation in text: [1] -
Bedford, R. B.; Engelhart, J. U.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Dalton Trans. 2010, 39, 10464–10472. doi:10.1039/c0dt00385a
Return to citation in text: [1] -
Song, B.; Zheng, X.; Mo, J.; Xu, B. Adv. Synth. Catal. 2010, 352, 329–335. doi:10.1002/adsc.200900778
Return to citation in text: [1] -
Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c
Return to citation in text: [1] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A
Return to citation in text: [1] -
Zhu, S.-E.; Li, F.; Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7535–7570. doi:10.1039/c3cs35494f
Return to citation in text: [1] [2] -
Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h
Return to citation in text: [1] [2] -
Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887
Return to citation in text: [1] -
Achar, T. K.; Bose, A.; Mal, P. Beilstein J. Org. Chem. 2017, 13, 1907–1931. doi:10.3762/bjoc.13.186
Return to citation in text: [1] -
Hernández, J. G. Chem. – Eur. J. 2017, 23, in press. doi:10.1002/chem.201786861
Return to citation in text: [1] -
Bose, A.; Mal, P. Tetrahedron Lett. 2014, 55, 2154–2156. doi:10.1016/j.tetlet.2014.02.064
Return to citation in text: [1] -
Maiti, S.; Mal, P. Synth. Commun. 2014, 44, 3461–3469. doi:10.1080/00397911.2014.946995
Return to citation in text: [1] -
Juribašić, M.; Užarević, K.; Gracin, D.; Ćurić, M. Chem. Commun. 2014, 50, 10287–10290. doi:10.1039/C4CC04423A
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. Chem. Commun. 2015, 51, 12582–12584. doi:10.1039/C5CC04423E
Return to citation in text: [1] [2] -
Hermann, G. N.; Becker, P.; Bolm, C. Angew. Chem., Int. Ed. 2015, 54, 7414–7417. doi:10.1002/anie.201502536
Return to citation in text: [1] -
Hermann, G. N.; Becker, P.; Bolm, C. Angew. Chem., Int. Ed. 2016, 55, 3781–3784. doi:10.1002/anie.201511689
Return to citation in text: [1] -
Lou, S.-J.; Mao, Y.-J.; Xu, D.-Q.; He, J.-Q.; Chen, Q.; Xu, Z.-Y. ACS Catal. 2016, 6, 3890–3894. doi:10.1021/acscatal.6b00861
Return to citation in text: [1] -
Hermann, G. N.; Bolm, C. ACS Catal. 2017, 7, 4592–4596. doi:10.1021/acscatal.7b00582
Return to citation in text: [1] -
Hermann, G. N.; Jung, C. L.; Bolm, C. Green Chem. 2017, 19, 2520–2523. doi:10.1039/C7GC00499K
Return to citation in text: [1] -
Jia, K.-Y.; Yu, J.-B.; Jiang, Z.-J.; Su, W.-K. J. Org. Chem. 2016, 81, 6049–6055. doi:10.1021/acs.joc.6b01138
Return to citation in text: [1] -
Jiang, X.; Chen, J.; Zhu, W.; Cheng, K.; Liu, Y.; Su, W.-K.; Yu, C. J. Org. Chem. 2017, 82, 10665–10672. doi:10.1021/acs.joc.7b01695
Return to citation in text: [1] -
Cheng, H.; Hernández, J. G.; Bolm, C. Org. Lett. 2017, 19, 6284–6287. doi:10.1021/acs.orglett.7b02973
Return to citation in text: [1] -
Li, L.; Wang, J.-J.; Wang, G.-W. J. Org. Chem. 2016, 81, 5433–5439. doi:10.1021/acs.joc.6b00786
Return to citation in text: [1] -
Li, H.-G.; Wang, G.-W. J. Org. Chem. 2017, 82, 6341–6348. doi:10.1021/acs.joc.7b00912
Return to citation in text: [1] -
Xu, H.; Liu, H.-W.; Lin, H.-S.; Wang, G.-W. Chem. Commun. 2017, 53, 12477–12480. doi:10.1039/C7CC08306H
Return to citation in text: [1] -
Wang, G.-W. Top. Organomet. Chem. 2016, 55, 119–136. doi:10.1007/3418_2015_128
Return to citation in text: [1] -
Wang, G.-W.; Yuan, T.-T.; Li, D.-D. Angew. Chem., Int. Ed. 2011, 50, 1380–1383. doi:10.1002/anie.201005874
Return to citation in text: [1] -
Li, Z.-Y.; Li, L.; Li, Q.-L.; Jing, K.; Xu, H.; Wang, G.-W. Chem. – Eur. J. 2017, 23, 3285–3290. doi:10.1002/chem.201700354
Return to citation in text: [1] -
Liu, Z., Two Organic Reactions under Mechanochemical Conditions, Master thesis, University of Science and Technology of China, 2013.
Return to citation in text: [1] -
Zhu, B.; Wang, G.-W. Org. Lett. 2009, 11, 4334–4337 and references cited therein. doi:10.1021/ol901675t
Return to citation in text: [1] [2] -
Boele, M. D. K.; van Strijdonck, G. P. F.; de Vries, A. H. M.; Kamer, P. C. J.; de Vries, J. G.; van Leeuwen, P. W. N. M. J. Am. Chem. Soc. 2002, 124, 1586–1587. doi:10.1021/ja0176907
Return to citation in text: [1]
| 1. | Evans, D. A.; Katz, J. L.; Peterson, G. S.; Hintermann, T. J. Am. Chem. Soc. 2001, 123, 12411–12413. doi:10.1021/ja011943e |
| 2. | Pelletier, J. C.; Youssefyeh, R. D.; Campbell, H. F. Substituted Saturated and Unsaturated Indole Quinoline and Benzazepine Carboxamides and Their Use as Pharmacological Agents. U.S. Patent 4920219 A, April 24, 1990. |
| 19. | Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c |
| 20. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A |
| 21. | Zhu, S.-E.; Li, F.; Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7535–7570. doi:10.1039/c3cs35494f |
| 22. | Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h |
| 23. | Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277 |
| 24. | Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887 |
| 25. | Achar, T. K.; Bose, A.; Mal, P. Beilstein J. Org. Chem. 2017, 13, 1907–1931. doi:10.3762/bjoc.13.186 |
| 26. | Hernández, J. G. Chem. – Eur. J. 2017, 23, in press. doi:10.1002/chem.201786861 |
| 27. | Bose, A.; Mal, P. Tetrahedron Lett. 2014, 55, 2154–2156. doi:10.1016/j.tetlet.2014.02.064 |
| 28. | Maiti, S.; Mal, P. Synth. Commun. 2014, 44, 3461–3469. doi:10.1080/00397911.2014.946995 |
| 5. | Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Org. Lett. 2006, 8, 2523–2526. doi:10.1021/ol060747f |
| 6. | Bedford, R. B.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Angew. Chem., Int. Ed. 2011, 50, 5524–5527. doi:10.1002/anie.201101606 |
| 7. | Dubost, E.; Fossey, C.; Cailly, T.; Rault, S.; Fabis, F. J. Org. Chem. 2011, 76, 6414–6420. doi:10.1021/jo200853j |
| 8. | Schröder, N.; Wencel-Delord, J.; Glorius, F. J. Am. Chem. Soc. 2012, 134, 8298–8301. doi:10.1021/ja302631j |
| 9. | John, A.; Nicholas, K. M. J. Org. Chem. 2012, 77, 5600–5605. doi:10.1021/jo300713h |
| 10. | Ma, X.-T.; Tian, S.-K. Adv. Synth. Catal. 2013, 355, 337–340. doi:10.1002/adsc.201200902 |
| 11. | Zhao, X.; Dimitrijević, E.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 3466–3467. doi:10.1021/ja900200g |
| 12. | Wang, W.; Pan, C.; Chen, F.; Cheng, J. Chem. Commun. 2011, 47, 3978–3980. doi:10.1039/c0cc05557c |
| 13. | Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215–5219. doi:10.1002/anie.200705613 |
| 14. | Mei, T.-S.; Wang, D.-H.; Yu, J.-Q. Org. Lett. 2010, 12, 3140–3143. doi:10.1021/ol1010483 |
| 15. | Li, J.-J.; Mei, T.-S.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 6452–6455. doi:10.1002/anie.200802187 |
| 16. | Wan, X.; Ma, Z.; Li, B.; Zhang, K.; Cao, S.; Zhang, S.; Shi, Z. J. Am. Chem. Soc. 2006, 128, 7416–7417. doi:10.1021/ja060232j |
| 17. | Bedford, R. B.; Engelhart, J. U.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Dalton Trans. 2010, 39, 10464–10472. doi:10.1039/c0dt00385a |
| 18. | Song, B.; Zheng, X.; Mo, J.; Xu, B. Adv. Synth. Catal. 2010, 352, 329–335. doi:10.1002/adsc.200900778 |
| 3. | de la Mare, P. B. D., Ed. Electrophilic Halogenation; Cambridge University Press: New York, 1976. |
| 42. | Wang, G.-W. Top. Organomet. Chem. 2016, 55, 119–136. doi:10.1007/3418_2015_128 |
| 43. | Wang, G.-W.; Yuan, T.-T.; Li, D.-D. Angew. Chem., Int. Ed. 2011, 50, 1380–1383. doi:10.1002/anie.201005874 |
| 44. | Li, Z.-Y.; Li, L.; Li, Q.-L.; Jing, K.; Xu, H.; Wang, G.-W. Chem. – Eur. J. 2017, 23, 3285–3290. doi:10.1002/chem.201700354 |
| 46. | Zhu, B.; Wang, G.-W. Org. Lett. 2009, 11, 4334–4337 and references cited therein. doi:10.1021/ol901675t |
| 21. | Zhu, S.-E.; Li, F.; Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7535–7570. doi:10.1039/c3cs35494f |
| 22. | Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h |
| 39. | Li, L.; Wang, J.-J.; Wang, G.-W. J. Org. Chem. 2016, 81, 5433–5439. doi:10.1021/acs.joc.6b00786 |
| 40. | Li, H.-G.; Wang, G.-W. J. Org. Chem. 2017, 82, 6341–6348. doi:10.1021/acs.joc.7b00912 |
| 41. | Xu, H.; Liu, H.-W.; Lin, H.-S.; Wang, G.-W. Chem. Commun. 2017, 53, 12477–12480. doi:10.1039/C7CC08306H |
| 46. | Zhu, B.; Wang, G.-W. Org. Lett. 2009, 11, 4334–4337 and references cited therein. doi:10.1021/ol901675t |
| 47. | Boele, M. D. K.; van Strijdonck, G. P. F.; de Vries, A. H. M.; Kamer, P. C. J.; de Vries, J. G.; van Leeuwen, P. W. N. M. J. Am. Chem. Soc. 2002, 124, 1586–1587. doi:10.1021/ja0176907 |
| 30. | Hernández, J. G.; Bolm, C. Chem. Commun. 2015, 51, 12582–12584. doi:10.1039/C5CC04423E |
| 29. | Juribašić, M.; Užarević, K.; Gracin, D.; Ćurić, M. Chem. Commun. 2014, 50, 10287–10290. doi:10.1039/C4CC04423A |
| 30. | Hernández, J. G.; Bolm, C. Chem. Commun. 2015, 51, 12582–12584. doi:10.1039/C5CC04423E |
| 31. | Hermann, G. N.; Becker, P.; Bolm, C. Angew. Chem., Int. Ed. 2015, 54, 7414–7417. doi:10.1002/anie.201502536 |
| 32. | Hermann, G. N.; Becker, P.; Bolm, C. Angew. Chem., Int. Ed. 2016, 55, 3781–3784. doi:10.1002/anie.201511689 |
| 33. | Lou, S.-J.; Mao, Y.-J.; Xu, D.-Q.; He, J.-Q.; Chen, Q.; Xu, Z.-Y. ACS Catal. 2016, 6, 3890–3894. doi:10.1021/acscatal.6b00861 |
| 34. | Hermann, G. N.; Bolm, C. ACS Catal. 2017, 7, 4592–4596. doi:10.1021/acscatal.7b00582 |
| 35. | Hermann, G. N.; Jung, C. L.; Bolm, C. Green Chem. 2017, 19, 2520–2523. doi:10.1039/C7GC00499K |
| 36. | Jia, K.-Y.; Yu, J.-B.; Jiang, Z.-J.; Su, W.-K. J. Org. Chem. 2016, 81, 6049–6055. doi:10.1021/acs.joc.6b01138 |
| 37. | Jiang, X.; Chen, J.; Zhu, W.; Cheng, K.; Liu, Y.; Su, W.-K.; Yu, C. J. Org. Chem. 2017, 82, 10665–10672. doi:10.1021/acs.joc.7b01695 |
| 38. | Cheng, H.; Hernández, J. G.; Bolm, C. Org. Lett. 2017, 19, 6284–6287. doi:10.1021/acs.orglett.7b02973 |
| 45. | Liu, Z., Two Organic Reactions under Mechanochemical Conditions, Master thesis, University of Science and Technology of China, 2013. |
© 2018 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)