Abstract
A straightforward and one-pot synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones via Ag(I)-catalyzed 1,3-dipolar cycloaddition of azomethine ylides with N-alkyl maleimide, followed by readily complete oxidation with DDQ, has been successfully developed. Further transformation with alkylamine/sodium alkoxide alcohol solution conveniently afforded novel polysubstituted pyrroles in good to excellent yields. This methodology for highly functionalized pyrroles performed well over a broad scope of substrates. It is conceivable that this efficient construction method for privileged pyrrole scaffolds could deliver more active compounds for medicinal chemistry research.

Graphical Abstract
Introduction
Pyrroles are an important class of five-membered nitrogen containing heterocycles which are widely used in numerous medically relevant fields. Pyrrolo[3,4-c]pyrrole-1,3-diones and highly substituted pyrroles with amide groups are important frameworks of pyrrole compounds that play important roles in medicinal chemistry (Figure 1), such as analgesic agent 1 [1], BET bromodomain inhibitor 2 [2], selective PARP-1 inhibitor 3 [3], histone deacetylase inhibitor with antitumor activity 4 [4], Flavivirus inhibitor 5 [5], and for treating cardiovascular diseases (atorvastatin, 6) [6].
![[1860-5397-14-75-1]](/bjoc/content/figures/1860-5397-14-75-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative pyrrolo[3,4-c]pyrrole-1,3-diones and polysubstituted pyrrole derivatives.
Figure 1: Representative pyrrolo[3,4-c]pyrrole-1,3-diones and polysubstituted pyrrole derivatives.
A number of methods have been reported till now to synthesize pyrroles and pyrrole containing analogs [6]. In the past decade, 1,3-dipolar cycloaddition has become a fundamental synthetic method for the construction of nitrogen-containing five-membered heterocycles including pyrroles. It is worth to note that considerable efforts have been expended to build stereogenic centers of pyrrolidine derivatives using chiral catalysts [7].
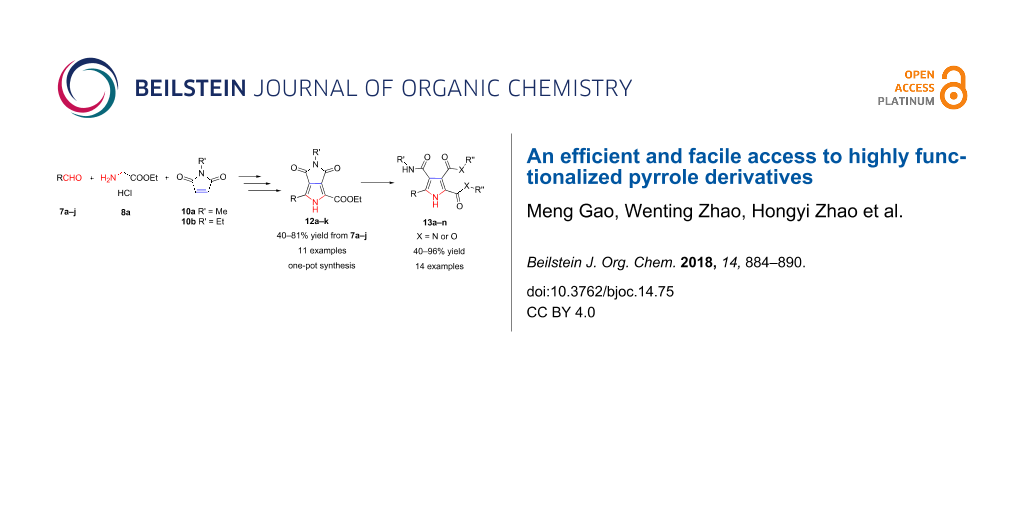
As our aim was to construct a small library of polysubstituted pyrroles for antibacterial screening, it prompted us to develop a concise and efficient synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones [8-10] via 1,3-dipolar cycloaddition without any chiral catalysts/ligands and a facile access to highly substituted pyrroles with amide groups or ester groups. Herein, we propose a straightforward and one-pot synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones via Ag(I)-catalyzed 1,3-dipolar cycloaddition of azomethine ylides with N-alkyl maleimide, followed by a facile oxidation using DDQ as oxidant. Further manipulation with alkylamine/sodium alkoxide alcohol solution conveniently led to novel polysubstituted pyrroles in good to excellent yields (Scheme 1).
![[1860-5397-14-75-i1]](/bjoc/content/inline/1860-5397-14-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathway for the preparation of pyrrolo[3,4-c]pyrrole-1,3-diones and highly substituted pyrroles. Reagents and conditions: (i) Et3N, MgSO4, CH2Cl2, rt, 8 h; (ii) AgOAc, N-methylmaleimide (10a) or N-ethylmaleimide (10b), CH2Cl2, rt, 3 h; (iii) DDQ, toluene, rt, 12–80 h; (iv) a: MeNH2 or EtNH2 in EtOH, 80 °C, in a sealed tube for 3 h; b: MeONa/MeOH, rt, 24 h or EtONa/EtOH, rt, 36 h.
Scheme 1: Synthetic pathway for the preparation of pyrrolo[3,4-c]pyrrole-1,3-diones and highly substituted py...
Results and Discussion
As shown in Scheme 1, one advantage of this 1,3-dipolar cycloaddition for constructing privileged pyrrole scaffolds is the broad scope of azomethine ylides as substrates. To our surprise, although there were many reports [11-16] about the synthesis of ethyl N-(phenylmethylidene)glycinate (9a, azomethine ylide) as the key substrate for 1,3-dipolar cycloadditions, the reaction conditions were very different exemplified by the wide range of reaction times from 0.3 h to 120 h, and the variable yields from 68% to 100% in different solvents. In order to establish the optimal experimental conditions suitable for the one-pot synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones 12a–k, we investigated the reaction conditions step by step.
Initially, benzaldehyde (7a) and ethyl glycinate hydrochloride (8a) were chosen as the model substrates for obtaining azomethine ylide 9a, and the results are summarized in Table 1. Based on reported methods, Et3N as base, MgSO4 as desiccant and CH2Cl2 as solvent were used for the synthesis of the azomethine ylide [13,14,17-24]. Because 9a easily decomposed to benzaldehyde on silica gel when monitored by TLC, we used 1H NMR to monitor the reaction process. Table 1 shows that the reaction time was crucial to the extent of the reaction. 9a could be obtained in excellent yield (94% total yield of 7a and 9a, of which 98% was 9a) without any further purification when the reaction time was prolonged to 8 h. The outcome of a 8 h reaction is favorable compared to the results of 2 h and 4 h reaction time (Table 1, entry 3 vs entries 1 and 2).
Table 1: Optimization of the reaction conditions for the synthesis of the azomethine ylidea.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-75-i5.svg?max-width=637&scale=1.0)
|
|||
| entry | time | yield (%)b | ratio of 9a/7ac |
|---|---|---|---|
| 1 | 2 h | 99 | 12:1 |
| 2 | 4 h | 98 | 24:1 |
| 3 | 8 h | 94 | 47:1 |
aGeneral reaction conditions: benzaldehyde (2.5 mmol), ethyl glycinate hydrochloride (3.75 mmol), Et3N (3.75 mmol), and MgSO4 (3.75 mmol), rt. bThe mixture yields (9a + 7a) were obtained after work-up. cThe ratio of 9a/7a was determined by 1H NMR.
Encouraged by the achievements on catalytic asymmetric 1,3-dipolar cycloaddition to build octahydropyrrolo[3,4-c]pyrrole frameworks [7,9], we screened the optimal reaction conditions for preparing the racemic pyrrolidine 11a using N-methylmaleimide (10a) as the simple dipolarophile without any chiral catalysts/ligands. To our delight, in the presence of 10 mol % of AgOAc, the reaction reached almost completion in CH2Cl2 within 1 h at room temperature and afforded the desired cycloadduct in moderate yield (64%, Table 2, entry 1). The annulation process did not occur without the catalyst (Table 2, entry 2). Since most of the 1,3-dipolar cycloadditions are known to work best in nonpolar solvents, toluene instead of CH2Cl2 was selected as the reaction solvent. Unfortunately, toluene was not a good choice even the reaction time was prolonged to 2 h (36% yield, Table 2, entry 3). Switching the base Et3N to DBU resulted in a significant decrease of product yield (Table 2, entry 4). Given that the crude azomethine ylide 9a was used without any purification, and 9a might be sensitive to other decomposition pathways, we increased the amount of 9a up to 1.5 equivalents, which delivered the bicyclic pyrrolidine 11a in excellent yield (82%, Table 2, entry 5). Prolonging the reaction time from 3 h to 6 h had no influence on the yield (Table 2, entry 6), which demonstrated that appropriate increase on the ratio of 9a/10a was beneficial to the completion of this cycloaddition. Finally, the use of Et3N as the base and CH2Cl2 as the solvent, and the suitable ratio of 9a/10a at 1.5:1 catalyzed by 10 mol % of AgOAc were identified as the optimal reaction conditions.
Table 2: Optimization of the reaction conditions of the cycloaddition reactiona.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-75-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | base | catalyst | solvent | time | yield (%)b |
|---|---|---|---|---|---|
| 1 | Et3N | AgOAc | CH2Cl2 | 1 h | 64 |
| 2 | Et3N | – | CH2Cl2 | 24 h | trace |
| 3 | Et3N | AgOAc | toluene | 2 h | 36 |
| 4 | DBU | AgOAc | CH2Cl2 | 1 h | 39 |
| 5c | Et3N | AgOAc | CH2Cl2 | 3 h | 82 |
| 6c | Et3N | AgOAc | CH2Cl2 | 6 h | 80 |
aGeneral reaction conditions: 9a (2.5 mmol), 10a (2.5 mmol), AgOAc (0.25 mmol) and base (0.25 mmol), rt. bIsolated yield. c9a/10a/AgOAC/Et3N = 1.5:1:0.1:0.1.
Finally, the transformation of the cycloadduct 11a to pyrrolo[3,4-c]pyrrole-1,3-dione 12a was investigated (Table 3). For the operational simplicity and continuity of this one-pot condensation–cycloaddition–aromatization reaction, CH2Cl2 was firstly selected as solvent instead of the generally used toluene [10,25,26] for this oxidation step. Unfortunately, the desired pyrrole product 12a was obtained only in 41% yield with DDQ (4 equiv) as oxidant at room temperature for 48 h (Table 3, entry 1). As expected, toluene as solvent improved the reaction outcome to afford 12a in a good yield up to 71% (Table 3, entry 2). Subsequently, reducing the amount of DDQ (2 equiv) led to a significantly decreased yield (Table 3, entry 3), and also the addition of silica gel [9] as an additive had no considerable effect on the yield (Table 3, entry 4 vs entry 2). The results demonstrated that using 4 equiv DDQ in toluene were the best conditions for this oxidation protocol.
Table 3: Optimization of the reaction conditions of the oxidation reaction.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-75-i7.svg?max-width=637&scale=1.0)
|
||||
| entry | catalyst | solvent | ratio of 11a/DDQ | yield (%)a |
|---|---|---|---|---|
| 1 | – | CH2Cl2 | 1:4 | 41 |
| 2 | – | toluene | 1:4 | 71 |
| 3 | – | toluene | 1:2 | 43 |
| 4 | silica gelb | toluene | 1:4 | 68 |
aIsolated yield. b10% Amount of 11a.
With the optimized reaction conditions for each step in hand, we prepared 12a starting from benzaldehyde and ethyl glycinate hydrochloride through three steps but via a one-pot process without any purification for intermediates 9a and 11a (see Supporting Information File 1). To our delight, the reaction outcome was satisfactory with a total yield of 47% for three-steps which was equivalent to the stepwise reactions (Scheme 2, 12a). In order to evaluate the generality of this process, we explored a variety of aldehydes 7b–j, and the results are shown in Scheme 2. All tested benzaldehydes bearing electron-withdrawing or electron-donating substituents on the phenyl ring reacted smoothly via condensation–cycloaddition–oxidation in the one-pot process, delivering the corresponding pyrrolo[3,4-c]pyrrole-1,3-diones in moderate to good yields (53–77%). Heteroaromatic aldehydes 7i and 7j afforded the products 12i and 12j in excellent total yields of 81%. Additionally, cinnamaldehyde (7h) was also well tolerated, giving the product 12h in moderate yield (40%). Finally, to further explore the scope and generality of this protocol, N-ethylmaleimide (10b) instead of N-methylmaleimide (10a) as another simple dipolarophile was also examined; the target product 12k was prepared smoothly in high yield (74%). This concise and efficient protocol displays the potentiality for scale-up synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones.
![[1860-5397-14-75-i2]](/bjoc/content/inline/1860-5397-14-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of one-pot synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones 12a–k. General reaction conditions: (i) aldehydes 7a–j (3 mmol), ethyl glycinate hydrochloride (8a, 4.5 mmol), Et3N, MgSO4, CH2Cl2, rt, 8 h; (ii) N-methylmaleimide (10a, 2 mmol) or N-ethylmaleimide (10b, 2 mmol), AgOAc (0.2 mmol), CH2Cl2, rt, 3 h; (iii) DDQ (8 mmol), toluene, rt, 12–80 h.
Scheme 2: Scope of one-pot synthesis of pyrrolo[3,4-c]pyrrole-1,3-diones 12a–k. General reaction conditions: ...
Having the pyrrolo[3,4-c]pyrrole-1,3-diones with diversified substituents in hand, polysubstituted pyrrole derivatives with three amide groups 13a–l were prepared smoothly by treating 12a–k with methylamine or ethylamine in EtOH at 80 °C in good to excellent yields (72–96%, Scheme 3). The structures of highly substituted pyrrole derivatives prompted us to envisage the reasonable mechanism for the formation of 3,4-diamide groups, besides the amidation of ethyl ester group on the 2-position to give one amide group on the pyrrole ring. Due to the electron-withdrawing effect of the ester group at the 2-position in 12a–k, the carbonyl group at the 3-position of the pyrrole was easily attacked by the nucleophilic amine (compared with the carbonyl at the 4-position, shown in Scheme 4) exemplified by the reaction of 12d with ethylamine to afford 13k. NOESY analysis of 13k was used to confirm the relative positions of 3,4-diamide groups formed. NOE correlations between an H of the methylamide at the 4-position and H2 or H6 of the 4-chlorophenyl moiety supported the reaction result (see Supporting Information File 1). Additionally, the corresponding pyrroles with more functionalized substituents with both amide and ester group 13m,n could also be readily achieved by treatment of 12a with MeONa in MeOH or EtONa in EtOH at room temperature in moderate to good yields (40–66%, Scheme 3). It was noted that the ethyl ester in 12a was replaced by methyl ester through transesterification when 12a was treated with MeONa in MeOH to afford 13m. Therefore, this efficient protocol developed could meet our requirement to basically construct a small library of highly functionalized pyrrole derivatives for antibacterial screening or for further chemical manipulations to get more promising compounds.
![[1860-5397-14-75-i3]](/bjoc/content/inline/1860-5397-14-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the synthesis of highly substituted pyrrole derivatives 13a–n. General reaction conditions: (i) 12a–k (0.17 mmol), methylamine or ethylamine solution in EtOH (3 mL), 80 °C, in a sealed tube for 3 h; (ii) a12a (0.17 mmol), MeONa (3 equiv), MeOH, rt, 24 h; b12a (0.17 mmol), EtONa (3 equiv), EtOH, rt, 36 h.
Scheme 3: Scope of the synthesis of highly substituted pyrrole derivatives 13a–n. General reaction conditions...
![[1860-5397-14-75-i4]](/bjoc/content/inline/1860-5397-14-75-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 13k from 12d and ethylamine.
Scheme 4: Synthesis of 13k from 12d and ethylamine.
Conclusion
In summary, we have developed an efficient, operationally simple protocol to afford pyrrolo[3,4-c]pyrrole-1,3-diones via 1,3-dipolar cycloaddition of azomethine ylides from diverse commercially available aldehydes in good to excellent yields and a facile access to highly functionalized pyrroles. Notably, the versatile conversion of the cycloaddition adducts leads to fascinating polysubstituted pyrrole compounds, which can deliver potentially valuable building blocks serving as precursors for drug discovery. Further research on the development of more diversified pyrrolo[3,4-c]pyrrole-1,3-diones and more complicated polysubstituted pyrroles is currently underway in our laboratory.
Supporting Information
| Supporting Information File 1: General information, experimental details, characterization data, copies of 1H and 13C NMR spectra of 12a–k and 13a–n, and NOESY spectra of compound 13k. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Malinka, W.; Kaczmarz, M.; Redzicka, A.; Filipek, B.; Sapa, J. Farmaco 2005, 60, 15–22. doi:10.1016/j.farmac.2004.10.002
Return to citation in text: [1] -
Ayoub, A. M.; Hawk, L. M. L.; Herzig, R. J.; Jiang, J.; Wisniewski, A. J.; Gee, C. T.; Zhao, P.; Zhu, J.-Y.; Berndt, N.; Offei-Addo, N. K.; Scott, T. G.; Qi, J.; Bradner, J. E.; Ward, T. R.; Schönbrunn, E.; Georg, G. I.; Pomerantz, W. C. K. J. Med. Chem. 2017, 60, 4805–4817. doi:10.1021/acs.jmedchem.6b01336
Return to citation in text: [1] -
Srivastava, B. K.; Desai, R. C.; Patel, P. R. Substituted Phthalazin-1-(2H)-one Derivatives as Selective Inhibitors of Poly (ADP-ribose) Polymerase-1. U.S. Pat. Appl. US20150291603A1, Oct 15, 2015.
Return to citation in text: [1] -
Zubia, A.; Ropero, S.; Otaegui, D.; Ballestar, E.; Fraga, M. F.; Boix-Chornet, M.; Berdasco, M.; Martinez, A.; Coll-Mulet, L.; Gil, J.; Cossío, F. P.; Esteller, M. Oncogene 2009, 28, 1477–1484. doi:10.1038/onc.2008.501
Return to citation in text: [1] -
Padmanabhan, R.; Pattabiraman, N.; Mueller, N.; Nagarajan, K. Flavivirus Inhibitors and Methods for Their Use. WO Patent WO2010039538, April 8, 2010.
Return to citation in text: [1] -
Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. RSC Adv. 2015, 5, 15233–15266. doi:10.1039/c4ra15710a
Return to citation in text: [1] [2] -
Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366–5412. doi:10.1021/cr5007182
Return to citation in text: [1] [2] -
Martinez-Ariza, G.; Dietrich, J.; De Moliner, F.; Hulme, C. Synlett 2013, 24, 1801–1804. doi:10.1055/s-0033-1338872
Return to citation in text: [1] -
Liu, H.-C.; Tao, H.-Y.; Cong, H.; Wang, C.-J. J. Org. Chem. 2016, 81, 3752–3760. doi:10.1021/acs.joc.6b00396
Return to citation in text: [1] [2] [3] -
Oikonomou, K.; Georgiou, D.; Katsamakas, S.; Hadjipavlou-Litina, D.; Elemes, Y. ARKIVOC 2015, No. iii, 214–231. doi:10.3998/ark.5550190.0016.315
Return to citation in text: [1] [2] -
Baumann, M.; Baxendale, I. R. J. Org. Chem. 2016, 81, 11898–11908. doi:10.1021/acs.joc.6b02346
Return to citation in text: [1] -
Sugimoto, K.; Yamamoto, N.; Tominaga, D.; Matsuya, Y. Org. Lett. 2015, 17, 1320–1323. doi:10.1021/acs.orglett.5b00320
Return to citation in text: [1] -
Gonzalez, K. A.; Wilson, L. J.; Wu, W.; Nancollas, G. H. Bioorg. Med. Chem. 2002, 10, 1991–1997. doi:10.1016/S0968-0896(02)00049-4
Return to citation in text: [1] [2] -
Sun, P.; Zhang, Y. Synth. Commun. 1997, 27, 4173–4179. doi:10.1080/00397919708005465
Return to citation in text: [1] [2] -
Merino, I.; Laxmi, S.; Flórez, J.; Barluenga, J. J. Org. Chem. 2002, 67, 648–655. doi:10.1021/jo010485p
Return to citation in text: [1] -
Katagiri, N.; Miura, Y.; Niwa, R.; Kato, T. Chem. Pharm. Bull. 1983, 31, 538–544. doi:10.1248/cpb.31.538
Return to citation in text: [1] -
Cabrera, S.; Arrayás, R. G.; Carretero, J. C. J. Am. Chem. Soc. 2005, 127, 16394–16395. doi:10.1021/ja0552186
Return to citation in text: [1] -
Jia, H.; Liu, H.; Guo, Z.; Huang, J.; Guo, H. Org. Lett. 2017, 19, 5236–5239. doi:10.1021/acs.orglett.7b02512
Return to citation in text: [1] -
Zhang, D.-J.; Xie, M.-S.; Qu, G.-R.; Gao, Y.-W.; Guo, H.-M. Org. Lett. 2016, 18, 820–823. doi:10.1021/acs.orglett.6b00108
Return to citation in text: [1] -
Yuan, C.; Liu, H.; Gao, Z.; Zhou, L.; Feng, Y.; Xiao, Y.; Guo, H. Org. Lett. 2015, 17, 26–29. doi:10.1021/ol503169d
Return to citation in text: [1] -
Udry, G. A. O.; Repetto, E.; Varela, O. J. Org. Chem. 2014, 79, 4992–5006. doi:10.1021/jo500547y
Return to citation in text: [1] -
Liu, H.; Wu, Y.; Zhao, Y.; Li, Z.; Zhang, L.; Yang, W.; Jiang, H.; Jing, C.; Yu, H.; Wang, B.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2014, 136, 2625–2629. doi:10.1021/ja4122268
Return to citation in text: [1] -
Yang, Q.-L.; Xie, M.-S.; Xia, C.; Sun, H.-L.; Zhang, D.-J.; Huang, K.-X.; Guo, Z.; Qu, G.-R.; Guo, H.-M. Chem. Commun. 2014, 50, 14809–14812. doi:10.1039/c4cc06632d
Return to citation in text: [1] -
Guo, H.; Liu, H.; Zhu, F.-L.; Na, R.; Jiang, H.; Wu, Y.; Zhang, L.; Li, Z.; Yu, H.; Wang, B.; Xiao, Y.; Hu, X.-P.; Wang, M. Angew. Chem., Int. Ed. 2013, 52, 12641–12645. doi:10.1002/anie.201307317
Return to citation in text: [1] -
Nájera, C.; de Gracia Retamosa, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Tetrahedron: Asymmetry 2008, 19, 2913–2923. doi:10.1016/j.tetasy.2008.12.021
Return to citation in text: [1] -
Mancebo-Aracil, J.; Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Costa, P. R. R.; de Lima, E. C.; Dias, A. G. Tetrahedron: Asymmetry 2012, 23, 1596–1606. doi:10.1016/j.tetasy.2012.10.015
Return to citation in text: [1]
| 1. | Malinka, W.; Kaczmarz, M.; Redzicka, A.; Filipek, B.; Sapa, J. Farmaco 2005, 60, 15–22. doi:10.1016/j.farmac.2004.10.002 |
| 5. | Padmanabhan, R.; Pattabiraman, N.; Mueller, N.; Nagarajan, K. Flavivirus Inhibitors and Methods for Their Use. WO Patent WO2010039538, April 8, 2010. |
| 4. | Zubia, A.; Ropero, S.; Otaegui, D.; Ballestar, E.; Fraga, M. F.; Boix-Chornet, M.; Berdasco, M.; Martinez, A.; Coll-Mulet, L.; Gil, J.; Cossío, F. P.; Esteller, M. Oncogene 2009, 28, 1477–1484. doi:10.1038/onc.2008.501 |
| 3. | Srivastava, B. K.; Desai, R. C.; Patel, P. R. Substituted Phthalazin-1-(2H)-one Derivatives as Selective Inhibitors of Poly (ADP-ribose) Polymerase-1. U.S. Pat. Appl. US20150291603A1, Oct 15, 2015. |
| 10. | Oikonomou, K.; Georgiou, D.; Katsamakas, S.; Hadjipavlou-Litina, D.; Elemes, Y. ARKIVOC 2015, No. iii, 214–231. doi:10.3998/ark.5550190.0016.315 |
| 25. | Nájera, C.; de Gracia Retamosa, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Tetrahedron: Asymmetry 2008, 19, 2913–2923. doi:10.1016/j.tetasy.2008.12.021 |
| 26. | Mancebo-Aracil, J.; Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Costa, P. R. R.; de Lima, E. C.; Dias, A. G. Tetrahedron: Asymmetry 2012, 23, 1596–1606. doi:10.1016/j.tetasy.2012.10.015 |
| 2. | Ayoub, A. M.; Hawk, L. M. L.; Herzig, R. J.; Jiang, J.; Wisniewski, A. J.; Gee, C. T.; Zhao, P.; Zhu, J.-Y.; Berndt, N.; Offei-Addo, N. K.; Scott, T. G.; Qi, J.; Bradner, J. E.; Ward, T. R.; Schönbrunn, E.; Georg, G. I.; Pomerantz, W. C. K. J. Med. Chem. 2017, 60, 4805–4817. doi:10.1021/acs.jmedchem.6b01336 |
| 9. | Liu, H.-C.; Tao, H.-Y.; Cong, H.; Wang, C.-J. J. Org. Chem. 2016, 81, 3752–3760. doi:10.1021/acs.joc.6b00396 |
| 8. | Martinez-Ariza, G.; Dietrich, J.; De Moliner, F.; Hulme, C. Synlett 2013, 24, 1801–1804. doi:10.1055/s-0033-1338872 |
| 9. | Liu, H.-C.; Tao, H.-Y.; Cong, H.; Wang, C.-J. J. Org. Chem. 2016, 81, 3752–3760. doi:10.1021/acs.joc.6b00396 |
| 10. | Oikonomou, K.; Georgiou, D.; Katsamakas, S.; Hadjipavlou-Litina, D.; Elemes, Y. ARKIVOC 2015, No. iii, 214–231. doi:10.3998/ark.5550190.0016.315 |
| 13. | Gonzalez, K. A.; Wilson, L. J.; Wu, W.; Nancollas, G. H. Bioorg. Med. Chem. 2002, 10, 1991–1997. doi:10.1016/S0968-0896(02)00049-4 |
| 14. | Sun, P.; Zhang, Y. Synth. Commun. 1997, 27, 4173–4179. doi:10.1080/00397919708005465 |
| 17. | Cabrera, S.; Arrayás, R. G.; Carretero, J. C. J. Am. Chem. Soc. 2005, 127, 16394–16395. doi:10.1021/ja0552186 |
| 18. | Jia, H.; Liu, H.; Guo, Z.; Huang, J.; Guo, H. Org. Lett. 2017, 19, 5236–5239. doi:10.1021/acs.orglett.7b02512 |
| 19. | Zhang, D.-J.; Xie, M.-S.; Qu, G.-R.; Gao, Y.-W.; Guo, H.-M. Org. Lett. 2016, 18, 820–823. doi:10.1021/acs.orglett.6b00108 |
| 20. | Yuan, C.; Liu, H.; Gao, Z.; Zhou, L.; Feng, Y.; Xiao, Y.; Guo, H. Org. Lett. 2015, 17, 26–29. doi:10.1021/ol503169d |
| 21. | Udry, G. A. O.; Repetto, E.; Varela, O. J. Org. Chem. 2014, 79, 4992–5006. doi:10.1021/jo500547y |
| 22. | Liu, H.; Wu, Y.; Zhao, Y.; Li, Z.; Zhang, L.; Yang, W.; Jiang, H.; Jing, C.; Yu, H.; Wang, B.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2014, 136, 2625–2629. doi:10.1021/ja4122268 |
| 23. | Yang, Q.-L.; Xie, M.-S.; Xia, C.; Sun, H.-L.; Zhang, D.-J.; Huang, K.-X.; Guo, Z.; Qu, G.-R.; Guo, H.-M. Chem. Commun. 2014, 50, 14809–14812. doi:10.1039/c4cc06632d |
| 24. | Guo, H.; Liu, H.; Zhu, F.-L.; Na, R.; Jiang, H.; Wu, Y.; Zhang, L.; Li, Z.; Yu, H.; Wang, B.; Xiao, Y.; Hu, X.-P.; Wang, M. Angew. Chem., Int. Ed. 2013, 52, 12641–12645. doi:10.1002/anie.201307317 |
| 7. | Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366–5412. doi:10.1021/cr5007182 |
| 7. | Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366–5412. doi:10.1021/cr5007182 |
| 9. | Liu, H.-C.; Tao, H.-Y.; Cong, H.; Wang, C.-J. J. Org. Chem. 2016, 81, 3752–3760. doi:10.1021/acs.joc.6b00396 |
| 6. | Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. RSC Adv. 2015, 5, 15233–15266. doi:10.1039/c4ra15710a |
| 6. | Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. RSC Adv. 2015, 5, 15233–15266. doi:10.1039/c4ra15710a |
| 11. | Baumann, M.; Baxendale, I. R. J. Org. Chem. 2016, 81, 11898–11908. doi:10.1021/acs.joc.6b02346 |
| 12. | Sugimoto, K.; Yamamoto, N.; Tominaga, D.; Matsuya, Y. Org. Lett. 2015, 17, 1320–1323. doi:10.1021/acs.orglett.5b00320 |
| 13. | Gonzalez, K. A.; Wilson, L. J.; Wu, W.; Nancollas, G. H. Bioorg. Med. Chem. 2002, 10, 1991–1997. doi:10.1016/S0968-0896(02)00049-4 |
| 14. | Sun, P.; Zhang, Y. Synth. Commun. 1997, 27, 4173–4179. doi:10.1080/00397919708005465 |
| 15. | Merino, I.; Laxmi, S.; Flórez, J.; Barluenga, J. J. Org. Chem. 2002, 67, 648–655. doi:10.1021/jo010485p |
| 16. | Katagiri, N.; Miura, Y.; Niwa, R.; Kato, T. Chem. Pharm. Bull. 1983, 31, 538–544. doi:10.1248/cpb.31.538 |
© 2018 Gao et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)