Abstract
The combination of the trityl cation and a chiral weakly coordinating Fe(III)-based bisphosphate anion was used to develop a new type of a highly active carbocation Lewis acid catalyst. The stereocontrol potential of the chiral tritylium ion pair was demonstrated by its application in an enantioselective Diels–Alder reaction of anthracene.

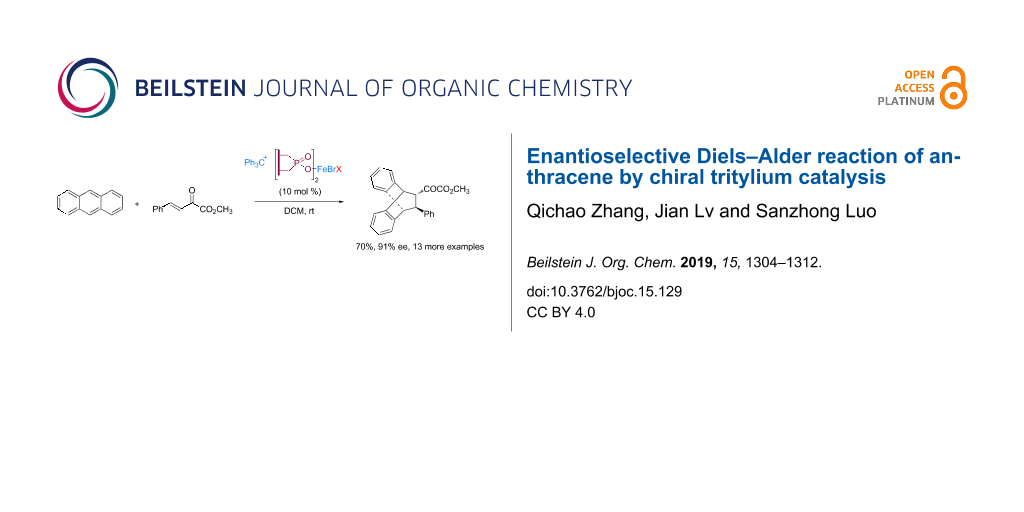
Graphical Abstract
Introduction
Carbocation Lewis acid catalysis has grown significantly over the last two decades [1-13]. The development of asymmetric carbocation catalysts has been long pursued but remains a challenging task. One strategy is to design and synthesize stabilized chiral carbocations with chirality installed onto their backbones. Pioneering efforts along this line by Kagan, Sammakia, and Chen have shown that chiral catalysis with such chiral carbocations was indeed plausible to achieve stereocontrol (Scheme 1a). [14-19]. However, the enantioselectivity was low in most cases. In addition, the synthetic efforts to access these chiral cations were generally non-trivial which limited their further development. Recently, we developed a chiral ion-pair strategy for asymmetric carbocation catalysis, with chiral trityl phosphate as the carbocation precursor [20,21]. In this latent strategy, the carbocation precursor can undergo facile ionic dissociation upon mild external stimulation such as polar substrates (such as α-ketoesters) to form a catalytically active chiral ion pair for substrate activation and chiral induction (Scheme 1b). In our further explorations, we noticed that the dissociation of trityl phosphate was generally sluggish, thus limiting its applicability. To expand its utility, we report herein a metal-complexed phosphate anion for chiral carbocation catalysis.
![[1860-5397-15-129-i1]](/bjoc/content/inline/1860-5397-15-129-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric carbocation catalysis.
Scheme 1: Asymmetric carbocation catalysis.
Weakly coordinating anions [22,23] have been widely used in inorganic and organic chemistry [24-27] as well as in polymer chemistry [28-33]. Although tritylium salts with various types of these counter anions based on B(III), Al(III), Ga(III), Fe(III), Nb(III), Ta(III), Y(III) and La(III) centers and ligands have been investigated in Lewis acid catalysis over the past decades, a chiral counter anion [34,35] with metal elements as the central atom, however, was seldom reported. Typically, the tritylium salts with weakly coordinating anions can be synthesized through a simple halide abstraction from the trityl halide in the presence of strong Lewis acids [36]. We herein report the design and exploration of a new trityl carbocation that has a chiral weakly coordinating Fe(III)-based phosphate anion for the effective asymmetric catalysis in the Diels–Alder reaction of anthracenes.
Results and Discussion
In our previous work, we found that less than 6% of trityl phosphate (TP) dissociated to trityl cations in the presence of a polar substrate such trifluoropyruvate [20]. In order to improve the efficiency of the dissociation, we started by first studying the properties of tritylium salts with a weakly coordinating metal-based phosphate anion (Scheme 2). Upon in situ mixing the chiral trityl phosphate (TP, 0.05 mM) and different Lewis acids (0.05 mM), such as InCl3, InBr3, InI3, In(OTf)3, Sc(OTf)3, Hf(OTf)3, GaCl3, and FeBr3, the originally colorless solution of the chiral trityl phosphate TP turned orange, suggesting the formation of tritylium ions (Scheme 2a). The stimulated trityl cation generation was probed by UV–vis spectroscopy. As shown in Figure 1a, when treated with different Lewis acids, trityl phosphate TP showed a variable tendency to dissociate into the free tritylium ion pair with InBr3 as the most active Lewis acid. An estimation based on UV absorption showed that approximately 76% of TP dissociated into trityl cations in the presence of InBr3. On the other hand, tritylium salts with a weakly coordinating metal-based monophosphate or bisphosphate anion could also be obtained when trityl bromide was treated with the corresponding metal phosphate, which can be prepared in situ following our previously described procedure (Scheme 2b,c) [37,38]. UV analysis indicated that the indium salt 1a or gallium salt 1b (0.05 mM) could induce ca. 92% dissociation of trityl bromide (0.05 mM) to generate the trityl cation. Also, FeBr3, a chiral Fe(III) monophosphate (M = FeBr2) 1c or even the bulky Fe(III) bisphosphate 2a promoted the dissociation of trityl bromide. In the latter case, the dissociation was estimated to be 54% by in-situ IR spectroscopy (UV–vis spectra were not applicable due to absorption overlap; see Supporting Information File 1 for details).
![[1860-5397-15-129-i2]](/bjoc/content/inline/1860-5397-15-129-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of new carbocation catalysts with weakly coordinating metal-based phosphate anion.
Scheme 2: Synthesis of new carbocation catalysts with weakly coordinating metal-based phosphate anion.
![[1860-5397-15-129-1]](/bjoc/content/figures/1860-5397-15-129-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Dissociation of latent carbocation by the use of Lewis acids. a) UV–vis absorption spectra of TP (0.05 mM) upon the addition of Lewis acids (0.05 mM), such as InCl3, InBr3, InI3, In(OTf)3, Sc(OTf)3, Hf(OTf)4, and GaCl3. b) UV–vis absorption spectra of trityl bromide (Ph3CBr, 0.05 mM) upon the addition of the chiral Lewis acids (0.05 mM), such as 1a, b, and d–g.
Figure 1: Dissociation of latent carbocation by the use of Lewis acids. a) UV–vis absorption spectra of TP (0...
We next tested the metal phosphate strategy in the Diels–Alder reaction of anthracene, for which a catalytic asymmetric version has not been achieved yet. Recently, we reported that the tritylium salt [Ph3C][BArF], in situ generated by Ph3CBr and NaBArF, could promote the Diels–Alder reaction with anthracenes and various unsaturated carbonyl compounds under mild conditions [13]. The use of latent carbocation catalysis with TP was examined in order to achieve enantioselective control. To our delight, TP catalyzed the asymmetric reaction affording cycloadduct 5a in excellent enantioselectivity (97% ee), however, with only 9% yield (Table 1, entry 1). Subsequent efforts to improve the activity by enhancing the dissociation efficiency of latent carbocation through heating or photolysis did not lead to any improvement. We next investigated whether the tritylium salts with a chiral weakly coordinating metal-based phosphate anion could facilitate the asymmetric catalytic Diels–Alder reaction. To implement this strategy, different trityl phosphates or halides, Lewis acids, chiral metal phosphate and their combinations were examined in the model reaction of anthracene (3a) and β,γ-unsaturated α-ketoester 4a. When TP was first treated with metal Lewis acid (Scheme 2a, and Table S1 in Supporting Information File 1), the reaction showed good reactivity but no enantioselectivity at all, indicating a strong background reaction (Table 1, entry 2). We next examined the second strategy in which trityl bromide was treated with preformed chiral metal phosphate to their equilibration before they were subjected to the catalytic test. When metal monophosphates 1a–c (Table 1, entries 3–5) were applied, the reaction started showing some enantioselectivity with decent activity maintained. The combined use of trityl bromide and 1a (10 mol %) led to the desired adduct 5a with 55% yield and in 14% ee at 50 °C (Table 1, entry 3). This is in contrast to the TP/InBr3 combination where the reaction was much faster but racemic (Table 1, entry 3 vs 2), suggesting that the preformed metal phosphate is critical to effect catalysis and chiral induction. Among the metals screened, Fe(III) phosphate gave the optimal results in terms of both activity and enantioselectivity (79% yield, 35% ee, Table 1, entry 5). Fe(III)-based bisphosphate anions were also tested. To our delight, when trityl bromide and 2a (10 mol %) were used, the reaction gave a slightly increased enantioselectivity (Table 1, entry 6). Further improvement on activity and enantioselectivity could be achieved by conducting the reaction in DCM as the solvent (Table 1, entries 7 vs 6, 8–10). Next, we screened different chiral Fe(III) bisphosphates 2a–f and the best results were obtained in the presence of 2f, whereas others resulted in either low activity or poor enantioselectivity (Table 1, entries 15 vs 7, 11–14). Eventually, trityl chloride and chiral Fe(III) bisphosphate 2f were identified to be the optimal combination, affording adduct 5a in 91% ee and 70% yield at room temperature (Table 1, entries 17 and 18).
Table 1: Screening and optimization for the asymmetric catalyzed Diels–Alder reaction of anthracene by carbocations.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-129-i4.svg?max-width=637&scale=1.0)
|
|||||
| entrya | carbocation | solvent | yield (%)b | eec | |
| TrX | Lewis acid | ||||
| 1 | TP | none | DCE | 9 | 97 |
| metal-based monophosphate anion | |||||
| 2 | TP | InBr3 | DCE | 94 | rac |
| 3 | Ph3CBr | 1a | DCE | 55 | 14 |
| 4 | Ph3CBr | 1b | DCE | 49 | −16 |
| 5 | Ph3CBr | 1c | DCE | 79 | 36 |
| Fe(III)-based bisphosphate anion | |||||
| 6 | Ph3CBr | 2a | DCE | 46 | 40 |
| 7 | Ph3CBr | 2a | DCM | 58 | 56 |
| 8 | Ph3CBr | 2a | CHCl3 | 36 | 42 |
| 9 | Ph3CBr | 2a | toluene | 20 | 46 |
| 10 | Ph3CBr | 2a | CH3CN | nr | – |
| 11 | Ph3CBr | 2b | DCM | 17 | 14 |
| 12 | Ph3CBr | 2c | DCM | 55 | 28 |
| 13 | Ph3CBr | 2d | DCM | 67 | 26 |
| 14 | Ph3CBr | 2e | DCM | 22 | 68 |
| 15 | Ph3CBr | 2f | DCM | 70 | 74 |
| 16d | Ph3CBr | 2f | DCM | 55 | 90 |
| 17d | Ph3CCl | 2f | DCM | 57 | 91 |
| 18d,e | Ph3CCl | 2f | DCM | 70 | 91 |
| 19d | none | 2f | DCM | nr | – |
| 20d | Ph3CCl | none | DCM | nr | – |
aGeneral conditions: 3a (0.4 mmol), 4a (0.2 mmol), TrX (10 mol %), and Lewis acid (10 mol %) in 2 mL solvent at 50 °C. bYield of isolated product. cDetermined by HPLC analysis on a chiral stationary phase. dRoom temperature. e48 h.
In a control experiment, we found that chiral iron salt 2f itself turned out to be ineffective to catalyze the reaction in the absence of trityl chloride (Table 1, entry 19), indicating that the reaction is catalyzed by tritylium salts with Fe(III)-complexed bisphosphate as the chirality-inducing anion.
With the optimal reactions conditions established, the scope was next explored with Ph3CCl/2f in CH2Cl2 (DCM) at room temperature and the results are presented in Table 2. A variety of β,γ-unsaturated α-ketoesters 4 was subjected to the reaction with anthracene (3a) to give the desired cycloadducts 5a–n in moderate to good yields and with up to 93% ee. The bulkier isopropyl ketoester resulted in a lower yield and enantioselectivity (Table 2, entry 3 vs 1 and 2). Variations on the aromatic group of the ketoesters were well tolerated, giving the products in decent yields and high enantioselectivities. Unfortunately, no reaction was observed when an aliphatic substituted β,γ-unsaturated α-ketoester was used (data not shown).
Table 2: Scope for the asymmetric catalyzed Diels–Alder reaction of anthracene (3a) with ketoesters 4 by carbocations.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-129-i5.svg?max-width=637&scale=1.0)
|
||||
| entrya | α-ketoesters | product | yield (%)b | ee (%)c |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-129-i6.svg?max-width=637&scale=1.0)
4a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-129-i7.svg?max-width=637&scale=1.0)
5a |
70 | 91 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-129-i8.svg?max-width=637&scale=1.0)
4b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-129-i9.svg?max-width=637&scale=1.0)
5b |
82 | 74 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-129-i10.svg?max-width=637&scale=1.0)
4c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-129-i11.svg?max-width=637&scale=1.0)
5c |
46 | 55 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-129-i12.svg?max-width=637&scale=1.0)
4d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-129-i13.svg?max-width=637&scale=1.0)
5d |
74 | 80 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-129-i14.svg?max-width=637&scale=1.0)
4e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-129-i15.svg?max-width=637&scale=1.0)
5e |
68 | 75 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-129-i16.svg?max-width=637&scale=1.0)
4f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-129-i17.svg?max-width=637&scale=1.0)
5f |
66 | 81 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-129-i18.svg?max-width=637&scale=1.0)
4g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-129-i19.svg?max-width=637&scale=1.0)
5g |
77 | 76 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-129-i20.svg?max-width=637&scale=1.0)
4h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-129-i21.svg?max-width=637&scale=1.0)
5h |
48 | 80 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-129-i22.svg?max-width=637&scale=1.0)
4i |
![[Graphic 20]](/bjoc/content/inline/1860-5397-15-129-i23.svg?max-width=637&scale=1.0)
5i |
68 | 93 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-15-129-i24.svg?max-width=637&scale=1.0)
4j |
![[Graphic 22]](/bjoc/content/inline/1860-5397-15-129-i25.svg?max-width=637&scale=1.0)
5j |
92 | 91 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-15-129-i26.svg?max-width=637&scale=1.0)
4k |
![[Graphic 24]](/bjoc/content/inline/1860-5397-15-129-i27.svg?max-width=637&scale=1.0)
5k |
86 | 87 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-15-129-i28.svg?max-width=637&scale=1.0)
4l |
![[Graphic 26]](/bjoc/content/inline/1860-5397-15-129-i29.svg?max-width=637&scale=1.0)
5l |
76 | 89 |
| 13 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-15-129-i30.svg?max-width=637&scale=1.0)
4m |
![[Graphic 28]](/bjoc/content/inline/1860-5397-15-129-i31.svg?max-width=637&scale=1.0)
5m |
85 | 73 |
| 14 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-15-129-i32.svg?max-width=637&scale=1.0)
4n |
![[Graphic 30]](/bjoc/content/inline/1860-5397-15-129-i33.svg?max-width=637&scale=1.0)
5n |
42 | 83 |
aGeneral conditions: 3a (0.4 mmol), 4 (0.2 mmol), TrCl (10 mol %), and 2a (10 mol %) in DCM (2 mL) at room temperature. bYield of isolated product. cDetermined by HPLC analysis on a chiral stationary phase.
The Diels–Alder reaction of substituted anthracenes has been well-developed and we next examined the scope with substituted anthracenes. Unfortunately, these well-explored substrates did not work in our chiral catalysis system giving either no activity or poor enantioselectivity, particularly in cases of 9-monosubstituted anthracenes. When 9,10-dimethylanthracene (3b) was used, the reaction showed high yield (93% for 5o) but low enantioselectivity (23% ee, Scheme 3a). Surprisingly, the chiral iron salt 2f itself in the absence of trityl chloride also promoted the reaction, showing a relatively lower activity with 85% yield of 5o but opposite chiral induction (−65% ee, Scheme 3a). The electron-rich nature of dimethylanthracene may account for catalysis with the iron salts. On the other hand, an opposite chiral induction in this case is a clear indication of distinctive carbocation catalysis instead of metal Lewis acid catalysis in the presence of trityl chloride.
![[1860-5397-15-129-i3]](/bjoc/content/inline/1860-5397-15-129-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: a) The reaction with 9,10-dimethylanthracene (3b). b) Gram-scale reaction of 3a and 4k, and transformation of cycloadduct 5k.
Scheme 3: a) The reaction with 9,10-dimethylanthracene (3b). b) Gram-scale reaction of 3a and 4k, and transfo...
In addition, we tested the current carbocation catalytic system to prepare cycloadduct 5k in a large scale (Scheme 3b). When using 10 mol % Ph3CCl/2f, the reaction afforded cycloadduct 5k in 88% yield of isolated product and with 87% ee. In the presence of MgSO4 (5 equiv), treatment of 5k (1 equiv) with sulfonylhydrazine 6 (1.2 equiv) in CH2Cl2 led to the desired N-tosylhydrazone 7 in 83% yield and with 82% ee (Scheme 3b). The absolute configuration was assigned on the basis of the structure of 7, which was confirmed unambiguously by an X-ray crystallographic study [39]. Tentative transition states to account for the observed stereoselectivity are provided in Supporting Information File 1, Figure S3.
Conclusion
In summary, we have introduced a new motif of chiral weakly coordinating Fe(III)-based bisphosphate anion for high performance asymmetric carbocation Lewis acid catalysis. The introduction of a metal-coordinated phosphonate anion with balanced association ability with tritylium ions provided a new opportunity in pursuing chiral ion pair-type carbocation catalysis. The resulted asymmetric tritylium catalysis has enabled the so-far challenging Diels–Alder reactions of unsubstituted anthracene with good activity and up to 93% ee. Further studies are currently underway to elucidate the mechanistic details and to extend the chiral tritylium salt catalysis to other reactions.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of all products, copies of 1H and 13C NMR, IR, HRMS, and HPLC spectra of all compounds. | ||
| Format: PDF | Size: 3.1 MB | Download |
Acknowledgements
We thank the Natural Science Foundation of China (21390400, 21521002, 21472193) and the Chinese Academy of Sciences (QYZDJ-SSW-SLU023) for financial support. S.L. is supported by the National Program of Top-notch Yong Professionals, and J. L. is supported by the Taishan Scholarship Project of Shandong Province.
References
-
Mukaiyama, T.; Kobayashi, S.; Shoda, S.-i. Chem. Lett. 1984, 13, 907–910. doi:10.1246/cl.1984.907
Return to citation in text: [1] -
Mukaiyama, T.; Kobayashi, S.; Murakami, M. Chem. Lett. 1985, 14, 447–450. doi:10.1246/cl.1985.447
Return to citation in text: [1] -
Kobayashi, S.; Matsui, S.; Mukaiyama, T. Chem. Lett. 1988, 17, 1491–1494. doi:10.1246/cl.1988.1491
Return to citation in text: [1] -
Yanagisawa, M.; Mukaiyama, T. Chem. Lett. 2001, 30, 224–225. doi:10.1246/cl.2001.224
Return to citation in text: [1] -
Roy, S. R.; Nijamudheen, A.; Pariyar, A.; Ghosh, A.; Vardhanapu, P. K.; Mandal, P. K.; Mandal, P. K.; Datta, A.; Mandal, S. K. ACS Catal. 2014, 4, 4307–4319. doi:10.1021/cs5010695
Return to citation in text: [1] -
Bah, J.; Franzén, J. Chem. – Eur. J. 2014, 20, 1066–1072. doi:10.1002/chem.201304160
Return to citation in text: [1] -
Bah, J.; Naidu, V. R.; Teske, J.; Franzén, J. Adv. Synth. Catal. 2015, 357, 148–158. doi:10.1002/adsc.201400609
Return to citation in text: [1] -
Naidu, V. R.; Ni, S.; Franzén, J. ChemCatChem 2015, 7, 1896–1905. doi:10.1002/cctc.201500225
Return to citation in text: [1] -
Moosavi-Zare, A. R.; Zolfigol, M. A.; Rezanejad, Z. Can. J. Chem. 2016, 94, 626–630. doi:10.1139/cjc-2015-0629
Return to citation in text: [1] -
Zhang, Q.; Lü, J.; Luo, S. Acta Chim. Sin. (Chin. Ed.) 2016, 74, 61–66. doi:10.6023/a15090587
Return to citation in text: [1] -
Lyons, D. J. M.; Crocker, R. D.; Enders, D.; Nguyen, T. V. Green Chem. 2017, 19, 3993–3996. doi:10.1039/c7gc01519d
Return to citation in text: [1] -
Liu, J.; Xu, J.; Li, Z.; Huang, Y.; Wang, H.; Gao, Y.; Guo, T.; Ouyang, P.; Guo, K. Eur. J. Org. Chem. 2017, 3996–4003. doi:10.1002/ejoc.201700634
Return to citation in text: [1] -
Zhang, Q.; Lv, J.; Li, S.; Luo, S. Org. Lett. 2018, 20, 2269–2272. doi:10.1021/acs.orglett.8b00619
Return to citation in text: [1] [2] -
Riant, O.; Samuel, O.; Kagan, H. B. J. Am. Chem. Soc. 1993, 115, 5835–5836. doi:10.1021/ja00066a066
Return to citation in text: [1] -
Taudien, S.; Riant, O.; Kagan, H. B. Tetrahedron Lett. 1995, 36, 3513–3516. doi:10.1016/0040-4039(95)00531-g
Return to citation in text: [1] -
Sammakia, T.; Latham, H. A. Tetrahedron Lett. 1995, 36, 6867–6870. doi:10.1016/0040-4039(95)01440-s
Return to citation in text: [1] -
Chen, C.-T.; Chao, S.-D.; Yen, K.-C.; Chen, C.-H.; Chou, I.-C.; Hon, S.-W. J. Am. Chem. Soc. 1997, 119, 11341–11342. doi:10.1021/ja970900o
Return to citation in text: [1] -
Magdziak, D.; Pettus, L. H.; Pettus, T. R. R. Org. Lett. 2001, 3, 557–559. doi:10.1021/ol006963k
Return to citation in text: [1] -
Pommerening, P.; Mohr, J.; Friebel, J.; Oestreich, M. Eur. J. Org. Chem. 2017, 2312–2316. doi:10.1002/ejoc.201700239
Return to citation in text: [1] -
Lv, J.; Zhang, Q.; Zhong, X.; Luo, S. J. Am. Chem. Soc. 2015, 137, 15576–15583. doi:10.1021/jacs.5b11085
Return to citation in text: [1] [2] -
Ni, S.; Ramesh Naidu, V.; Franzén, J. Eur. J. Org. Chem. 2016, 1708–1713. doi:10.1002/ejoc.201501621
Return to citation in text: [1] -
Krossing, I.; Raabe, I. Angew. Chem., Int. Ed. 2004, 43, 2066–2090. doi:10.1002/anie.200300620
Return to citation in text: [1] -
Strauss, S. H. Chem. Rev. 1993, 93, 927–942. doi:10.1021/cr00019a005
Return to citation in text: [1] -
Moss, S.; King, B. T.; de Meijere, A.; Kozhushkov, S. I.; Eaton, P. E.; Michl, J. Org. Lett. 2001, 3, 2375–2377. doi:10.1021/ol0161864
Return to citation in text: [1] -
Anderson, L. L.; Arnold, J.; Bergman, R. G. J. Am. Chem. Soc. 2005, 127, 14542–14543. doi:10.1021/ja053700i
Return to citation in text: [1] -
Nguyen, T.-T.-T.; Türp, D.; Wagner, M.; Müllen, K. Angew. Chem., Int. Ed. 2013, 52, 669–673. doi:10.1002/anie.201206010
Return to citation in text: [1] -
Fischer, S.; Schmidt, J.; Strauch, P.; Thomas, A. Angew. Chem., Int. Ed. 2013, 52, 12174–12178. doi:10.1002/anie.201303045
Return to citation in text: [1] -
Roberts, J. A. S.; Chen, M.-C.; Seyam, A. M.; Li, L.; Zuccaccia, C.; Stahl, N. G.; Marks, T. J. J. Am. Chem. Soc. 2007, 129, 12713–12733. doi:10.1021/ja0680360
Return to citation in text: [1] -
Chen, M.-C.; Roberts, J. A. S.; Seyam, A. M.; Li, L.; Zuccaccia, C.; Stahl, N. G.; Marks, T. J. Organometallics 2006, 25, 2833–2850. doi:10.1021/om0508334
Return to citation in text: [1] -
Metz, M. V.; Sun, Y.; Stern, C. L.; Marks, T. J. Organometallics 2002, 21, 3691–3702. doi:10.1021/om020087s
Return to citation in text: [1] -
Mager, M.; Becke, S.; Windisch, H.; Denninger, U. Angew. Chem., Int. Ed. 2001, 40, 1898–1902. doi:10.1002/1521-3773(20010518)40:10<1898::aid-anie1898>3.0.co;2-k
Return to citation in text: [1] -
Zhou, J.; Lancaster, S. J.; Walker, D. A.; Beck, S.; Thornton-Pett, M.; Bochmann, M. J. Am. Chem. Soc. 2001, 123, 223–237. doi:10.1021/ja002820h
Return to citation in text: [1] -
Sun, Y.; Metz, M. V.; Stern, C. L.; Marks, T. J. Organometallics 2000, 19, 1625–1627. doi:10.1021/om990946l
Return to citation in text: [1] -
Mahlau, M.; List, B. Angew. Chem., Int. Ed. 2013, 52, 518–533. doi:10.1002/anie.201205343
Return to citation in text: [1] -
Phipps, R. J.; Hamilton, G. L.; Toste, F. D. Nat. Chem. 2012, 4, 603–614. doi:10.1038/nchem.1405
Return to citation in text: [1] -
Hinz, A.; Labbow, R.; Reiß, F.; Schulz, A.; Sievert, K.; Villinger, A. Struct. Chem. 2015, 26, 1641–1650. doi:10.1007/s11224-015-0638-0
Return to citation in text: [1] -
Lv, J.; Zhang, L.; Luo, S.; Cheng, J.-P. Angew. Chem., Int. Ed. 2013, 52, 9786–9790. doi:10.1002/anie.201304561
Return to citation in text: [1] -
Wang, L.; Lv, J.; Zhang, L.; Luo, S. Angew. Chem., Int. Ed. 2017, 56, 10867–10871. doi:10.1002/anie.201704020
Return to citation in text: [1] -
CCDC 1863542 (7) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Return to citation in text: [1]
| 1. | Mukaiyama, T.; Kobayashi, S.; Shoda, S.-i. Chem. Lett. 1984, 13, 907–910. doi:10.1246/cl.1984.907 |
| 2. | Mukaiyama, T.; Kobayashi, S.; Murakami, M. Chem. Lett. 1985, 14, 447–450. doi:10.1246/cl.1985.447 |
| 3. | Kobayashi, S.; Matsui, S.; Mukaiyama, T. Chem. Lett. 1988, 17, 1491–1494. doi:10.1246/cl.1988.1491 |
| 4. | Yanagisawa, M.; Mukaiyama, T. Chem. Lett. 2001, 30, 224–225. doi:10.1246/cl.2001.224 |
| 5. | Roy, S. R.; Nijamudheen, A.; Pariyar, A.; Ghosh, A.; Vardhanapu, P. K.; Mandal, P. K.; Mandal, P. K.; Datta, A.; Mandal, S. K. ACS Catal. 2014, 4, 4307–4319. doi:10.1021/cs5010695 |
| 6. | Bah, J.; Franzén, J. Chem. – Eur. J. 2014, 20, 1066–1072. doi:10.1002/chem.201304160 |
| 7. | Bah, J.; Naidu, V. R.; Teske, J.; Franzén, J. Adv. Synth. Catal. 2015, 357, 148–158. doi:10.1002/adsc.201400609 |
| 8. | Naidu, V. R.; Ni, S.; Franzén, J. ChemCatChem 2015, 7, 1896–1905. doi:10.1002/cctc.201500225 |
| 9. | Moosavi-Zare, A. R.; Zolfigol, M. A.; Rezanejad, Z. Can. J. Chem. 2016, 94, 626–630. doi:10.1139/cjc-2015-0629 |
| 10. | Zhang, Q.; Lü, J.; Luo, S. Acta Chim. Sin. (Chin. Ed.) 2016, 74, 61–66. doi:10.6023/a15090587 |
| 11. | Lyons, D. J. M.; Crocker, R. D.; Enders, D.; Nguyen, T. V. Green Chem. 2017, 19, 3993–3996. doi:10.1039/c7gc01519d |
| 12. | Liu, J.; Xu, J.; Li, Z.; Huang, Y.; Wang, H.; Gao, Y.; Guo, T.; Ouyang, P.; Guo, K. Eur. J. Org. Chem. 2017, 3996–4003. doi:10.1002/ejoc.201700634 |
| 13. | Zhang, Q.; Lv, J.; Li, S.; Luo, S. Org. Lett. 2018, 20, 2269–2272. doi:10.1021/acs.orglett.8b00619 |
| 24. | Moss, S.; King, B. T.; de Meijere, A.; Kozhushkov, S. I.; Eaton, P. E.; Michl, J. Org. Lett. 2001, 3, 2375–2377. doi:10.1021/ol0161864 |
| 25. | Anderson, L. L.; Arnold, J.; Bergman, R. G. J. Am. Chem. Soc. 2005, 127, 14542–14543. doi:10.1021/ja053700i |
| 26. | Nguyen, T.-T.-T.; Türp, D.; Wagner, M.; Müllen, K. Angew. Chem., Int. Ed. 2013, 52, 669–673. doi:10.1002/anie.201206010 |
| 27. | Fischer, S.; Schmidt, J.; Strauch, P.; Thomas, A. Angew. Chem., Int. Ed. 2013, 52, 12174–12178. doi:10.1002/anie.201303045 |
| 22. | Krossing, I.; Raabe, I. Angew. Chem., Int. Ed. 2004, 43, 2066–2090. doi:10.1002/anie.200300620 |
| 23. | Strauss, S. H. Chem. Rev. 1993, 93, 927–942. doi:10.1021/cr00019a005 |
| 20. | Lv, J.; Zhang, Q.; Zhong, X.; Luo, S. J. Am. Chem. Soc. 2015, 137, 15576–15583. doi:10.1021/jacs.5b11085 |
| 21. | Ni, S.; Ramesh Naidu, V.; Franzén, J. Eur. J. Org. Chem. 2016, 1708–1713. doi:10.1002/ejoc.201501621 |
| 14. | Riant, O.; Samuel, O.; Kagan, H. B. J. Am. Chem. Soc. 1993, 115, 5835–5836. doi:10.1021/ja00066a066 |
| 15. | Taudien, S.; Riant, O.; Kagan, H. B. Tetrahedron Lett. 1995, 36, 3513–3516. doi:10.1016/0040-4039(95)00531-g |
| 16. | Sammakia, T.; Latham, H. A. Tetrahedron Lett. 1995, 36, 6867–6870. doi:10.1016/0040-4039(95)01440-s |
| 17. | Chen, C.-T.; Chao, S.-D.; Yen, K.-C.; Chen, C.-H.; Chou, I.-C.; Hon, S.-W. J. Am. Chem. Soc. 1997, 119, 11341–11342. doi:10.1021/ja970900o |
| 18. | Magdziak, D.; Pettus, L. H.; Pettus, T. R. R. Org. Lett. 2001, 3, 557–559. doi:10.1021/ol006963k |
| 19. | Pommerening, P.; Mohr, J.; Friebel, J.; Oestreich, M. Eur. J. Org. Chem. 2017, 2312–2316. doi:10.1002/ejoc.201700239 |
| 20. | Lv, J.; Zhang, Q.; Zhong, X.; Luo, S. J. Am. Chem. Soc. 2015, 137, 15576–15583. doi:10.1021/jacs.5b11085 |
| 13. | Zhang, Q.; Lv, J.; Li, S.; Luo, S. Org. Lett. 2018, 20, 2269–2272. doi:10.1021/acs.orglett.8b00619 |
| 36. | Hinz, A.; Labbow, R.; Reiß, F.; Schulz, A.; Sievert, K.; Villinger, A. Struct. Chem. 2015, 26, 1641–1650. doi:10.1007/s11224-015-0638-0 |
| 39. | CCDC 1863542 (7) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre. |
| 34. | Mahlau, M.; List, B. Angew. Chem., Int. Ed. 2013, 52, 518–533. doi:10.1002/anie.201205343 |
| 35. | Phipps, R. J.; Hamilton, G. L.; Toste, F. D. Nat. Chem. 2012, 4, 603–614. doi:10.1038/nchem.1405 |
| 28. | Roberts, J. A. S.; Chen, M.-C.; Seyam, A. M.; Li, L.; Zuccaccia, C.; Stahl, N. G.; Marks, T. J. J. Am. Chem. Soc. 2007, 129, 12713–12733. doi:10.1021/ja0680360 |
| 29. | Chen, M.-C.; Roberts, J. A. S.; Seyam, A. M.; Li, L.; Zuccaccia, C.; Stahl, N. G.; Marks, T. J. Organometallics 2006, 25, 2833–2850. doi:10.1021/om0508334 |
| 30. | Metz, M. V.; Sun, Y.; Stern, C. L.; Marks, T. J. Organometallics 2002, 21, 3691–3702. doi:10.1021/om020087s |
| 31. | Mager, M.; Becke, S.; Windisch, H.; Denninger, U. Angew. Chem., Int. Ed. 2001, 40, 1898–1902. doi:10.1002/1521-3773(20010518)40:10<1898::aid-anie1898>3.0.co;2-k |
| 32. | Zhou, J.; Lancaster, S. J.; Walker, D. A.; Beck, S.; Thornton-Pett, M.; Bochmann, M. J. Am. Chem. Soc. 2001, 123, 223–237. doi:10.1021/ja002820h |
| 33. | Sun, Y.; Metz, M. V.; Stern, C. L.; Marks, T. J. Organometallics 2000, 19, 1625–1627. doi:10.1021/om990946l |
| 37. | Lv, J.; Zhang, L.; Luo, S.; Cheng, J.-P. Angew. Chem., Int. Ed. 2013, 52, 9786–9790. doi:10.1002/anie.201304561 |
| 38. | Wang, L.; Lv, J.; Zhang, L.; Luo, S. Angew. Chem., Int. Ed. 2017, 56, 10867–10871. doi:10.1002/anie.201704020 |
© 2019 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)