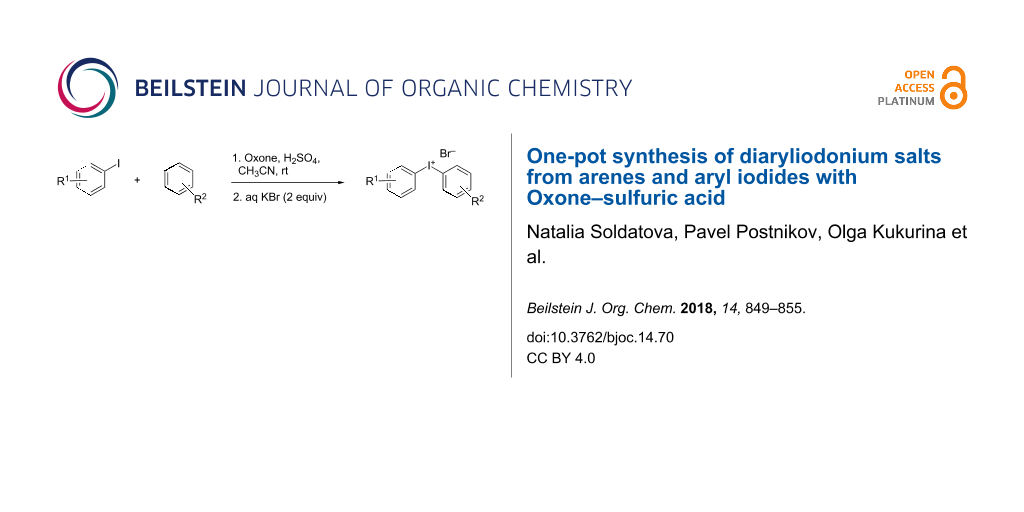
Abstract
A facile synthesis of diaryliodonium salts utilizing Oxone as versatile and cheap oxidant has been developed. This method shows wide applicability and can be used for the preparation of iodonium salts containing electron-donating or electron-withdrawing groups in good yields. In addition, this procedure can be applied to the preparation of symmetric iodonium salts directly from arenes via a one-pot iodination–oxidation sequence.

Graphical Abstract
Introduction
Diaryliodonium salts, which are also known as diaryl-λ3-iodanes, are widely considered to be an important and practically useful class of hypervalent iodine compounds [1-4]. Diaryliodonium salts have found broad synthetic application as electrophilic arylating reagents in reactions with various nucleophiles including electron-rich carbon-centered species [5-7]. The unique arylating reactivity of diaryliodonium salts has been demonstrated in many metal-catalyzed and also metal-free transformations [8-15].
The development of novel synthetic approaches to diaryliodonium salts based on the use of inexpensive, commercially available oxidants is an important and challenging goal. A vast majority of existing procedures involve the interaction of electrophilic hypervalent iodine(III) species with suitable arenes through ligand exchange processes [16-20]. The reactive hypervalent iodine(III) species can be used as stable reagents or can be generated in situ [21-25]. In particular, Olofsson and co-workers reported procedures based on the in situ generation of reactive λ3-iodane species directly from arenes, which was a significant achievement in this field [26-29]. However, these now well-established processes involve oxidations using mCPBA in the presence of strong organic acids [30-35]. Therefore, the development of new, convenient and inexpensive methods utilizing readily available and easy-to-handle oxidants still remains a highly desirable goal.
Previously, we have published the utilization of Oxone® (2KHSO5·KHSO4·K2SO4) as a readily available and effective oxidant for the preparation of various hypervalent iodine compounds [36-42]. Oxone is used as an efficient oxidant for the direct conversion of substituted 2-iodobenzoic acids to arylbenziodoxoles [37,38], 2-iodobiphenyl to dibenziodolium compounds [39], iodoarenes to iodylbenzenes and [bis(trifluoroacetoxy)iodo]arenes [40,41], and for the preparation of diaryliodonium trifluoroacetates and triflates [42]. Yakura also used Oxone® as an oxidant for the generation of iodine(III) species in the oxidation of phenols [43]. In the present work, we report the development of a reliable and convenient procedure for the preparation of diaryliodonium bromides using Oxone in the presence of sulfuric acid.
Results and Discussion
After having investigated previously described reaction conditions [37,38], initial optimization studies were performed using iodobenzene and toluene as reactants for the synthesis of diaryliodonium salt 3a (Table 1). A simple mixing of the starting materials with finely ground Oxone and sulfuric acid leads to the formation of a very viscous and dark reaction mixture (Table 1, entry 1). Full conversion of the starting materials could not be achieved even after 24 h stirring. The addition of aqueous KBr to the reaction mixture resulted in the formation of the desired bromide salt 3a in 30% isolated yield. The addition of KBr is necessary as the high solubility of the diaryliodonium sulfonates does not allow their isolation from the reaction mixture. Dilution of the reaction mixture with dichloromethane did not increase the yield of the target product significantly (Table 1, entry 2). Moreover, we observed the formation of 4-iodobenzenesulfonic acid in both cases, probably due to the high concentration of sulfuric acid in the reaction mixture. Mixing dichloromethane with acetonitrile resulted in an increased yield of 3a and a decreased amount of 4-iodobenzenesulfonic acid (Table 1, entry 3). When the reaction was carried out in pure acetonitrile, the iodonium salt 3a was formed in 75% yield (Table 1, entry 4). The addition of 2 equivalents of Oxone increased the yield to 92% (Table 1, entry 5). Surprisingly, a smaller amount of toluene did not affect the yield (Table 1, entry 6). In order to avoid the formation of undesired 4-iodobenzenesulfonic acid, the reaction was carried out using smaller amounts of acid (Table 1, entries 7 and 8). That reaction proceeds smoothly with only 7.5 equivalents of sulfuric acid producing the target compound 3a high yields. However, the use of 3.75 equivalents of sulfuric acid resulted in a significantly lower yield.
Table 1: Optimization studies.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-70-i1.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | toluene (equiv) | Oxone (equiv) | H2SO4 (equiv) | yieldb (%) |
|---|---|---|---|---|---|
| 1 | – | 2.8 | 1.3 | 15 | 30c |
| 2 | CH2Cl2 | 2.8 | 1.3 | 15 | 40c |
| 3 | CH2Cl2/MeCN | 2.8 | 1.3 | 15 | 60c |
| 4 | MeCN | 2.8 | 1.3 | 15 | 75c |
| 5 | MeCN | 2.8 | 2 | 15 | 92c |
| 6 | MeCN | 1.2 | 2 | 15 | 88c |
| 7 | MeCN | 1.2 | 2 | 7.5 | 86 |
| 8 | MeCN | 1.2 | 2 | 3.75 | 53 |
aReaction conditions: PhI (1 mmol), overnight, rt; bisolated yield; caccording to NMR data product contains up to 10% of (4-tolyl)phenyliodonium 4-iodobenzenesulfonate as an impurity.
With the optimized procedure, the synthetic utility of this method using various aryliodides and arenes was investigated (Table 2). Iodobenzene (1a) smoothly reacts with arenes containing electron-donating substituents to form the corresponding iodonium salts in high yields. 3-Trifluoromethyliodobenzene (1b) exhibited higher reactivity, and iodonium salts have been isolated in higher yields. Moreover, iodoarene 1b reacted with the moderately electron-poor chlorobenzene (2e) forming iodonium salt 3i in 51% yield. In contrast, 4-bromoiodobenzene (1c) was less reactive and afforded iodonium salts 3j–m in lower yields. Similar reactions of 3,5-bis(trifluoromethyl)iodobenzene (1d) with benzene and mesitylene formed the corresponding iodonium salts 3n and 3o in moderate yields.
Table 2: Synthesis of diaryliodonium bromides.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-70-i2.svg?max-width=637&scale=1.0)
|
|||
| R1 (1) | R2 (2) | product 3 | yieldb (%) |
|---|---|---|---|
| R1 = H (1a) | R2 = Me (2a) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-70-i3.svg?max-width=637&scale=1.0)
3a |
86c |
| R1 = H (1a) | R2 = H (2b) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-70-i4.svg?max-width=637&scale=1.0)
3b |
74d,e,f |
| R1 = H (1a) | R2 = 1,4-Me2 (2c) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-70-i5.svg?max-width=637&scale=1.0)
3c |
75 |
| R1 = H (1a) | R2 = 1,3,5-Me3 (2d) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-70-i6.svg?max-width=637&scale=1.0)
3d |
75 |
| R1 = 3-CF3 (1b) | R2 = H (2b) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-70-i7.svg?max-width=637&scale=1.0)
3e |
85e,f |
| R1 = 3-CF3 (1b) | R2 = Me (2a) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-70-i8.svg?max-width=637&scale=1.0)
3f |
76c |
| R1 = 3-CF3 (1b) | R2 = 1,4-Me2 (2c) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-70-i9.svg?max-width=637&scale=1.0)
3g |
95 |
| R1 = 3-CF3 (1b) | R2 = 1,3,5-Me3 (2d) |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-70-i10.svg?max-width=637&scale=1.0)
3h |
92 |
| R1 = 3-CF3 (1b) | R2 = Cl (2e) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-70-i11.svg?max-width=637&scale=1.0)
3i |
51g,h |
| R1 = 4-Br (1c) | R2 = Me (2a) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-70-i12.svg?max-width=637&scale=1.0)
3j |
70c |
| R1 = 4-Br (1c) | R2 = 1,4-Me2 (2c) |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-70-i13.svg?max-width=637&scale=1.0)
3k |
32 |
| R1 = 4-Br (1c) | R2 = 1,3,5-Me3 (2d) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-70-i14.svg?max-width=637&scale=1.0)
3l |
71 |
| R1 = 4-Br (1c) | R2 = Cl (2e) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-70-i15.svg?max-width=637&scale=1.0)
3m |
34g,h |
| R1 = 3,5-(CF3)2 (1d) | R2 = H (2b) |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-70-i16.svg?max-width=637&scale=1.0)
3n |
35e,f |
| R1 = 3,5-(CF3)2 (1d) | R2 = 1,3,5-Me3 (2d) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-70-i17.svg?max-width=637&scale=1.0)
3o |
62 |
aReaction conditions: 1 (1 mmol), 2 (1.1 mmol), H2SO4 (7.5 mmol), MeCN (2 mL) overnight, rt; bisolated yield; c1.2 mmol of 2a was used; daccording to NMR data product contains an up to 1.5% of diphenyliodonium 4-iodobenzenesulfonate as impurity; e11.3 mmol of H2SO4 was used; f1.3 mmol of 2b was used; g15 mmol of H2SO4 was used; h1.5 mmol of 2e was used.
With electron-deficient arenes 2 such as chlorobenzene (2e) and benzene, an excess of sulfuric acid and arene was used to improve the yields. Subsequently it was shown that the addition of aqueous potassium bromide can be modified and other counter anions can be introduced to prepare different diaryliodonium salts. This has been demonstrated in the preparation of diaryliodonium salts using 1-iodo-3-trifluoromethylbenzene (1b) and mesitylene (2d) as model substrates (Table 3).
Table 3: Preparation of diaryliodonium salts with different anions.a
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-70-i18.svg?max-width=637&scale=1.0)
|
||
| NaX / HX | product | yieldb (%) |
|---|---|---|
| NaBr | 3h | 92 |
| TsOH | 3p | 82 |
| TfOH | 3q | 89 |
| NaBF4 | 3r | 81 |
| NaPF6 | 3s | 80 |
aReaction conditions: 1b (1 mmol), 2d (1.1 mmol), Oxone (1 mmol), H2SO4 (7.5 mmol), MeCN (2 mL), NaX or HX (2 mmol) overnight, rt; bisolated yield.
The yield of the salts 3h and 3p–s do not depend on the nature of the anion and its source. Small differences in yield can be explained by different solubility of salts in acetonitrile/water. Diaryliodonium bromides were isolated in higher yields because of the low solubility of these products (Table 3).
Finally, a one-step procedure for the preparation of symmetric iodonium salts directly from arenes via an in situ iodination was developed (Table 4). Arenes 2b–e can be transformed to the symmetric iodonium salts 3b and 3t–v by the reaction with iodine, Oxone, and sulfuric acid. The attempted synthesis of the symmetric iodonium salt using toluene as substrate led to a regioisomeric mixture of products due to the low regioselectivity of iodination.
Table 4: Preparation of symmetric iodonium salts via a one-step iodination–oxidation procedure.a
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-70-i19.svg?max-width=637&scale=1.0)
|
||
| R2 (2) | product 3 | yieldb (%) |
|---|---|---|
| R2 = H (2b) |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-70-i20.svg?max-width=637&scale=1.0)
3b |
84c,d |
| R2 = 1,4-Me2 (2c) |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-70-i21.svg?max-width=637&scale=1.0)
3t |
80 |
| R2 = 1,3,5-Me3 (2d) |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-70-i22.svg?max-width=637&scale=1.0)
3u |
77 |
| R2 = Cl (2e) |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-70-i23.svg?max-width=637&scale=1.0)
3v |
40e |
aReaction conditions: ArH (2.2 mmol), Oxone (2 mmol), H2SO4 (7.5 mmol), MeCN (2 mL) overnight, rt; bisolated yield; caccording to NMR data the product contains up to 1.5% of diphenyliodonium 4-iodobenzenesulfonate as impurity; d11.3 mmol of H2SO4 and 2.6 mmol of 2b were used; e15 mmol of H2SO4 and 3.0 mmol of 2e were used.
This procedure allowed the synthesis of iodonium salts with arenes containing electron-donating groups. Unfortunately, electron-poor arenes exhibited a lower reactivity and bis-(4-chlorophenyl)iodonium bromide have been isolated in only 40% yield. Nevertheless, the developed procedure is characterized by important advantages, such as simplicity, the use of inexpensive and available reagents, and typically good yields of iodonium salts. It is a versatile addition to the methodology toolbox for the preparation of diaryliodonium salts.
Conclusion
In conclusion, a new facile protocol for the preparation of diaryliodonium salts using cheap and readily available Oxone as an oxidant in the presence of sulfuric acid has been developed. The procedure allows the synthesis of a wide range of iodonium salts containing electron-donating and electron-withdrawing substituents. Particularly attractive is the possibility of the one-pot synthesis of symmetric bis-aryliodonium salts directly from arenes via an iodination–oxidation sequence.
Supporting Information
| Supporting Information File 1: Experimental details and NMR spectra. | ||
| Format: PDF | Size: 4.8 MB | Download |
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (RFBR-mol_nr 17-33-50111), the Tomsk Polytechnic University Competitiveness Enhancement Program and the Royal Society (IE160304). We thank the EPSRC National Mass Spectrometry Facility, Swansea, for mass spectrometric data.
References
-
Yusubov, M. S.; Maskaev, A. V.; Zhdankin, V. V. ARKIVOC 2011, i, 370–409. doi:10.3998/ark.5550190.0012.107
Return to citation in text: [1] -
Zhdankin, V. V. Hypervalent Iodine Chemistry; Wiley: Chichester, 2014.
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Wirth, T., Ed. Hypervalent Iodine Chemistry in Topics in Current Chemistry; Springer: Berlin, 2016; Vol. 373.
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] -
Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Fañanás-Mastral, M. Synthesis 2017, 49, 1905–1930. doi:10.1055/s-0036-1589483
Return to citation in text: [1] -
Gonda, Z.; Novák, Z. Chem. – Eur. J. 2015, 21, 16801–16806. doi:10.1002/chem.201502995
Return to citation in text: [1] -
Dey, C.; Lindstedt, E.; Olofsson, B. Org. Lett. 2015, 17, 4554–4557. doi:10.1021/acs.orglett.5b02270
Return to citation in text: [1] -
Tinnis, F.; Stridfeldt, E.; Lundberg, H.; Adolfsson, H.; Olofsson, B. Org. Lett. 2015, 17, 2688–2691. doi:10.1021/acs.orglett.5b01079
Return to citation in text: [1] -
Cahard, E.; Male, H. P. J.; Tissot, M.; Gaunt, M. J. J. Am. Chem. Soc. 2015, 137, 7986–7989. doi:10.1021/jacs.5b03937
Return to citation in text: [1] -
Modha, S. G.; Greaney, M. F. J. Am. Chem. Soc. 2015, 137, 1416–1419. doi:10.1021/ja5124754
Return to citation in text: [1] -
Beaud, R.; Phipps, R. J.; Gaunt, M. J. J. Am. Chem. Soc. 2016, 138, 13183–13186. doi:10.1021/jacs.6b09334
Return to citation in text: [1] -
Yin, K.; Zhang, R. Org. Lett. 2017, 19, 1530–1533. doi:10.1021/acs.orglett.7b00310
Return to citation in text: [1] -
Seidl, T. L.; Stuart, D. R. J. Org. Chem. 2017, 82, 11765–11771. doi:10.1021/acs.joc.7b01599
Return to citation in text: [1] -
Margida, A. J.; Koser, G. F. J. Org. Chem. 1984, 49, 3643–3646. doi:10.1021/jo00193a039
Return to citation in text: [1] -
Kitamura, T.; Matsuyuki, J.-i.; Nagata, K.; Furuki, R.; Taniguchi, H. Synthesis 1992, 945–946. doi:10.1055/s-1992-26272
Return to citation in text: [1] -
Kitamura, T.; Matsuyuki, J.-i.; Taniguchi, H. Synthesis 1994, 147–148. doi:10.1055/s-1994-25423
Return to citation in text: [1] -
Pike, V. W.; Butt, F.; Shah, A.; Widdowson, D. A. J. Chem. Soc., Perkin Trans. 1 1999, 245–248. doi:10.1039/a809349k
Return to citation in text: [1] -
Carroll, M. A.; Pike, V. W.; Widdowson, D. A. Tetrahedron Lett. 2000, 41, 5393–5396. doi:10.1016/S0040-4039(00)00861-3
Return to citation in text: [1] -
Kaźmierczak, P.; Skulski, L. Synthesis 1995, 1027–1032. doi:10.1055/s-1995-4045
Return to citation in text: [1] -
Kryska, A.; Skulski, L. Molecules 2001, 6, 875–880. doi:10.3390/61100875
Return to citation in text: [1] -
Hossain, M. D.; Kitamura, T. Tetrahedron 2006, 62, 6955–6960. doi:10.1016/j.tet.2006.04.073
Return to citation in text: [1] -
Hossain, M. D.; Ikegami, Y.; Kitamura, T. J. Org. Chem. 2006, 71, 9903–9905. doi:10.1021/jo061889q
Return to citation in text: [1] -
Dohi, T.; Yamaoka, N.; Itani, I.; Kita, Y. Aust. J. Chem. 2011, 64, 529–535. doi:10.1071/CH11057
Return to citation in text: [1] -
Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373
Return to citation in text: [1] -
Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a
Return to citation in text: [1] -
Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974
Return to citation in text: [1] -
Zhu, M.; Jalalian, N.; Olofsson, B. Synlett 2008, 592–596. doi:10.1055/s-2008-1032050
Return to citation in text: [1] -
Jalalian, N.; Olofsson, B. Tetrahedron 2010, 66, 5793–5800. doi:10.1016/j.tet.2010.05.004
Return to citation in text: [1] -
Seidl, T. L.; Sundalam, S. K.; McCullough, B.; Stuart, D. R. J. Org. Chem. 2016, 81, 1998–2009. doi:10.1021/acs.joc.5b02833
Return to citation in text: [1] -
Lindstedt, E.; Reitti, M.; Olofsson, B. J. Org. Chem. 2017, 82, 11909–11914. doi:10.1021/acs.joc.7b01652
Return to citation in text: [1] -
Carreras, V.; Sandtorv, A. H.; Stuart, D. R. J. Org. Chem. 2017, 82, 1279–1284. doi:10.1021/acs.joc.6b02811
Return to citation in text: [1] -
Gemoets, H. P. L.; Laudadio, G.; Verstraete, K.; Hessel, V.; Noël, T. Angew. Chem., Int. Ed. 2017, 56, 7161–7165. doi:10.1002/anie.201703369
Return to citation in text: [1] -
Laudadio, G.; Gemoets, H. P. L.; Hessel, V.; Noël, T. J. Org. Chem. 2017, 82, 11735–11741. doi:10.1021/acs.joc.7b01346
Return to citation in text: [1] -
Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537–4538. doi:10.1021/jo9824596
Return to citation in text: [1] -
Yusubov, M. S.; Yusubova, R. Y.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2013, 78, 3767–3773. doi:10.1021/jo400212u
Return to citation in text: [1] [2] [3] -
Yusubov, M. S.; Soldatova, N. S.; Postnikov, P. S.; Valiev, R. R.; Svitich, D. Y.; Yusubova, R. Y.; Yoshimura, A.; Wirth, T.; Zhdankin, V. V. Eur. J. Org. Chem. 2018, 640–647. doi:10.1002/ejoc.201701595
Return to citation in text: [1] [2] [3] -
Postnikov, P. S.; Guselnikova, O. A.; Yusubov, M. S.; Yoshimura, A.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2015, 80, 5783–5788. doi:10.1021/acs.joc.5b00741
Return to citation in text: [1] [2] -
Zagulyaeva, A. A.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2010, 75, 2119–2122. doi:10.1021/jo902733f
Return to citation in text: [1] [2] -
Soldatova, N.; Postnikov, P.; Troyan, A. A.; Yoshimura, A.; Yusubov, M. S.; Zhdankin, V. V. Tetrahedron Lett. 2016, 57, 4254–4256. doi:10.1016/j.tetlet.2016.08.038
Return to citation in text: [1] [2] -
Soldatova, N. S.; Postnikov, P. S.; Kukurina, O. S.; Zhdankin, V. V.; Yoshimura, A.; Wirth, T.; Yusubov, M. S. ChemistryOpen 2017, 6, 18–20. doi:10.1002/open.201600129
Return to citation in text: [1] [2] -
Yakura, T.; Omoto, M.; Yamauchi, Y.; Tian, Y.; Ozono, A. Tetrahedron 2010, 66, 5833–5840. doi:10.1016/j.tet.2010.04.124
Return to citation in text: [1]
| 1. | Yusubov, M. S.; Maskaev, A. V.; Zhdankin, V. V. ARKIVOC 2011, i, 370–409. doi:10.3998/ark.5550190.0012.107 |
| 2. | Zhdankin, V. V. Hypervalent Iodine Chemistry; Wiley: Chichester, 2014. |
| 3. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 4. | Wirth, T., Ed. Hypervalent Iodine Chemistry in Topics in Current Chemistry; Springer: Berlin, 2016; Vol. 373. |
| 21. | Kaźmierczak, P.; Skulski, L. Synthesis 1995, 1027–1032. doi:10.1055/s-1995-4045 |
| 22. | Kryska, A.; Skulski, L. Molecules 2001, 6, 875–880. doi:10.3390/61100875 |
| 23. | Hossain, M. D.; Kitamura, T. Tetrahedron 2006, 62, 6955–6960. doi:10.1016/j.tet.2006.04.073 |
| 24. | Hossain, M. D.; Ikegami, Y.; Kitamura, T. J. Org. Chem. 2006, 71, 9903–9905. doi:10.1021/jo061889q |
| 25. | Dohi, T.; Yamaoka, N.; Itani, I.; Kita, Y. Aust. J. Chem. 2011, 64, 529–535. doi:10.1071/CH11057 |
| 16. | Margida, A. J.; Koser, G. F. J. Org. Chem. 1984, 49, 3643–3646. doi:10.1021/jo00193a039 |
| 17. | Kitamura, T.; Matsuyuki, J.-i.; Nagata, K.; Furuki, R.; Taniguchi, H. Synthesis 1992, 945–946. doi:10.1055/s-1992-26272 |
| 18. | Kitamura, T.; Matsuyuki, J.-i.; Taniguchi, H. Synthesis 1994, 147–148. doi:10.1055/s-1994-25423 |
| 19. | Pike, V. W.; Butt, F.; Shah, A.; Widdowson, D. A. J. Chem. Soc., Perkin Trans. 1 1999, 245–248. doi:10.1039/a809349k |
| 20. | Carroll, M. A.; Pike, V. W.; Widdowson, D. A. Tetrahedron Lett. 2000, 41, 5393–5396. doi:10.1016/S0040-4039(00)00861-3 |
| 8. | Gonda, Z.; Novák, Z. Chem. – Eur. J. 2015, 21, 16801–16806. doi:10.1002/chem.201502995 |
| 9. | Dey, C.; Lindstedt, E.; Olofsson, B. Org. Lett. 2015, 17, 4554–4557. doi:10.1021/acs.orglett.5b02270 |
| 10. | Tinnis, F.; Stridfeldt, E.; Lundberg, H.; Adolfsson, H.; Olofsson, B. Org. Lett. 2015, 17, 2688–2691. doi:10.1021/acs.orglett.5b01079 |
| 11. | Cahard, E.; Male, H. P. J.; Tissot, M.; Gaunt, M. J. J. Am. Chem. Soc. 2015, 137, 7986–7989. doi:10.1021/jacs.5b03937 |
| 12. | Modha, S. G.; Greaney, M. F. J. Am. Chem. Soc. 2015, 137, 1416–1419. doi:10.1021/ja5124754 |
| 13. | Beaud, R.; Phipps, R. J.; Gaunt, M. J. J. Am. Chem. Soc. 2016, 138, 13183–13186. doi:10.1021/jacs.6b09334 |
| 14. | Yin, K.; Zhang, R. Org. Lett. 2017, 19, 1530–1533. doi:10.1021/acs.orglett.7b00310 |
| 15. | Seidl, T. L.; Stuart, D. R. J. Org. Chem. 2017, 82, 11765–11771. doi:10.1021/acs.joc.7b01599 |
| 43. | Yakura, T.; Omoto, M.; Yamauchi, Y.; Tian, Y.; Ozono, A. Tetrahedron 2010, 66, 5833–5840. doi:10.1016/j.tet.2010.04.124 |
| 5. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 6. | Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 7. | Fañanás-Mastral, M. Synthesis 2017, 49, 1905–1930. doi:10.1055/s-0036-1589483 |
| 37. | Yusubov, M. S.; Yusubova, R. Y.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2013, 78, 3767–3773. doi:10.1021/jo400212u |
| 38. | Yusubov, M. S.; Soldatova, N. S.; Postnikov, P. S.; Valiev, R. R.; Svitich, D. Y.; Yusubova, R. Y.; Yoshimura, A.; Wirth, T.; Zhdankin, V. V. Eur. J. Org. Chem. 2018, 640–647. doi:10.1002/ejoc.201701595 |
| 37. | Yusubov, M. S.; Yusubova, R. Y.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2013, 78, 3767–3773. doi:10.1021/jo400212u |
| 38. | Yusubov, M. S.; Soldatova, N. S.; Postnikov, P. S.; Valiev, R. R.; Svitich, D. Y.; Yusubova, R. Y.; Yoshimura, A.; Wirth, T.; Zhdankin, V. V. Eur. J. Org. Chem. 2018, 640–647. doi:10.1002/ejoc.201701595 |
| 40. | Zagulyaeva, A. A.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2010, 75, 2119–2122. doi:10.1021/jo902733f |
| 41. | Soldatova, N.; Postnikov, P.; Troyan, A. A.; Yoshimura, A.; Yusubov, M. S.; Zhdankin, V. V. Tetrahedron Lett. 2016, 57, 4254–4256. doi:10.1016/j.tetlet.2016.08.038 |
| 36. | Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537–4538. doi:10.1021/jo9824596 |
| 37. | Yusubov, M. S.; Yusubova, R. Y.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2013, 78, 3767–3773. doi:10.1021/jo400212u |
| 38. | Yusubov, M. S.; Soldatova, N. S.; Postnikov, P. S.; Valiev, R. R.; Svitich, D. Y.; Yusubova, R. Y.; Yoshimura, A.; Wirth, T.; Zhdankin, V. V. Eur. J. Org. Chem. 2018, 640–647. doi:10.1002/ejoc.201701595 |
| 39. | Postnikov, P. S.; Guselnikova, O. A.; Yusubov, M. S.; Yoshimura, A.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2015, 80, 5783–5788. doi:10.1021/acs.joc.5b00741 |
| 40. | Zagulyaeva, A. A.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2010, 75, 2119–2122. doi:10.1021/jo902733f |
| 41. | Soldatova, N.; Postnikov, P.; Troyan, A. A.; Yoshimura, A.; Yusubov, M. S.; Zhdankin, V. V. Tetrahedron Lett. 2016, 57, 4254–4256. doi:10.1016/j.tetlet.2016.08.038 |
| 42. | Soldatova, N. S.; Postnikov, P. S.; Kukurina, O. S.; Zhdankin, V. V.; Yoshimura, A.; Wirth, T.; Yusubov, M. S. ChemistryOpen 2017, 6, 18–20. doi:10.1002/open.201600129 |
| 42. | Soldatova, N. S.; Postnikov, P. S.; Kukurina, O. S.; Zhdankin, V. V.; Yoshimura, A.; Wirth, T.; Yusubov, M. S. ChemistryOpen 2017, 6, 18–20. doi:10.1002/open.201600129 |
| 30. | Jalalian, N.; Olofsson, B. Tetrahedron 2010, 66, 5793–5800. doi:10.1016/j.tet.2010.05.004 |
| 31. | Seidl, T. L.; Sundalam, S. K.; McCullough, B.; Stuart, D. R. J. Org. Chem. 2016, 81, 1998–2009. doi:10.1021/acs.joc.5b02833 |
| 32. | Lindstedt, E.; Reitti, M.; Olofsson, B. J. Org. Chem. 2017, 82, 11909–11914. doi:10.1021/acs.joc.7b01652 |
| 33. | Carreras, V.; Sandtorv, A. H.; Stuart, D. R. J. Org. Chem. 2017, 82, 1279–1284. doi:10.1021/acs.joc.6b02811 |
| 34. | Gemoets, H. P. L.; Laudadio, G.; Verstraete, K.; Hessel, V.; Noël, T. Angew. Chem., Int. Ed. 2017, 56, 7161–7165. doi:10.1002/anie.201703369 |
| 35. | Laudadio, G.; Gemoets, H. P. L.; Hessel, V.; Noël, T. J. Org. Chem. 2017, 82, 11735–11741. doi:10.1021/acs.joc.7b01346 |
| 26. | Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373 |
| 27. | Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a |
| 28. | Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974 |
| 29. | Zhu, M.; Jalalian, N.; Olofsson, B. Synlett 2008, 592–596. doi:10.1055/s-2008-1032050 |
| 39. | Postnikov, P. S.; Guselnikova, O. A.; Yusubov, M. S.; Yoshimura, A.; Nemykin, V. N.; Zhdankin, V. V. J. Org. Chem. 2015, 80, 5783–5788. doi:10.1021/acs.joc.5b00741 |
© 2018 Soldatova et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)