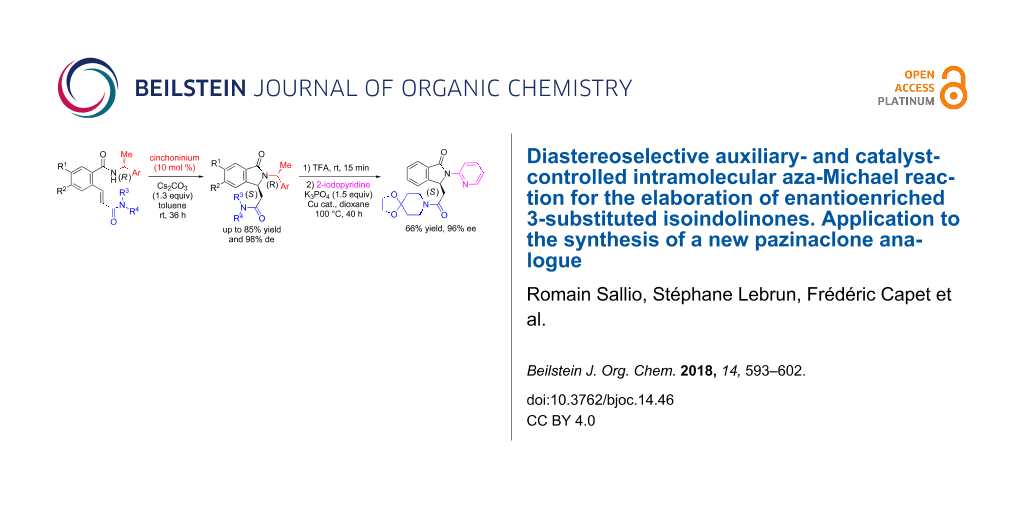
Abstract
A new asymmetric organocatalyzed intramolecular aza-Michael reaction by means of both a chiral auxiliary and a catalyst for stereocontrol is reported for the synthesis of optically active isoindolinones. A selected cinchoninium salt was used as phase-transfer catalyst in combination with a chiral nucleophile, a Michael acceptor and a base to provide 3-substituted isoindolinones in good yields and diastereomeric excesses. This methodology was applied to the asymmetric synthesis of a new pazinaclone analogue which is of interest in the field of benzodiazepine-receptor agonists.

Graphical Abstract
Introduction
Isoindolinones I (Figure 1), e.g., 2,3-dihydro-1H-isoindol-1-ones, also called phthalimidines are bicyclic lactams whose molecular structure is the basis of a wide range of alkaloids and biologically active compounds [1-11]. Among the latter, optically pure compounds functionalized at C-3 by acetamido groups play an important role as key targets for the pharmaceutical industry. For example, substituted 3-isoindolinones, such as JM-1232 (II) [12-14] and pazinaclone (III) [15,16] (Figure 1), have shown sedative-hypnotic activities used for the treatment of anxiety by acting as partial agonists at GABAA (γ-aminobutyric acid type A) benzodiazepine receptors [17]. All these studies have highlighted a strong correlation between the compound pharmacological activities and the absolute configurations of their stereocenter [14]. Hence, the asymmetric synthesis of functionalized 3-substituted isoindolinones using short, versatile and selective procedures is clearly a topic of current interest.
![[1860-5397-14-46-1]](/bjoc/content/figures/1860-5397-14-46-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of synthetic pharmacologically active chiral 3-substituted isoindolinones.
Figure 1: Examples of synthetic pharmacologically active chiral 3-substituted isoindolinones.
Two strategies can be applied for the asymmetric synthesis of 3-substituted isoindolinones. First, diastereoselective reactions implying the use of a chiral auxiliary resulted effectively in various optically pure compounds [10,18-20]. Second, enantioselective syntheses of these bicylic lactams were performed by using chiral transition metal- or organocatalysts which control the configuration of the trisubstituted carbon stereocenter alpha to the nitrogen [10,20-34]. Though various metal or organic catalysts were used to promote the aza-Michael reaction in different syntheses for the creation of nitrogen–carbon bonds, phase-transfer catalysts were less studied (see reviews [35-38]) in intermolecular [39-43] and intramolecular [44-46] sequences. Among the latter, a short regio- and stereoselective organocatalyzed intramolecular aza-Michael reaction was reported by us for the asymmetric synthesis of several isoindolinones [20,34]. Indeed, we noticed along our studies some intramolecular aza-Michael reactions were effectively catalysed by cinchoninium phase-transfer catalysts (PCT) affording the targeted 3-substituted isoindolinones with promising enantioselectivities (up to 91%) [20]. However, high enantioselectivities were reached only for specific substitution patterns on the amide nitrogen atom and to a lesser extent on the Michael acceptor. In order to overcome these limitations, we decided to incorporate a chiral auxiliary in our substrates combined with a proper chiral phase-transfer organocatalyst to operate an efficient stereocontrol. To the best of our knowledge such approach involving a double auxiliary and catalyst stereocontrol was never applied before to asymmetric synthesis of enantioenriched isoindolinones.
Results and Discussion
Retrosynthetic analysis
From a retrosynthetic point of view, (3S)-NH free 3-substituted isoindolinones 1 and 2 could be obtained in high enantioselectivities from the intermediates (2R,3S)-3–5 after removal of the chiral auxiliary (Scheme 1). (2R,3S)-bicyclic lactams 3–5 could be prepared by the asymmetric intramolecular organo-catalyzed aza-Michael reaction of (R)-benzamides 6–8 bearing an acrylamide group at the ortho-position of the benzene ring.
![[1860-5397-14-46-i1]](/bjoc/content/inline/1860-5397-14-46-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of NH free chiral 3-substituted isoindolinones (3S)-1 and (3S)-2.
Scheme 1: Retrosynthetic analysis of NH free chiral 3-substituted isoindolinones (3S)-1 and (3S)-2.
Synthesis of parent chiral benzamides 6–8
The use of a stereoselective chiral auxiliary which could be incorporated and removed easily without racemization was crucial for the success of our strategy. These requirements prompted us to incorporate α-methylbenzylamine-type chiral auxiliaries, which have been extensively used by Davies et al. to gain access to a wide range of chiral N-heterocycles via intermolecular aza-Michael reactions [34,47-51]. The starting unsaturated benzoic acids 14a–e and 15 were readily prepared via a two steps sequence involving first a palladium-catalyzed Heck cross coupling between 2-bromobenzoic tert-butyl esters 9 and 10 with acrylamides 11a–e (69–72% isolated yields, Scheme 2, Figure 2). The subsequent removal of the t-butyl group in esters 12a–e and 13 (Figure 2) was then achieved by treatment with trifluoroacetic acid to provide in-situ the corresponding benzoic acids 14a–e and 15. The direct coupling of these functionalized carboxylic acids with chiral benzylic primary amines, (R) or (S)-16 (NH2-CH(Me)Ph) and (R)-17 (NH2CH(Me)p-MeO-C6H4), afforded the required parent amides 6a–d, 7a–e and 8 in 61–75% isolated yields after work-up (Scheme 2, Figure 3).
![[1860-5397-14-46-i2]](/bjoc/content/inline/1860-5397-14-46-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of parent benzamides 6–8.
Scheme 2: Synthesis of parent benzamides 6–8.
![[1860-5397-14-46-2]](/bjoc/content/figures/1860-5397-14-46-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Esters 12a–e, 13 prepared, isolated yield.
Figure 2: Esters 12a–e, 13 prepared, isolated yield.
![[1860-5397-14-46-3]](/bjoc/content/figures/1860-5397-14-46-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Benzamides 6a–d, 7a–e, 8 prepared, isolated yield.
Figure 3: Benzamides 6a–d, 7a–e, 8 prepared, isolated yield.
Diastereoselective intramolecular aza-Michael reaction
First, the study of the diastereoselective intramolecular aza-Michael reaction of benzamide substrate (S)-6a allowed us to optimize the reaction conditions (Table 1) and latter to screen various privileged phase-transfer catalysts (Figure 4, Table 2). As some aza-Michael reactions were shown to be performed without the use of any catalyst or additional reagent [52-60], we performed control experiments (Table 1). The reaction of reagent (S)-6a led to product (S)-3a solely by using a base like Cs2CO3 in toluene with a good yield (74%) and a modest diastereomeric excess (37% de, Table 1, entry 1). Increasing the reaction time from 16 h to 36 h led to higher diastereomeric excess but no further improvement was noticed with longer reaction times (Table 1, entries 2 and 3). Such chiral amplification versus time was already found to operate through a retro-aza-Michael reaction [61,62]. Indeed, through an equilibration of aza-Michael and retro-aza-Michael reactions, the minor diastereoisomer of 3a may lead back to a racemic starting material and subsequently favour the major diastereoisomer (Table 1, entries 1–3). The use of a catalytic amount of base led to product 3a in a good yield (Table 1, entry 4) but with a loss of diastereoselectivity. Because the optically pure auxiliary and the conjugated ketone were not interacting well, a significant diastereoselectivity could not be obtained and we looked for improvements through the use of an appropriate chiral organocatalyst [63-65]. Indeed, within the same reaction conditions, the use of cinchoninium catalyst 18a afforded isoindolinone (S)-3a with higher de (54%), (Table 1, entry 5). We assumed such de increase resulted from a match effect [65-67] of the diastereomeric ion pair formed by the chiral nucleophile, the conjugated ketone and the cinchoninium salt. Hence, in our case, the chirality of the new stereochemical center was shown to be controlled by both Michael acceptor and donor interacting with the chiral ammonium. Surprisingly, the diastereoselectivity initially obtained for 3a (Table 1, entry 5) was not improved by a decrease of the temperature to −10 °C (Table 1, entry 6) or by the use of polar solvents (Table 1, entries 7 and 8) or of another base (Table 1, entry 9).
Table 1: Identification of the optimum reaction conditions for the diastereoselective intramolecular aza-Michael reaction of (S)-6a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-46-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst | solvent | base (equiv) | conditions | yield (%)a | de (%)b |
|---|---|---|---|---|---|---|
| 1 | – | toluene | Cs2CO3 (1.3) | rt, 16 h | (2S)-3a (74) | 37 |
| 2 | – | toluene | Cs2CO3 (1.3) | rt, 24 h | (2S)-3a (80) | 40 |
| 3 | – | toluene | Cs2CO3 (1.3) | rt, 36 h | (2S)-3a (80) | 44c |
| 4 | – | toluene | Cs2CO3 (0.1) | rt, 36 h | (2S)-3a (82) | 30 |
| 5 | 18a | toluene | Cs2CO3 (1.3) | rt, 36 h | (2S)-3a (75) | 54 |
| 6 | 18a | toluene | Cs2CO3 (1.3) | −10 °C, 36 h | (2S)-3a (75) | 46 |
| 7 | 18a | THF | Cs2CO3 (1.3) | rt, 36 h | (2S)-3a (73) | 40 |
| 8 | 18a | CH2Cl2 | Cs2CO3 (1.3) | rt, 36 h | (2S)-3a (80) | 40 |
| 9 | 18a | toluene | Ba(OH)2 (1.3) | rt, 36 h | (2S)-3a (81) | 42 |
aAfter purification. bDetermined by HPLC and 1H NMR. cNo change with longer reaction times.
In order to identify the most active catalyst for the aza-Michael reaction of (S)-and (R)-6a, an array of phase-transfer catalysts was screened (Figure 4, Table 2). By comparing catalysts 18a and 18b, a bromide anion was shown to be preferred to a chloride one (Table 2, entries 1 and 2). Catalyst 18c para-substituted with a tert-butyl increased significantly the diastereoselectivity of the reaction with 62% de (Table 2, entry 3).
Table 2: Identification of the most active catalyst for the diastereoselective intramolecular aza-Michael reaction of (S)- and (R)-6a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-46-i7.svg?max-width=637&scale=1.0)
|
||||
| entry | reagent | catalyst | yield (%)a | de (%)b |
|---|---|---|---|---|
| 1 | (S)-6a | 18a | (2S)-3a (75) | 54 |
| 2 | (S)-6a | 18b | (2S)-3a (73) | 44 |
| 3 | (S)-6a | 18c | (2S)-3a (77) | 62 |
| 4 | (S)-6a | 18d | (2S)-3a (78) | 48 |
| 5 | (S)-6a | 18e | (2S)-3a (75) | 42 |
| 6 | (R)-6a | 18a | (2R)-3a (76) | 66 |
| 7 | (R)-6a | 18c | (2R)-3a (75) | 80 (>96%)c |
| 8 | (R)-6a | 18f | (2R)-3a (74) | 40 |
| 9 | (R)-6a | 18g | (2R)-3a (72) | 76 |
| 10 | (R)-6a | 18h | (2R)-3a (76) | 74 |
| 11 | (R)-6a | 18i | (2R)-3a (72) | 68 |
| 12 | (R)-6a | 18j | (2R)-3a (74) | 60 |
| 13 | (R)-6a | 18k | (2R)-3a (78) | 62 |
| 14 | (R)-6a | 18l | (2R)-3a (80) | 54 |
| 15 | (S)-6a | 19 | (2S)-3a (71) | 34 |
| 16 | (S)-6a | 20 | (2S)-3a (70) | 31 |
| 17 | (R)-6a | 21 | (2R)-3a (79) | 48 |
aAfter purification. bDetermined by HPLC and 1H NMR. cAfter flash chromatography on silica gel (EtOAc/hexanes 3:7) and crystallization from hexanes/toluene. Yield: 63%.
![[1860-5397-14-46-4]](/bjoc/content/figures/1860-5397-14-46-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Phase transfer catalysts (PTC) used in this study.
Figure 4: Phase transfer catalysts (PTC) used in this study.
No de improvements resulted from the use of catalysts 18d,e which were modified by methylation or allylation of the cinchoninium alcohol fragment (Table 2, entries 4 and 5). While using cinchoninium catalyst 18a and the same reaction conditions, we noticed amide reagent (R)-6a led to a higher diastereomeric excess (de) of 66% for product (R)-3a as compared to reagent (S)-6a for product (S)-3a, one configuration being preferred from the other (Table 2, entry 6). A quite similar effect was previously observed in other Michael-additions involving chiral auxiliaries on the nucleophile and on the Michael acceptor [62]. As for (S)-6a, catalyst 18c bearing a bulky tert-butyl group at the benzyl para-position gave the best results in term of stereoselectivity (80% de, Table 2, entry 7). However, the use of bulkier benzyl, naphthyl and anthracenyl fragments, e.g., catalysts 18i–l, did not enhance the reaction diastereoselectivity (Table 2, entries 11–14). Whereas catalyst 18f bearing an ortho-fluoro substituent led to a decrease of de (Table 2, entry 8), catalyst 18g and 18h, respectively, substituted at the benzyl para-position by isopropyl and CF3 groups, gave good results with 76 and 74% de (Table 2, entries 9 and 10). Dimeric and trimeric organocatalysts 19 and 20 based on a cinchonine core did not enhance the reaction diastereoselectivity (Table 2, entries 15 and 16). Finally, by comparison to all the studied cinchoninium catalysts, the use of cinchonidinium catalyst 21 proved to be less efficient with a 48% de (Table 2, entry 17).
With the optimized reaction conditions in hand, catalyst 18c was employed in the asymmetric intramolecular aza-Michael reaction of benzamides (R)-6a–d bearing an array of acrylamide groups (Scheme 3). Substrate 6a bearing a (R)-α-methylbenzyl chiral auxiliary led to isoindolinone 3a in 80% de and a pure diastereoisomer was recovered after chromatography on silica gel (EtOAc/hexanes 3:7) and crystallization from hexanes/toluene. Reactions of substrates 6b–d highlighted the diastereoselection of the reaction was highly dependent of the starting benzamide substitution, 44 to 60% de being obtained for 3b–d. Finally, whereas diastereoisomers issued from 6b could be separated by flash chromatography, this was not possible for products 3c and d. Cyclisation of chiral benzamides (R)-7a–e and (R)-8 led to isoindolinones (2R)-4a–e, (2R)-5 in good yields and average to good diastereoselectivities (Scheme 3). In some cases, purification by flash chromatography afforded products 4b, 4c and 5 in higher diastereomeric purity.
![[1860-5397-14-46-i3]](/bjoc/content/inline/1860-5397-14-46-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of isoindolinones 3a–d, 4a–e, 5; isolated yield, de by HPLC and 1H NMR. aAfter flash chromatography on silica gel (EtOAc/hexanes 3:7) and crystallization from hexanes/toluene; bafter flash chromatography.on silica gel (EtOAc/hexanes 3:7).
Scheme 3: Synthesis of isoindolinones 3a–d, 4a–e, 5; isolated yield, de by HPLC and 1H NMR. aAfter flash chro...
In order to access to the targeted NH-free isoindolinones, the cleavage of the (R)-α-methylbenzyl chiral auxiliary was performed in acidic conditions but the reactions proved to be ineffective. However, a change in our models for the more electron rich (R)-α-methyl-para-methoxybenzyl group resulted in a straightforward and selective cleavage in mild acidic conditions without racemization (Scheme 4). Indeed, further cleavage of the α-methyl-para-methoxyphenyl chiral auxiliary in protected isoindolinones 4a–c, 4e and 5 resulted in the corresponding NH-free lactams 1a–c, 1e and 2 without any racemization (Scheme 4).
![[1860-5397-14-46-i4]](/bjoc/content/inline/1860-5397-14-46-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Removal of the chiral auxiliary. Synthesis of isoindolinones 1a–c, 1e, 2; isolated yield, ee by HPLC.
Scheme 4: Removal of the chiral auxiliary. Synthesis of isoindolinones 1a–c, 1e, 2; isolated yield, ee by HPL...
A subsequent X-ray analysis of a single crystal allowed us to assert the (2R,3S) configuration of 3a (Figure 5). This result allowed for the determination of the absolute configurations of all isolated isoindolinones.
![[1860-5397-14-46-5]](/bjoc/content/figures/1860-5397-14-46-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP plot of isoindolinone (2R,3S)-3a (CCDC 1590565) [68].
Figure 5: ORTEP plot of isoindolinone (2R,3S)-3a (CCDC 1590565) [68].
Asymmetric synthesis of a new pazinaclone analogue
With this handful methodology in hands, we then turned our attention to the asymmetric synthesis of a new pazinaclone analogue, which could be of particular interest in the field of benzodiazepine-receptor agonists [8-17]. Indeed, pazinaclone produces its sedative and anxiolytic effects by acting as a partial agonist at GABAA (γ-aminobutyric acid type A) benzodiazepine receptors [17]. In order to circumvent any hydrolysis of the ketal group during the preparation of the starting benzamide (see Supporting Information File 1), the synthesis of intermediate 24 was performed according another pathway depicted in Scheme 5. Aldehyde 23 was first readily prepared via a Heck cross coupling reaction between 2-bromobenzaldehyde (22) and acrylamide 11f. Next, a Pinnick oxidation of the aldehyde 23 followed with a coupling reaction with chiral benzylamine 17 delivered the targeted benzamide 24 in good yield (65%).
![[1860-5397-14-46-i5]](/bjoc/content/inline/1860-5397-14-46-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of pazinaclone analogue (3S)-27.
Scheme 5: Synthesis of pazinaclone analogue (3S)-27.
The intramolecular aza-Michael reaction of acrylamide (R)-24 was then performed using the best phase-transfer catalyst 18c and the optimized experimental conditions to give isoindolinone (2R,3S)-25 as a mixture of diastereoisomers (82% yield, 71% de) which were separated by chromatography and purified by crystallization (70% yield, >96% de). Lactam (2R,3S)-25 bearing a α-methyl-para-methoxyphenyl chiral auxiliary was then deprotected with trifluoroacetic acid at room temperature to deliver the NH-free isoindolinone (3S)-26 (76% yield, 98% ee) which is a key building block in the synthesis of benzodiazepine-receptor agonists [8-16]. Indeed, the copper-catalyzed N-arylation of (3S)-26 was performed in dioxane with N,N-dimethylethylenediamine as ligand [27] to deliver the targeted pazinaclone analogue (3S)-27 in a fair yield (66%) without significant loss in enantiomeric purity. Moreover, it was worth to note compound (3S)-27 was not racemizing when heated in DMF at 150 °C for 48 h.
Conclusion
Herein, a new synthetic route towards optically active 3-substituted isoindolinones was developed. These organic compounds are useful for the development of agonists of GABAA (γ-aminobutyric acid type A) benzodiazepine-receptors. Various functionalized isoindolinones were prepared in good yields and diastereomeric excesses by intramolecular aza-Michael reactions using a double stereo-induction approach. The combined use of selected cinchoninium salts as phase-transfer catalysts and of nucleophiles bearing a chiral auxiliary enabled an effective match effect between the diastereomeric ion pair formed by the nucleophile, the Michael acceptor and the cinchoninium salt. Further investigations on this synthetic methodology will be reported in due course.
Supporting Information
| Supporting Information File 1:
File Name S1.pdf.
Experimental procedures, characterization data, copies of the 1H, 13C NMR spectra, HPLC chromatograms, ORTEP drawing of 3a and the summary of 3a crystallographic information. |
||
| Format: PDF | Size: 8.7 MB | Download |
| Supporting Information File 2:
File Name S1.cif.
Crystallographic information file of compound 3a. |
||
| Format: CIF | Size: 215.5 KB | Download |
Acknowledgements
The University Lille 1 is gratefully acknowledged for a PhD fellowship (R.S). The CNRS, the Chevreul Institute (FR 2638), the Ministère de l’Enseignement Supérieur et de la Recherche, the Région Hauts-de-France and the FEDER are acknowledged for supporting and funding partially this work. Mrs Céline Delabre (UCCS) is thanked for GC–MS and HPLC analyses. Dr M. Kouach and Ms A. Descat (Univ. Lille) are thanked for HRMS analyses. Ms M. Dubois (UCCS) and Mrs T. Donne (Dpt Chimie) are thanked for technical assistance. Dr. A. Couture is acknowledged for helpful comments on the manuscript.
References
-
Wrobel, J.; Dietrich, A.; Woolson, S. A.; Millen, J.; McCaleb, M.; Harrison, M. C.; Hohman, T. C.; Sredy, J.; Sullivan, D. J. Med. Chem. 1992, 35, 4613–4627. doi:10.1021/jm00102a016
Return to citation in text: [1] -
Pigeon, P.; Decroix, B. Tetrahedron Lett. 1996, 37, 7707–7710. doi:10.1016/0040-4039(96)01738-8
Return to citation in text: [1] -
Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7
Return to citation in text: [1] -
Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C. J. Org. Chem. 1998, 63, 3128–3132. doi:10.1021/jo972247t
Return to citation in text: [1] -
Belliotti, T. R.; Brink, W. A.; Kesten, S. R.; Rubin, J. R.; Wustrow, D. J.; Zoski, K. T.; Whetzel, S. Z.; Corbin, A. E.; Pugsley, T. A.; Heffner, T. G.; Wise, L. D. Bioorg. Med. Chem. Lett. 1998, 8, 1499–1502. doi:10.1016/S0960-894X(98)00252-2
Return to citation in text: [1] -
Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C. Tetrahedron 2000, 56, 1491–1499. doi:10.1016/S0040-4020(00)00067-3
Return to citation in text: [1] -
Riedinger, C.; Endicott, J. A.; Kemp, S. J.; Smyth, L. A.; Watson, A.; Valeur, E.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Noble, M. E.; McDonnel, J. M. J. Am. Chem. Soc. 2008, 130, 16038–16044. doi:10.1021/ja8062088
Return to citation in text: [1] -
Di Mola, A.; Palombi, L.; Massa, A. Curr. Org. Chem. 2012, 16, 2302–2320. doi:10.2174/138527212803520254
Return to citation in text: [1] [2] [3] -
Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243
Return to citation in text: [1] [2] [3] -
Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140.
Return to citation in text: [1] [2] [3] [4] [5] -
Liu, C.; Zhang, Q.; Li, H.; Guo, S.; Xiao, B.; Deng, W.; Liu, L.; He, W. Chem. – Eur. J. 2016, 22, 6208–6212. doi:10.1002/chem.201600107
Return to citation in text: [1] [2] [3] -
Uemura, S.; Fujita, T.; Sakaguchi, Y.; Kumamoto, E. Biochem. Biophys. Res. Commun. 2012, 418, 695–700. doi:10.1016/j.bbrc.2012.01.080
Return to citation in text: [1] [2] [3] -
Nishiyama, T.; Chiba, S.; Yamada, Y. Eur. J. Pharmacol. 2008, 596, 56–61. doi:10.1016/j.ejphar.2008.07.054
Return to citation in text: [1] [2] [3] -
Kanamitsu, N.; Osaki, T.; Itsuji, Y.; Yoshimura, M.; Tsujimoto, H.; Soga, M. Chem. Pharm. Bull. 2007, 55, 1682–1688. doi:10.1248/cpb.55.1682
Return to citation in text: [1] [2] [3] [4] -
Hussein, Z.; Mulford, D. J.; Bopp, B. A.; Granneman, G. R. Br. J. Clin. Pharmacol. 1993, 36, 357–361. doi:10.1111/j.1365-2125.1993.tb00376.x
Return to citation in text: [1] [2] [3] -
Kondo, T.; Yoshida, K.; Yamamoto, M.; Tanayama, S. Arzneim. Forsch. 1996, 46, 11–14.
Return to citation in text: [1] [2] [3] -
Wada, T.; Nakajima, R.; Kurihara, E.; Narumi, S.; Masuoka, Y.; Goto, G.; Saji, Y.; Fukuda, N. Jpn. J. Pharmacol. 1989, 49, 337–349. doi:10.1254/jjp.49.337
Return to citation in text: [1] [2] [3] -
Lamblin,, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014
and references therein.
Return to citation in text: [1] -
Zhang, L.; Kim, J. B.; Jang, D. O. Tetrahedron Lett. 2014, 55, 2654–2658. doi:10.1016/j.tetlet.2014.03.023
Return to citation in text: [1] -
Lebrun, S.; Sallio, R.; Dubois, M.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Eur. J. Org. Chem. 2015, 1995–2004. doi:10.1002/ejoc.201403573
and references therein.
Return to citation in text: [1] [2] [3] [4] -
Nishimura, M.; Sugawara, N.; Nigorikawa, Y.; Inomiya, N.; Ueda, K.; Ishii, A.; Kanemitsu, N. Synthetic methods for isoindoline intermediate derivatives. Jpn. Pat. JP 2010241770, Oct 28, 2010.
Return to citation in text: [1] -
More, V.; Rohlmann, R.; Mancheño, O. G.; Petronzi, C.; Palombi, L.; De Rosa, A.; Di Mola, A.; Massa, A. RSC Adv. 2012, 2, 3592–3595. doi:10.1039/c2ra20231j
Return to citation in text: [1] -
Fujioka, M.; Morimoto, T.; Tsumagari, T.; Tanimoto, H.; Nishiyama, Y.; Kakiuchi, K. J. Org. Chem. 2012, 77, 2911–2923. doi:10.1021/jo300201g
Return to citation in text: [1] -
Tiso, S.; Palombi, L.; Vignes, C.; Di Mola, A.; Massa, A. RSC Adv. 2013, 3, 19380–19387. doi:10.1039/c3ra43074j
Return to citation in text: [1] -
Zhou, J.-Q.; Sheng, W.-J.; Jia, J.-H.; Ye, Q.; Gao, J.-R.; Jia, Y.-X. Tetrahedron Lett. 2013, 54, 3082–3084. doi:10.1016/j.tetlet.2013.03.138
Return to citation in text: [1] -
Ye, B.; Cramer, N. Angew. Chem., Int. Ed. 2014, 53, 7896–7899. doi:10.1002/anie.201404895
Return to citation in text: [1] -
Bisai, V.; Suneja, A.; Singh, V. K. Angew. Chem., Int. Ed. 2014, 53, 10737–10741. doi:10.1002/anie.201405074
Return to citation in text: [1] [2] -
Di Mola, A.; Tiffner, M.; Scorzelli, F.; Palombi, L.; Filosa, R.; De Caprariis, P.; Waser, M.; Massa, A. Beilstein J. Org. Chem. 2015, 11, 2591–2599. doi:10.3762/bjoc.11.279
Return to citation in text: [1] -
Tiso, S.; Massa, A. J. Heterocycl. Chem. 2015, 52, 1570–1575. doi:10.1002/jhet.2170
Return to citation in text: [1] -
Barrio, P.; Ibáñez, I.; Herrera, L.; Román, R.; Catalán, S.; Fustero, S. Chem. – Eur. J. 2015, 21, 11579–11584. doi:10.1002/chem.201500773
Return to citation in text: [1] -
Scorzelli, F.; Di Mola, A.; Palombi, L.; Massa, A. Molecules 2015, 20, 8484–8498. doi:10.3390/molecules20058484
Return to citation in text: [1] -
Suneja, A.; Bisai, V.; Singh, V. K. J. Org. Chem. 2016, 81, 4779–4788. doi:10.1021/acs.joc.6b00770
Return to citation in text: [1] -
Capobianco, A.; Di Mola, A.; Intintoli, V.; Massa, A.; Capaccio, V.; Roiser, L.; Waser, M.; Palombi, L. RSC Adv. 2016, 6, 31861–31870. doi:10.1039/C6RA05488A
Return to citation in text: [1] -
Sallio, R.; Lebrun, S.; Schifano-Faux, N.; Goossens, J. F.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Synlett 2013, 24, 1785–1790. doi:10.1055/s-0033-1339487
Return to citation in text: [1] [2] [3] -
Xu, L.-W.; Xia, C.-G. Eur. J. Org. Chem. 2005, 633–639. doi:10.1002/ejoc.200400619
Return to citation in text: [1] -
Krishna, P. R.; Sreeshailam, A.; Srinivas, R. Tetrahedron 2009, 65, 9657–9672. doi:10.1016/j.tet.2009.08.021
Return to citation in text: [1] -
Enders, D.; Wang, C.; Liebich, J. X. Chem. – Eur. J. 2009, 15, 11058–11076. doi:10.1002/chem.200902236
Return to citation in text: [1] -
Wang, J.; Li, P.; Choy, P. Y.; Chan, A. S. C.; Kwong, F. Y. ChemCatChem 2012, 4, 917–925. doi:10.1002/cctc.201200135
Return to citation in text: [1] -
Mahé, O.; Dez, I.; Levacher, V.; Brière, J.-F. Org. Biomol. Chem. 2012, 10, 3946–3954. doi:10.1039/c2ob25227a
Return to citation in text: [1] -
Lee, H.-J.; Cho, C.-W. J. Org. Chem. 2015, 80, 11435–11440. doi:10.1021/acs.joc.5b02124
Return to citation in text: [1] -
Wang, L.; Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 5327–5330. doi:10.1002/anie.201101307
Return to citation in text: [1] -
Lee, S.-J.; Bae, J.-Y.; Cho, C.-W. Eur. J. Org. Chem. 2015, 6495–6502. doi:10.1002/ejoc.201500940
Return to citation in text: [1] -
Weiß, M.; Borchert, S.; Rémond, E.; Jugé, S.; Gröger, H. Heteroat. Chem. 2012, 23, 202–209. doi:10.1002/hc.21004
Return to citation in text: [1] -
Guo, J.; Yu, S. Org. Biomol. Chem. 2015, 13, 1179–1186. doi:10.1039/C4OB02227K
Return to citation in text: [1] -
Bandini, M.; Bottoni, A.; Eichholzer, A.; Miscione, G. P.; Stenta, M. Chem. – Eur. J. 2010, 16, 12462–12473. doi:10.1002/chem.201000560
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A.; Tragni, M.; Umani-Ronchi, A. Angew. Chem., Int. Ed. 2008, 47, 3238–3241. doi:10.1002/anie.200705685
Return to citation in text: [1] -
Davies, S. G.; Garner, A. C.; Goddard, E. C.; Kruchinin, D.; Roberts, P. M.; Rodriguez-Solla, H.; Smith, A. D. Chem. Commun. 2006, 2664–2666. doi:10.1039/b604835h
Return to citation in text: [1] -
Davies, S. G.; Fletcher, A. M.; Hermann, G. J.; Poce, G.; Roberts, P. M.; Smith, A. D.; Sweet, M. J.; Thomson, J. E. Tetrahedron: Asymmetry 2010, 21, 1635–1648. doi:10.1016/j.tetasy.2010.03.033
Return to citation in text: [1] -
Davies, S. G.; Hughes, D. G.; Price, P. D.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E.; Williams, O. M. H. Synlett 2010, 567–570. doi:10.1055/s-0029-1219346
Return to citation in text: [1] -
Davies, S. G.; Lee, J. A.; Roberts, P. M.; Stonehouse, J. P.; Thomson, J. E. Tetrahedron Lett. 2012, 53, 1119–1121. doi:10.1016/j.tetlet.2011.12.088
Return to citation in text: [1] -
Davies, S. G.; Huckvale, R.; Lee, J. A.; Lorkin, T. J. A.; Roberts, P. M.; Thomson, J. E. Tetrahedron 2012, 68, 3263–3275. doi:10.1016/j.tet.2011.12.084
Return to citation in text: [1] -
Jung, M. E. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I.; Semmelhack, M. F., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 30–41.
and references therein.
Return to citation in text: [1] -
Rulev, A. Y. Russ. Chem. Rev. 2011, 80, 197–218. doi:10.1070/RC2011v080n03ABEH004162
Return to citation in text: [1] -
De, K.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. J. Org. Chem. 2009, 74, 6260–6265. doi:10.1021/jo9012699
Return to citation in text: [1] -
Wang, J.; Li, P.-F.; Chan, S. H.; Chan, A. S. C.; Kwong, F. Y. Tetrahedron Lett. 2012, 53, 2887–2889. doi:10.1016/j.tetlet.2012.03.132
Return to citation in text: [1] -
Amara, Z.; Drège, E.; Troufflard, C.; Retailleau, P.; Joseph, D. Org. Biomol. Chem. 2012, 10, 7148–7157. doi:10.1039/c2ob25963j
Return to citation in text: [1] -
Medina, F.; Michon, C.; Agbossou-Niedercorn, F. Eur. J. Org. Chem. 2012, 6218–6227. doi:10.1002/ejoc.201200891
Return to citation in text: [1] -
Medina, F.; Duhal, N.; Michon, C.; Agbossou-Niedercorn, F. C. R. Chim. 2013, 16, 311–317. doi:10.1016/j.crci.2012.11.001
Return to citation in text: [1] -
Bosica, G.; Debono, A. J. Tetrahedron 2014, 70, 6607–6612. doi:10.1016/j.tet.2014.06.124
Return to citation in text: [1] -
March, J. Advanced Organic Chemistry: Reactions, Mechanisms and Structures, 4th ed.; Wiley-VCH: New-York, USA, 1992; 795, 1027.
Return to citation in text: [1] -
Cai, Y.-F.; Li, L.; Luo, M.-X.; Yang, K.-F.; Lai, G.-Q.; Jiang, J.-X.; Xu, L.-W. Chirality 2011, 23, 397–403. doi:10.1002/chir.20940
Return to citation in text: [1] -
Kolodiazhnyi, O. I. Tetrahedron 2003, 59, 5953–6018. doi:10.1016/S0040-4020(03)00911-6
Return to citation in text: [1] [2] -
Gelat, F.; Lebrun, S.; Henry, N.; Agbossou-Niedercorn, F.; Michon, C.; Deniau, E. Synlett 2017, 28, 225–230. doi:10.1055/s-0036-1588895
Return to citation in text: [1] -
Gelat, F.; Coffinet, M.; Lebrun, S.; Agbossou-Niedercorn, F.; Michon, C.; Deniau, E. Tetrahedron: Asymmetry 2016, 27, 980–989. doi:10.1016/j.tetasy.2016.07.010
Return to citation in text: [1] -
Munive, L.; Dzakuma, S. A.; Olivo, H. F. Tetrahedron Lett. 2013, 54, 1230–1232. doi:10.1016/j.tetlet.2012.12.074
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561
Return to citation in text: [1] -
Masamune, S.; Choy, W.; Petersen, J. S.; Sita, L. R. Angew. Chem., Int. Ed. Engl. 1985, 24, 1–30. doi:10.1002/anie.198500013
Return to citation in text: [1] -
CCDC 1590565: these data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1]
| 68. | CCDC 1590565: these data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 65. | Munive, L.; Dzakuma, S. A.; Olivo, H. F. Tetrahedron Lett. 2013, 54, 1230–1232. doi:10.1016/j.tetlet.2012.12.074 |
| 66. | Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561 |
| 67. | Masamune, S.; Choy, W.; Petersen, J. S.; Sita, L. R. Angew. Chem., Int. Ed. Engl. 1985, 24, 1–30. doi:10.1002/anie.198500013 |
| 62. | Kolodiazhnyi, O. I. Tetrahedron 2003, 59, 5953–6018. doi:10.1016/S0040-4020(03)00911-6 |
| 1. | Wrobel, J.; Dietrich, A.; Woolson, S. A.; Millen, J.; McCaleb, M.; Harrison, M. C.; Hohman, T. C.; Sredy, J.; Sullivan, D. J. Med. Chem. 1992, 35, 4613–4627. doi:10.1021/jm00102a016 |
| 2. | Pigeon, P.; Decroix, B. Tetrahedron Lett. 1996, 37, 7707–7710. doi:10.1016/0040-4039(96)01738-8 |
| 3. | Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7 |
| 4. | Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C. J. Org. Chem. 1998, 63, 3128–3132. doi:10.1021/jo972247t |
| 5. | Belliotti, T. R.; Brink, W. A.; Kesten, S. R.; Rubin, J. R.; Wustrow, D. J.; Zoski, K. T.; Whetzel, S. Z.; Corbin, A. E.; Pugsley, T. A.; Heffner, T. G.; Wise, L. D. Bioorg. Med. Chem. Lett. 1998, 8, 1499–1502. doi:10.1016/S0960-894X(98)00252-2 |
| 6. | Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C. Tetrahedron 2000, 56, 1491–1499. doi:10.1016/S0040-4020(00)00067-3 |
| 7. | Riedinger, C.; Endicott, J. A.; Kemp, S. J.; Smyth, L. A.; Watson, A.; Valeur, E.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Noble, M. E.; McDonnel, J. M. J. Am. Chem. Soc. 2008, 130, 16038–16044. doi:10.1021/ja8062088 |
| 8. | Di Mola, A.; Palombi, L.; Massa, A. Curr. Org. Chem. 2012, 16, 2302–2320. doi:10.2174/138527212803520254 |
| 9. | Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243 |
| 10. | Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140. |
| 11. | Liu, C.; Zhang, Q.; Li, H.; Guo, S.; Xiao, B.; Deng, W.; Liu, L.; He, W. Chem. – Eur. J. 2016, 22, 6208–6212. doi:10.1002/chem.201600107 |
| 14. | Kanamitsu, N.; Osaki, T.; Itsuji, Y.; Yoshimura, M.; Tsujimoto, H.; Soga, M. Chem. Pharm. Bull. 2007, 55, 1682–1688. doi:10.1248/cpb.55.1682 |
| 61. | Cai, Y.-F.; Li, L.; Luo, M.-X.; Yang, K.-F.; Lai, G.-Q.; Jiang, J.-X.; Xu, L.-W. Chirality 2011, 23, 397–403. doi:10.1002/chir.20940 |
| 62. | Kolodiazhnyi, O. I. Tetrahedron 2003, 59, 5953–6018. doi:10.1016/S0040-4020(03)00911-6 |
| 17. | Wada, T.; Nakajima, R.; Kurihara, E.; Narumi, S.; Masuoka, Y.; Goto, G.; Saji, Y.; Fukuda, N. Jpn. J. Pharmacol. 1989, 49, 337–349. doi:10.1254/jjp.49.337 |
| 63. | Gelat, F.; Lebrun, S.; Henry, N.; Agbossou-Niedercorn, F.; Michon, C.; Deniau, E. Synlett 2017, 28, 225–230. doi:10.1055/s-0036-1588895 |
| 64. | Gelat, F.; Coffinet, M.; Lebrun, S.; Agbossou-Niedercorn, F.; Michon, C.; Deniau, E. Tetrahedron: Asymmetry 2016, 27, 980–989. doi:10.1016/j.tetasy.2016.07.010 |
| 65. | Munive, L.; Dzakuma, S. A.; Olivo, H. F. Tetrahedron Lett. 2013, 54, 1230–1232. doi:10.1016/j.tetlet.2012.12.074 |
| 15. | Hussein, Z.; Mulford, D. J.; Bopp, B. A.; Granneman, G. R. Br. J. Clin. Pharmacol. 1993, 36, 357–361. doi:10.1111/j.1365-2125.1993.tb00376.x |
| 16. | Kondo, T.; Yoshida, K.; Yamamoto, M.; Tanayama, S. Arzneim. Forsch. 1996, 46, 11–14. |
| 34. | Sallio, R.; Lebrun, S.; Schifano-Faux, N.; Goossens, J. F.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Synlett 2013, 24, 1785–1790. doi:10.1055/s-0033-1339487 |
| 47. | Davies, S. G.; Garner, A. C.; Goddard, E. C.; Kruchinin, D.; Roberts, P. M.; Rodriguez-Solla, H.; Smith, A. D. Chem. Commun. 2006, 2664–2666. doi:10.1039/b604835h |
| 48. | Davies, S. G.; Fletcher, A. M.; Hermann, G. J.; Poce, G.; Roberts, P. M.; Smith, A. D.; Sweet, M. J.; Thomson, J. E. Tetrahedron: Asymmetry 2010, 21, 1635–1648. doi:10.1016/j.tetasy.2010.03.033 |
| 49. | Davies, S. G.; Hughes, D. G.; Price, P. D.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E.; Williams, O. M. H. Synlett 2010, 567–570. doi:10.1055/s-0029-1219346 |
| 50. | Davies, S. G.; Lee, J. A.; Roberts, P. M.; Stonehouse, J. P.; Thomson, J. E. Tetrahedron Lett. 2012, 53, 1119–1121. doi:10.1016/j.tetlet.2011.12.088 |
| 51. | Davies, S. G.; Huckvale, R.; Lee, J. A.; Lorkin, T. J. A.; Roberts, P. M.; Thomson, J. E. Tetrahedron 2012, 68, 3263–3275. doi:10.1016/j.tet.2011.12.084 |
| 12. | Uemura, S.; Fujita, T.; Sakaguchi, Y.; Kumamoto, E. Biochem. Biophys. Res. Commun. 2012, 418, 695–700. doi:10.1016/j.bbrc.2012.01.080 |
| 13. | Nishiyama, T.; Chiba, S.; Yamada, Y. Eur. J. Pharmacol. 2008, 596, 56–61. doi:10.1016/j.ejphar.2008.07.054 |
| 14. | Kanamitsu, N.; Osaki, T.; Itsuji, Y.; Yoshimura, M.; Tsujimoto, H.; Soga, M. Chem. Pharm. Bull. 2007, 55, 1682–1688. doi:10.1248/cpb.55.1682 |
| 52. |
Jung, M. E. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I.; Semmelhack, M. F., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 30–41.
and references therein. |
| 53. | Rulev, A. Y. Russ. Chem. Rev. 2011, 80, 197–218. doi:10.1070/RC2011v080n03ABEH004162 |
| 54. | De, K.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. J. Org. Chem. 2009, 74, 6260–6265. doi:10.1021/jo9012699 |
| 55. | Wang, J.; Li, P.-F.; Chan, S. H.; Chan, A. S. C.; Kwong, F. Y. Tetrahedron Lett. 2012, 53, 2887–2889. doi:10.1016/j.tetlet.2012.03.132 |
| 56. | Amara, Z.; Drège, E.; Troufflard, C.; Retailleau, P.; Joseph, D. Org. Biomol. Chem. 2012, 10, 7148–7157. doi:10.1039/c2ob25963j |
| 57. | Medina, F.; Michon, C.; Agbossou-Niedercorn, F. Eur. J. Org. Chem. 2012, 6218–6227. doi:10.1002/ejoc.201200891 |
| 58. | Medina, F.; Duhal, N.; Michon, C.; Agbossou-Niedercorn, F. C. R. Chim. 2013, 16, 311–317. doi:10.1016/j.crci.2012.11.001 |
| 59. | Bosica, G.; Debono, A. J. Tetrahedron 2014, 70, 6607–6612. doi:10.1016/j.tet.2014.06.124 |
| 60. | March, J. Advanced Organic Chemistry: Reactions, Mechanisms and Structures, 4th ed.; Wiley-VCH: New-York, USA, 1992; 795, 1027. |
| 39. | Mahé, O.; Dez, I.; Levacher, V.; Brière, J.-F. Org. Biomol. Chem. 2012, 10, 3946–3954. doi:10.1039/c2ob25227a |
| 40. | Lee, H.-J.; Cho, C.-W. J. Org. Chem. 2015, 80, 11435–11440. doi:10.1021/acs.joc.5b02124 |
| 41. | Wang, L.; Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 5327–5330. doi:10.1002/anie.201101307 |
| 42. | Lee, S.-J.; Bae, J.-Y.; Cho, C.-W. Eur. J. Org. Chem. 2015, 6495–6502. doi:10.1002/ejoc.201500940 |
| 43. | Weiß, M.; Borchert, S.; Rémond, E.; Jugé, S.; Gröger, H. Heteroat. Chem. 2012, 23, 202–209. doi:10.1002/hc.21004 |
| 20. |
Lebrun, S.; Sallio, R.; Dubois, M.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Eur. J. Org. Chem. 2015, 1995–2004. doi:10.1002/ejoc.201403573
and references therein. |
| 34. | Sallio, R.; Lebrun, S.; Schifano-Faux, N.; Goossens, J. F.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Synlett 2013, 24, 1785–1790. doi:10.1055/s-0033-1339487 |
| 8. | Di Mola, A.; Palombi, L.; Massa, A. Curr. Org. Chem. 2012, 16, 2302–2320. doi:10.2174/138527212803520254 |
| 9. | Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243 |
| 10. | Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140. |
| 11. | Liu, C.; Zhang, Q.; Li, H.; Guo, S.; Xiao, B.; Deng, W.; Liu, L.; He, W. Chem. – Eur. J. 2016, 22, 6208–6212. doi:10.1002/chem.201600107 |
| 12. | Uemura, S.; Fujita, T.; Sakaguchi, Y.; Kumamoto, E. Biochem. Biophys. Res. Commun. 2012, 418, 695–700. doi:10.1016/j.bbrc.2012.01.080 |
| 13. | Nishiyama, T.; Chiba, S.; Yamada, Y. Eur. J. Pharmacol. 2008, 596, 56–61. doi:10.1016/j.ejphar.2008.07.054 |
| 14. | Kanamitsu, N.; Osaki, T.; Itsuji, Y.; Yoshimura, M.; Tsujimoto, H.; Soga, M. Chem. Pharm. Bull. 2007, 55, 1682–1688. doi:10.1248/cpb.55.1682 |
| 15. | Hussein, Z.; Mulford, D. J.; Bopp, B. A.; Granneman, G. R. Br. J. Clin. Pharmacol. 1993, 36, 357–361. doi:10.1111/j.1365-2125.1993.tb00376.x |
| 16. | Kondo, T.; Yoshida, K.; Yamamoto, M.; Tanayama, S. Arzneim. Forsch. 1996, 46, 11–14. |
| 35. | Xu, L.-W.; Xia, C.-G. Eur. J. Org. Chem. 2005, 633–639. doi:10.1002/ejoc.200400619 |
| 36. | Krishna, P. R.; Sreeshailam, A.; Srinivas, R. Tetrahedron 2009, 65, 9657–9672. doi:10.1016/j.tet.2009.08.021 |
| 37. | Enders, D.; Wang, C.; Liebich, J. X. Chem. – Eur. J. 2009, 15, 11058–11076. doi:10.1002/chem.200902236 |
| 38. | Wang, J.; Li, P.; Choy, P. Y.; Chan, A. S. C.; Kwong, F. Y. ChemCatChem 2012, 4, 917–925. doi:10.1002/cctc.201200135 |
| 20. |
Lebrun, S.; Sallio, R.; Dubois, M.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Eur. J. Org. Chem. 2015, 1995–2004. doi:10.1002/ejoc.201403573
and references therein. |
| 27. | Bisai, V.; Suneja, A.; Singh, V. K. Angew. Chem., Int. Ed. 2014, 53, 10737–10741. doi:10.1002/anie.201405074 |
| 10. | Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140. |
| 20. |
Lebrun, S.; Sallio, R.; Dubois, M.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Eur. J. Org. Chem. 2015, 1995–2004. doi:10.1002/ejoc.201403573
and references therein. |
| 21. | Nishimura, M.; Sugawara, N.; Nigorikawa, Y.; Inomiya, N.; Ueda, K.; Ishii, A.; Kanemitsu, N. Synthetic methods for isoindoline intermediate derivatives. Jpn. Pat. JP 2010241770, Oct 28, 2010. |
| 22. | More, V.; Rohlmann, R.; Mancheño, O. G.; Petronzi, C.; Palombi, L.; De Rosa, A.; Di Mola, A.; Massa, A. RSC Adv. 2012, 2, 3592–3595. doi:10.1039/c2ra20231j |
| 23. | Fujioka, M.; Morimoto, T.; Tsumagari, T.; Tanimoto, H.; Nishiyama, Y.; Kakiuchi, K. J. Org. Chem. 2012, 77, 2911–2923. doi:10.1021/jo300201g |
| 24. | Tiso, S.; Palombi, L.; Vignes, C.; Di Mola, A.; Massa, A. RSC Adv. 2013, 3, 19380–19387. doi:10.1039/c3ra43074j |
| 25. | Zhou, J.-Q.; Sheng, W.-J.; Jia, J.-H.; Ye, Q.; Gao, J.-R.; Jia, Y.-X. Tetrahedron Lett. 2013, 54, 3082–3084. doi:10.1016/j.tetlet.2013.03.138 |
| 26. | Ye, B.; Cramer, N. Angew. Chem., Int. Ed. 2014, 53, 7896–7899. doi:10.1002/anie.201404895 |
| 27. | Bisai, V.; Suneja, A.; Singh, V. K. Angew. Chem., Int. Ed. 2014, 53, 10737–10741. doi:10.1002/anie.201405074 |
| 28. | Di Mola, A.; Tiffner, M.; Scorzelli, F.; Palombi, L.; Filosa, R.; De Caprariis, P.; Waser, M.; Massa, A. Beilstein J. Org. Chem. 2015, 11, 2591–2599. doi:10.3762/bjoc.11.279 |
| 29. | Tiso, S.; Massa, A. J. Heterocycl. Chem. 2015, 52, 1570–1575. doi:10.1002/jhet.2170 |
| 30. | Barrio, P.; Ibáñez, I.; Herrera, L.; Román, R.; Catalán, S.; Fustero, S. Chem. – Eur. J. 2015, 21, 11579–11584. doi:10.1002/chem.201500773 |
| 31. | Scorzelli, F.; Di Mola, A.; Palombi, L.; Massa, A. Molecules 2015, 20, 8484–8498. doi:10.3390/molecules20058484 |
| 32. | Suneja, A.; Bisai, V.; Singh, V. K. J. Org. Chem. 2016, 81, 4779–4788. doi:10.1021/acs.joc.6b00770 |
| 33. | Capobianco, A.; Di Mola, A.; Intintoli, V.; Massa, A.; Capaccio, V.; Roiser, L.; Waser, M.; Palombi, L. RSC Adv. 2016, 6, 31861–31870. doi:10.1039/C6RA05488A |
| 34. | Sallio, R.; Lebrun, S.; Schifano-Faux, N.; Goossens, J. F.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Synlett 2013, 24, 1785–1790. doi:10.1055/s-0033-1339487 |
| 8. | Di Mola, A.; Palombi, L.; Massa, A. Curr. Org. Chem. 2012, 16, 2302–2320. doi:10.2174/138527212803520254 |
| 9. | Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243 |
| 10. | Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140. |
| 11. | Liu, C.; Zhang, Q.; Li, H.; Guo, S.; Xiao, B.; Deng, W.; Liu, L.; He, W. Chem. – Eur. J. 2016, 22, 6208–6212. doi:10.1002/chem.201600107 |
| 12. | Uemura, S.; Fujita, T.; Sakaguchi, Y.; Kumamoto, E. Biochem. Biophys. Res. Commun. 2012, 418, 695–700. doi:10.1016/j.bbrc.2012.01.080 |
| 13. | Nishiyama, T.; Chiba, S.; Yamada, Y. Eur. J. Pharmacol. 2008, 596, 56–61. doi:10.1016/j.ejphar.2008.07.054 |
| 14. | Kanamitsu, N.; Osaki, T.; Itsuji, Y.; Yoshimura, M.; Tsujimoto, H.; Soga, M. Chem. Pharm. Bull. 2007, 55, 1682–1688. doi:10.1248/cpb.55.1682 |
| 15. | Hussein, Z.; Mulford, D. J.; Bopp, B. A.; Granneman, G. R. Br. J. Clin. Pharmacol. 1993, 36, 357–361. doi:10.1111/j.1365-2125.1993.tb00376.x |
| 16. | Kondo, T.; Yoshida, K.; Yamamoto, M.; Tanayama, S. Arzneim. Forsch. 1996, 46, 11–14. |
| 17. | Wada, T.; Nakajima, R.; Kurihara, E.; Narumi, S.; Masuoka, Y.; Goto, G.; Saji, Y.; Fukuda, N. Jpn. J. Pharmacol. 1989, 49, 337–349. doi:10.1254/jjp.49.337 |
| 10. | Di Mola, A.; Palombi, L.; Massa, A. Targets Heterocycl. Syst. 2014, 18, 113–140. |
| 18. |
Lamblin,, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014
and references therein. |
| 19. | Zhang, L.; Kim, J. B.; Jang, D. O. Tetrahedron Lett. 2014, 55, 2654–2658. doi:10.1016/j.tetlet.2014.03.023 |
| 20. |
Lebrun, S.; Sallio, R.; Dubois, M.; Agbossou-Niedercorn, F.; Deniau, E.; Michon, C. Eur. J. Org. Chem. 2015, 1995–2004. doi:10.1002/ejoc.201403573
and references therein. |
| 44. | Guo, J.; Yu, S. Org. Biomol. Chem. 2015, 13, 1179–1186. doi:10.1039/C4OB02227K |
| 45. | Bandini, M.; Bottoni, A.; Eichholzer, A.; Miscione, G. P.; Stenta, M. Chem. – Eur. J. 2010, 16, 12462–12473. doi:10.1002/chem.201000560 |
| 46. | Bandini, M.; Eichholzer, A.; Tragni, M.; Umani-Ronchi, A. Angew. Chem., Int. Ed. 2008, 47, 3238–3241. doi:10.1002/anie.200705685 |
| 17. | Wada, T.; Nakajima, R.; Kurihara, E.; Narumi, S.; Masuoka, Y.; Goto, G.; Saji, Y.; Fukuda, N. Jpn. J. Pharmacol. 1989, 49, 337–349. doi:10.1254/jjp.49.337 |
© 2018 Sallio et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)