Abstract
The one-pot reaction of 2,6-bis(diphenylmethyl)-4-methoxyaniline with tert-butylnitrite, BTEAC and DABSO in the presence of CuCl2 provided an unexpected 3H-indazole product 8. The structure of the compound was determined by HRMS, IR, NMR and further confirmed by single crystal X-ray crystallography. The compound crystallises in the triclinic P-1 space group, with unit cell parameters a = 9.2107 (4), b = 10.0413 (5), c = 14.4363 (6) Å, α = 78.183 (2), β = 87.625 (2), γ = 71.975 (2)°. The formation of 8 proceeded through a facile intramolecular [2 + 3] cycloaddition of the diazo intermediate 9. The molecules of 8 are organised by edge–face Ar–H···π, face–face π···π, and bifurcated OCH2–H···N interactions. In addition to these, there are Ar–H···H–Ar close contacts, (edge–edge and surrounding inversion centres) arranged as infinite tapes along the a direction.

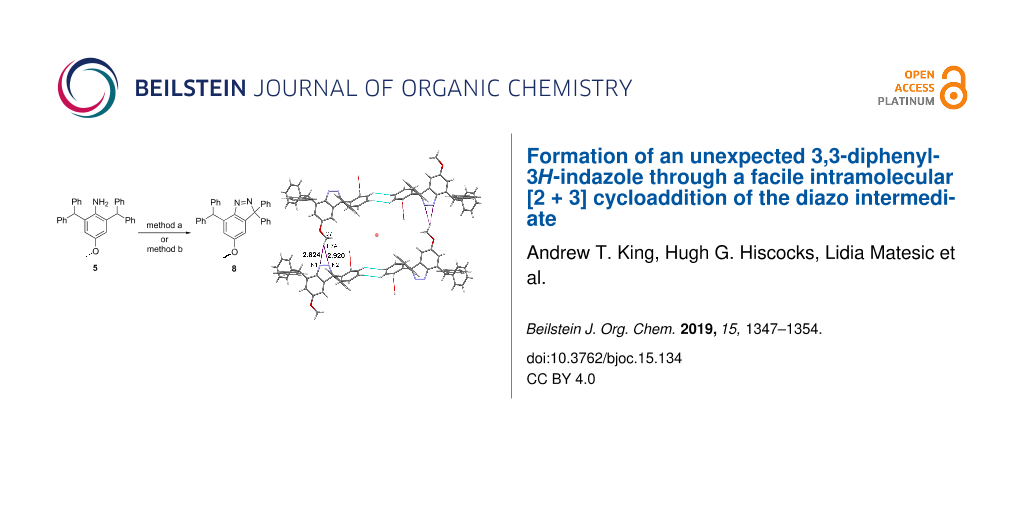
Graphical Abstract
Introduction
The use of sulfonyl fluorides in biological applications has been on the increase as evidenced by the number of recent publications [1-3]. One potential application of sulfonyl fluorides is as an 18F-radiolabelled synthon that can be conjugated to macromolecules and used to image cancer and other diseases in the body. Synthons are becoming more prevalent in imaging modalities like positron emission tomography (PET) as there is a greater need to further investigate lesions, and disease tissues on the molecular level. However, issues with the stability of sulfonyl fluorides have been reported in the literature [4,5].
Stability and radiochemical improvements were investigated by Matesic et al. [5]. For instance, electron-donating 18F-arylsulfonyl fluoride 1 was >98% stable in the buffer at 3 h, while the electron-withdrawing compound 2 was only 8% stable [5] (Figure 1). The more sterically hindered 18F-arylsulfonyl fluoride 3 was 95% intact in rat plasma after 120 min at 37 °C [5]. This indicated that the combination of electron-donating effects and steric hindrance of isopropyl groups provide greater stability to a sulfur–fluorine bond than groups that can only provide the steric hindrance in the molecule [6].
![[1860-5397-15-134-1]](/bjoc/content/figures/1860-5397-15-134-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of 18F-radiolabelled arylsulfonyl fluorides containing electron-donating 1, electron-withdrawing 2, and sterically bulky groups 3 [5] and 4.
Figure 1: Examples of 18F-radiolabelled arylsulfonyl fluorides containing electron-donating 1, electron-withd...
To further investigate the stability of 18F-sulfonyl fluorides, the sterically hindered compound 4 was selected with enough steric bulk in the 2,6-position. The initial chlorosulfonylation reaction was performed using 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct (DABSO) as a source of sulfur dioxide. DABSO was selected as it would provide a facile method and is a safer alternative to sulfur dioxide gas [7-10]. The target molecule 4 was envisaged via the diazonium salt of 5 [11] and 6 and was a modification of the microfluidic flow reaction reported by Malet-Sanz et al. [12] (Scheme 1).
![[1860-5397-15-134-i1]](/bjoc/content/inline/1860-5397-15-134-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction for the formation of sulfonyl chloride 6 using DABSO.
Scheme 1: Reaction for the formation of sulfonyl chloride 6 using DABSO.
Herein, we report the unexpected product derived from 5 under the reaction conditions described in Scheme 1. The unusual crystal packing present in this 3H-indazole product was also analysed.
Results and Discussion
The one-pot reaction of compound 5 with tert-BuONO and DABSO in the presence of benzyltriethylammonium chloride (BTEAC) and CuCl2 gave a crude product which was purified by crystallisation from dichloromethane/hexane to give a pale yellow crystalline material in 27% yield. HRMS–ESI analysis produced [M + H]+ m/z 467.2126 corresponding to the formula of C33H27N2O. Considering that the expected mass of the sulfonyl chloride 6 was m/z 539.1403 with the corresponding formula of C33H28ClO3S, it was concluded that 6 did not form. It was initially thought that the product was the diazonium intermediate 7 (Figure 2), as it was expected from the reaction conditions and the HRMS analysis of diazonium ion 7 in the form of [M]+ was C33H27N2O+.
![[1860-5397-15-134-2]](/bjoc/content/figures/1860-5397-15-134-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible compounds with the molecular formula C33H26N2O (structure 7 contains 27 hydrogen atoms).
Figure 2: Possible compounds with the molecular formula C33H26N2O (structure 7 contains 27 hydrogen atoms).
Spectroscopy analysis
A close analysis of the 1H NMR spectrum revealed the non-symmetrical substitution of the aromatic core. Two aromatic protons appeared as two doublets (J = 2 Hz) at 6.92 and 6.67 ppm, respectively. Furthermore, the spectrum showed only one benzylic proton, as a singlet at 6.82 ppm. 13C NMR and HSQC showed tertiary and quaternary resonances at 51.3 and 101.5 ppm, respectively. The first resonance is indicative of HC(Ph)2, while the one at 101.5 ppm is more likely to be the 3,3-diphenyl-substituted carbon of the indazole ring. The ambiguity was overcome through the X-ray structural analysis of the compound. The single crystals were easily obtained by recrystallisation from CH2Cl2.
Crystal structure analysis
The X-ray crystal analysis revealed that the product was not the diazonium intermediate 7, but rather the compound 8 containing the 3,3-diphenyl-3H-indazole core structure. The crystals were found to be triclinic, P-1 space group with cell constants a = 9.2107(4), b = 10.0413(5), c = 14.4363(6) Å, α = 78.183(2), β = 87.625(2), γ = 71.975(2)°. The structure of 8 and the atom-labelling scheme are presented in Figure 3. The crystal and refinement data are given in Table 1. The 3H-indazole 8 (–N=N–CRR–) has a break in its conjugation at the sp3 carbon atom when it is compared to the 1H-indazole isomer –NH–N=CH– that is fully conjugated with the benzo group. Therefore, the 3H-indazole isomer is higher in energy and thus much less common. The bond lengths within this 3H-indazole ring structure confirm the evidence for sp2 N=N. The N1–C1 and N2–C9 distances are 1.426(2) Å and 1.530(2) Å. The N1=N2 length was found to be 1.266(2) Å, a value that agrees with literature reports [13].
![[1860-5397-15-134-3]](/bjoc/content/figures/1860-5397-15-134-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP view of the molecule 8 showing the atom labelling (ellipsoids are drawn at 50% probability level).
Figure 3: ORTEP view of the molecule 8 showing the atom labelling (ellipsoids are drawn at 50% probability le...
Table 1: Crystal data and structure refinement for compound 8.
| crystal data | |
| chemical formula | C33H26N2O |
| Mr | 466.56 |
| crystal system, space group | triclinic, P-1 |
| temperature (K) | 150 |
| a, b, c (Å) | 9.2107 (4), 10.0413 (5), 14.4363 (6) |
| α, β, γ (°) | 78.183 (2), 87.625 (2), 71.975 (2) |
| V (Å3) | 1242.40 (10) |
| Z | 2 |
| radiation type | Mo Kα |
| µ (mm−1) | 0.08 |
| crystal size (mm) | 0.22 × 0.19 × 0.11 |
| data collection | |
| diffractometer | Bruker APEX-II CCD |
| absorption correction | – |
| no. of measured, independent and observed [I > 2σ(I)] reflections | 40887, 5423, 4236 |
| Rint | 0.039 |
| (sin θ/λ)max (Å−1) | 0.640 |
| refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.054, 0.157, 1.04 |
| no. of reflections | 5423 |
| no. of parameters | 326 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.51, −0.20 |
| CCDC deposition number | 1902859 |
The 3,3-diphenyl-3H-indazole core of 8 carries both benzhydryl and germinal diphenyl groups. These aromatic substituents make significantly different intermolecular contacts in the crystal. The phenyl rings of the former (C1A to C6A, and C1B to C6B) make complementary C–H···π interactions with the related molecule across a centre of inversion. Hydrogen atoms C2A–H2A and C3A–H3A make an almost perpendicular approach to ring B at C2B and C6B, respectively: H2A···C2B = 3.04 Å and H3A···C6B = 2.96 Å (Figure 4a). One of the gem-diphenyl rings (C1C to C6C) also employs C-H···π interactions across a centre of inversion. Its partner is, however, the aromatic ring of the core (C1 to C6). Thus, the hydrogens C5C–H5C and C4C–H4C make nearly perpendicular approaches to atoms C5 and C3, respectively: H5C···C5 = 2.95 Å and H4C···C3 = 3.08 Å. This phenyl ring also participates in a slipped π···π contact with its inversion-related counterpart: C6C···C5C = 3.50 Å (Figure 4b).
![[1860-5397-15-134-4]](/bjoc/content/figures/1860-5397-15-134-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Significant intermolecular interactions made by the benzhydryl group (a, upper) and the gem-diphenyl group (b, lower). Crystallographic colour code: C–H···π (dark blue), π···π (light blue), inversion centre (orange sphere), and incomplete contacts (red).
Figure 4: Significant intermolecular interactions made by the benzhydryl group (a, upper) and the gem-dipheny...
The C4 methoxy and the heterocyclic diazo functionality of 8 are linked through an interesting arrangement along the b axis that can be described either as a OCH2–H···π (N=N) or as a bifurcated N···H(CH2O)···N interaction. This motif is close to symmetrical with H···N distances of C7–H7A···N1 = 2.82 Å and C7–H7A···N2 = 2.92 Å. The second of the gem-diphenyl rings (C1D to C6D) surrounds an inversion centre along with its counterpart through a cycle of two C4D–H4D···H5D–C5D contacts. Four molecules of 8, involving two C–H···N and two cyclic H···H motifs, surround another inversion centre (Figure 5a). Consideration of additional molecules reveals a stepladder assembly along b, in which the C–H···N interactions form the side rails and the cyclic H···H motifs the rungs (Figure 5b).
![[1860-5397-15-134-5]](/bjoc/content/figures/1860-5397-15-134-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Relationship of the C–H···N and cyclic C–H···H-C contacts in the crystal structure of 8. The centrosymmetric array of four molecules (5a, upper) and a stepladder association of eight molecules (5b, lower). Crystallographic colour code: C–H···N purple), C–H···H–C (light blue), inversion centre (orange sphere), and incomplete contacts (red).
Figure 5: Relationship of the C–H···N and cyclic C–H···H-C contacts in the crystal structure of 8. The centro...
As well as the centrosymmetric cycle of C4D–H4D···H5D–C5D contacts (H···H = 2.48 Å) there is also a linear C6D–H6D···H6D–C6D close contact (H···H = 2.37 Å) around a further inversion centre. These two motifs alternate along the a axis to produce infinite hydrocarbon tapes with inversion centres separated by a/2 (Figure 6).
![[1860-5397-15-134-6]](/bjoc/content/figures/1860-5397-15-134-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Part of a hydrocarbon tape along a formed by a combination of alternating linear and cyclic C–H···H–C close contacts. Crystallographic colour code: C–H···H–C (light blue), inversion centre (orange sphere), and incomplete contacts (red).
Figure 6: Part of a hydrocarbon tape along a formed by a combination of alternating linear and cyclic C–H···H...
The molecular structure of 8 precludes Pauling-type hydrogen bonding, and therefore weaker attractions must be used in the crystal packing. Its interaction between the methoxy and diazo functionalities is uncommon, though entirely reasonable in the context of weak hydrogen bonding [14]. The presence of close C–H···H–C contacts in this structure was, however, unexpected and required careful consideration. First, one of us has reported previously a short C–H···H–C contact in another structure [15]. This was a linear centrosymmetric motif formed by Ar–H (H···H = 2.35 Å) and is directly analogous in structure to the linear example (2.37 Å) found here. Both distances are less than the van der Waals (VDW) separation of 2.40 Å [16]. The new cyclic example is slightly longer (2.48 Å), but well under the value of 2.80 Å (= VDW + 0.40) discussed by Dance [17]. All three distance values must be treated with caution since their H atoms are in calculated positions. However, since these are well-defined Ar–H groups, their positional errors will be relatively small. Over recent years much understanding has been gained of dihydrogen bonding X–H···H–Y [18,19]. Close examination has been made of the alkane C–H···H–C contact, particularly for reactive structures stabilised by multiple tert-butyl groups [20,21], multi-ring cage hydrocarbons [22,23], and linear alkanes [22,23]. The interaction was found to be attractive in all these cases, and computational justifications have been published [21,23]. It, therefore, appears probable that, despite their unfamiliarity, the C–H···H–C contacts illustrated in Figure 6 are similarly attractive in nature.
Proposed mechanism for the formation of 8
Indazoles are well-recognised for their important biological activities. They are known to be used as building blocks in drug development. A review by Gaikwad et al. describes reliable routes to particularly fused aromatic 1H and 3H-indazoles [24]. The common synthetic routes for the formation of cyclic 1H-indazoles are diazotisation of corresponding o-alkylanilines [25] and nitrosation of the N-acetyl derivatives of 2-alkylanilines (Jacobson modification) [26-28]. More recently, the formation of cyclic 3,3-disubstituted 3H-indazoles was reported to form mainly through the [2 + 3] cycloaddition of diazo compounds with arynes under mild reaction conditions [29-31]. However, none of these contained a 2-diphenylmethyl (benzhydryl) aniline system, as found in our compound 5.
The formation of compound 8 can be explained by the rearrangement of 7 through a hydrogen-shift to form the diazo intermediate 9 which underwent a concerted [2 + 3] intramolecular cyclisation to 3,3-diphenyl-3H-indazole 8 (Scheme 2). The process was facilitated by the presence of a highly activated benzylic carbon and further expedited by three phenyl groups, to form the resonance stabilisation of 9 [32]. Furthermore, 8 cannot attain planarity of the methylene–benzhydryl fragment due to steric hindrance. These reactions appear to be also encouraged by the reduction of the crowding originally present in the diazonium salt.
![[1860-5397-15-134-i2]](/bjoc/content/inline/1860-5397-15-134-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the formation of 8.
Scheme 2: Proposed mechanism for the formation of 8.
The direct diazotisation of 5 was performed using anhydrous organic solvents and t-BuONO at −10 °C in the presence of CuCl2 (method a) and the classical NaNO2/HCl method b in the absence of CuCl2, as shown in Scheme 3. Compound 8 was obtained in 89% and 96% from method a or b, respectively. The results confirm our initial hypothesis that the cyclisation occurred through the diazo species 9 (Scheme 2) and the Cu2+ plays no part in the reaction process.
![[1860-5397-15-134-i3]](/bjoc/content/inline/1860-5397-15-134-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Direct preparation of compound 8. method a: t-BuONO, CuCl2, dry CH3CN, −10 °C, 89%; method b: NaNO2, HCl/acetic acid, −10 °C, 96%.
Scheme 3: Direct preparation of compound 8. method a: t-BuONO, CuCl2, dry CH3CN, −10 °C, 89%; method b: NaNO2...
Conclusion
The formation of the unexpected product 8 from 5 under anhydrous diazotisation conditions has led to the discovery of a facile synthetic pathway to 3,3-diphenyl-3H-indazoles. Formation of 8 has been proposed to proceed through the diazonium intermediate. This was further confirmed by reactions of 5 using t-BuONO, and the conventional NaNO2/HCl method to give 8 in almost quantitative yields. The crystal structure of 8 involves edge–face Ar–H···π, face–face π···π, bifurcated OCH2–H···N, plus edge–edge centrosymmetric Ar–H···H–Ar interactions. These rather uncommon examples of dihydrogen contact assemble as infinite tapes along the a direction.
Experimental
Materials and physical measurements
All chemical reagents and analytical grade solvents were obtained from commercial sources such as Sigma-Aldrich, Cambridge Isotope Laboratories Inc. and Merck Millipore. All reactions were monitored using either TLC aluminium oxide 60 F254 neutral or TLC Silica gel 60 F254 with UV detection at 254 nm. 1H NMR and 13C NMR spectra were recorded on an Agilent 500 MHz spectrometer (500 MHz 1H, 125 MHz 13C) in deuterated chloroform (CDCl3). The mass spectral data were obtained using the attached Agilent 5973n MS (EI) spectrometer. High-resolution mass spectra were obtained using an Agilent 6510 Q-TOF Mass Spectrometer (ESI). The infrared spectra were recorded on an Agilent Cary 630 FTIR with a diamond window using 16 background and sample scans. Melting points were measured on a Gallenkamp Melting Point Apparatus equipment and were uncorrected.
Synthesis of 7-benzhydryl-5-methoxy-3,3-diphenyl-3H-indazole (8)
2,6-Bis(diphenylmethyl)-4-methoxyaniline (5, 0.23 g, 0.505 mmol, 1 equiv) DABSO (0.57 g, 2.37 mmol, 4.75 equiv) and BTEAC (0.12 g, 1 mmol, 1 equiv) were dissolved in DCM (5 mL, solution A) that had been cooled to 0 °C. tert-Butyl nitrite (0.15 g, 170 μL) was dissolved in DCM (6 mL) and cooled to 0 °C (solution B). Copper(II) chloride (0.02 g, 0.10 mmol, 0.1 equiv) was dissolved in acetonitrile (6 mL) with sonication and cooled to 0 °C (solution C). Once all solutions had been cooled the following order of addition was used: solution C was added to solution A dropwise. Solution B was added dropwise to solutions A and C. The reaction was allowed to react at room temperature for 24 h before the organic product was extracted using DCM (3 × 10 mL) and brine (10 mL). The organic layers were combined, dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. The solid crude product was crystallised from dichloromethane to give a brown crystalline solid (0.064 g, 0.137 mmol, 27%); mp 183–184 °C; IR (neat) vmax/cm−1: 3057, 3025, 2959, 2926, 2650, 2322, 2112, 1943, 1591, 1490, 1470, 1445, 1349, 1263, 1205, 1127, 1023, 843, 748, 607; 1H NMR (500 MHz, CDCl3) δ 7.28–7.31 (m, H-Ar, 10H), 7.26–7.21 (m, H-Ar, 10H), 6.92 (d, J = 2.0 Hz, H-Ar, 1H), 6.82 (s, HC(Ph)2, 1H), 6.67 (d, J = 2.0 Hz, H-Ar, 1H), 3.75 (s, O-CH3, 3H); 13C NMR (125 MHz, CDCl3) δ 161.6 (C), 149.9 (C), 146.6 (C), 142.9 (2C), 139.9 (C), 138.3 (2C) 129.7 (4CH), 128.6 (4CH), 128.4 (4CH), 128.0 (2CH), 127.8 (4CH), 126.6 (2CH), 115.9 (CH), 107.7 (CH), 101.5 (C(Ph)2), 55.8 (CH3), 51.3 (HC(Ph)2); HRMS–ESI (m/z): [M + H]+ calcd for C33H27N2O, 467.2118; found, 467.2126.
Direct preparation of compound 8 - method a
Under a nitrogen atmosphere, compound 5 (0.23 g, 0.505 mmol, 1 equiv) was dissolved in acetonitrile (20 mL) then cooled to 0 °C (solution A). tert-Butyl nitrite (0.15 g, 170 μL) was dissolved in DCM (6 mL) and cooled to 0 °C (solution B). Copper(II) chloride (0.02 g, 0.10 mmol, 0.2 equiv), was dissolved in acetonitrile (6 mL) with sonication and cooled to 0 °C (solution C). Once all solutions had been cooled, the following order of addition was used: solution C was added to solution A dropwise. Solution B was added dropwise to solutions A and C. The reaction was allowed to react at room temperature for 24 h before the organic product was extracted using DCM (3 × 10 mL) and brine (10 mL). The organic layers were combined, dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure to give the crude product which was purified by filtering through a short column flash silica gel using DCM as the mobile phase to give 8 (0.210 g, 0.449 mmol, 89%).
Direct preparation of compound 8 - method b
Compound 5 (0.23 g, 0.505 mmol, 1 equiv) was dissolved in acetonitrile (10 mL) at room temperature before acetic acid (0.96 mL, 14.76 mmol, 29 equiv) and concentrated hydrochloric acid (0.98 mL, 27 mmol, 54 equiv) was added dropwise to the solution over 2 min at −10 °C. Sodium nitrite (0.123 g, 1.72 mmol, 3.4 equiv) in water (5 mL) was added to the stirring solution over 1 min at −10 °C. The resulting solution was stirred for 1 h at −10 °C followed by 24 h at room temperature. The organic product was extracted using DCM (3 × 20 mL) and brine (30 mL). The organic layers were combined, dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure to give the pure product 8 (0.226 g, 0.485 mmol, 96%). The 1H NMR spectrum of the crude product indicates that it is pure and required no further purification.
X-ray crystallography
A colourless plate-like crystal of 8 with dimensions of 0.11 × 0.19 × 0.22 mm, selected under the polarising microscope (Leica M165Z), was mounted on a MicroMount (MiTeGen, USA) consisting of a thin polymer tip with a wicking aperture. The X-ray diffraction measurements were carried out on a Bruker kappa-II CCD diffractometer at 150 K using IµS Incoatec Microfocus Source with Mo Kα radiation (λ = 0.710723 Å). The single crystal, mounted on the goniometer using a cryo-loop for intensity measurements, was coated with immersion oil type NVH and then quickly transferred to the cold nitrogen stream generated by an Oxford Cryostream 700 series. Symmetry-related absorption corrections using the program SADABS [33] were applied, and the data were corrected for Lorentz and polarisation effects using Bruker APEX3 software [33]. The structure was solved by program SHELXT [34] (with intrinsic phasing), and the full-matrix least-square refinements were carried out using SHELXL-2014 [35] through Olex2 [36] suite of software. The non-hydrogen atoms were refined anisotropically. Molecular graphics were generated using Mercury [37]. Key crystallographic data and refinement details are presented in Table 1.
Crystal structure data
Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number 1902859. The data can be obtained free of charge via http://www.ccdc.cam.ac.uk or by e-mailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223/336-033, Tel.: (+44) 1223/336-408.
Supporting Information
| Supporting Information File 1: Copies of 1H NMR, 13C NMR and IR of compound 8. | ||
| Format: PDF | Size: 491.1 KB | Download |
Acknowledgements
We are grateful to the University of Technology Sydney for providing financial support to this project and Drs Maxine Roberts, Benjamin Fraser and Giancarlo Pascali at ANSTO for technical assistance and useful discussions. We also would like to thank the Australian Institute of Nuclear Science and Engineering (AINSE) for providing Andrew King with the AINSE Honours scholarship.
References
-
Brouwer, A. J.; Jonker, A.; Werkhoven, P.; Kuo, E.; Li, N.; Gallastegui, N.; Kemmink, J.; Florea, B. I.; Groll, M.; Overkleeft, H. S.; Liskamp, R. M. J. J. Med. Chem. 2012, 55, 10995–11003. doi:10.1021/jm301443r
Return to citation in text: [1] -
Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650–2659. doi:10.1039/c5sc00408j
Return to citation in text: [1] -
Grimster, N. P.; Connelly, S.; Baranczak, A.; Dong, J.; Krasnova, L. B.; Sharpless, K. B.; Powers, E. T.; Wilson, I. A.; Kelly, J. W. J. Am. Chem. Soc. 2013, 135, 5656–5668. doi:10.1021/ja311729d
Return to citation in text: [1] -
Inkster, J. A. H.; Liu, K.; Ait-Mohand, S.; Schaffer, P.; Guérin, B.; Ruth, T. J.; Storr, T. Chem. – Eur. J. 2012, 18, 11079–11087. doi:10.1002/chem.201103450
Return to citation in text: [1] -
Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z
Return to citation in text: [1] [2] [3] [4] [5] -
Smith, T. A. D. J. Labelled Compd. Radiopharm. 2012, 55, 281–288. doi:10.1002/jlcr.2940
Return to citation in text: [1] -
Emmett, E. J.; Willis, M. C. Asian J. Org. Chem. 2015, 4, 602–611. doi:10.1002/ajoc.201500103
Return to citation in text: [1] -
Woolven, H.; González-Rodríguez, C.; Marco, I.; Thompson, A. L.; Willis, M. C. Org. Lett. 2011, 13, 4876–4878. doi:10.1021/ol201957n
Return to citation in text: [1] -
Deeming, A. S.; Russell, C. J.; Hennessy, A. J.; Willis, M. C. Org. Lett. 2014, 16, 150–153. doi:10.1021/ol403122a
Return to citation in text: [1] -
Hu, T.; Baxendale, I. R.; Baumann, M. Molecules 2016, 21, 918. doi:10.3390/molecules21070918
Return to citation in text: [1] -
Meiries, S.; Speck, K.; Cordes, D. B.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2013, 32, 330–339. doi:10.1021/om3011867
Return to citation in text: [1] -
Malet-Sanz, L.; Madrzak, J.; Ley, S. V.; Baxendale, I. R. Org. Biomol. Chem. 2010, 8, 5324–5332. doi:10.1039/c0ob00450b
Return to citation in text: [1] -
Zhou, J.; Liu, L. L.; Cao, L. L.; Stephan, D. W. Angew. Chem., Int. Ed. 2018, 57, 3322–3326. doi:10.1002/anie.201713118
Return to citation in text: [1] -
Desiraju, G. R.; Steiner, T. The Weak Hydrogen Bond: In Structure Chemistry and Biology; Oxford University Press: Oxford, United Kingdom, 1999.
Return to citation in text: [1] -
Bhosekar, G.; Murali, C.; Gonnade, R. G.; Shashidhar, M. S.; Bhadbhade, M. M. Cryst. Growth Des. 2005, 5, 1977–1982. doi:10.1021/cg050272j
Return to citation in text: [1] -
Bondi, A. J. Phys. Chem. 1964, 68, 441–451. doi:10.1021/j100785a001
Return to citation in text: [1] -
Dance, I. New J. Chem. 2003, 27, 22–27. doi:10.1039/b206867b
Return to citation in text: [1] -
Crabtree, R. H. Science 1998, 282, 2000–2001. doi:10.1126/science.282.5396.2000
Return to citation in text: [1] -
Custelcean, R.; Jackson, J. E. Chem. Rev. 2001, 101, 1963–1980. doi:10.1021/cr000021b
Return to citation in text: [1] -
Matta, C. F.; Hernández-Trujillo, J.; Tang, T.-H.; Bader, R. F. W. Chem. – Eur. J. 2003, 9, 1940–1951. doi:10.1002/chem.200204626
Return to citation in text: [1] -
Wang, C.-C.; Tang, T.-H.; Wu, L.-C.; Wang, Y. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004, 60, 488–493. doi:10.1107/s0108767304015375
Return to citation in text: [1] [2] -
Echeverría, J.; Aullón, G.; Danovich, D.; Shaik, S.; Alvarez, S. Nat. Chem. 2011, 3, 323–330. doi:10.1038/nchem.1004
Return to citation in text: [1] [2] -
Danovich, D.; Shaik, S.; Neese, F.; Echeverría, J.; Aullón, G.; Alvarez, S. J. Chem. Theory Comput. 2013, 9, 1977–1991. doi:10.1021/ct400070j
Return to citation in text: [1] [2] [3] -
Gaikwad, D. D.; Chapolikar, A. D.; Devkate, C. G.; Warad, K. D.; Tayade, A. P.; Pawar, R. P.; Domb, A. J. Eur. J. Med. Chem. 2015, 90, 707–731. doi:10.1016/j.ejmech.2014.11.029
Return to citation in text: [1] -
Stadlbauer, W. Indazoles. In Science of Synthesis; Neier, R., Ed.; Georg-Thieme Verlag: Stuttgart, New York, 2002; Vol. 12, pp 227–324.
Return to citation in text: [1] -
Jacobson, P.; Huber, L. Ber. Dtsch. Chem. Ges. 1908, 41, 660–671. doi:10.1002/cber.190804101127
Return to citation in text: [1] -
Rüchardt, C.; Hassmann, V. Liebigs Ann. Chem. 1980, 908–927. doi:10.1002/jlac.198019800611
Return to citation in text: [1] -
Yoshida, T.; Matsuura, N.; Yamamoto, K.; Doi, M.; Shimada, K.; Morie, T.; Kato, S. Heterocycles 1996, 43, 2701–2712. doi:10.3987/com-96-7614
Return to citation in text: [1] -
Jin, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2007, 46, 3323–3325. doi:10.1002/anie.200700101
Return to citation in text: [1] -
Liu, Z.; Shi, F.; Martinez, P. D. G.; Raminelli, C.; Larock, R. C. J. Org. Chem. 2008, 73, 219–226. doi:10.1021/jo702062n
Return to citation in text: [1] -
Cheng, B.; Bao, B.; Zu, B.; Duan, X.; Duan, S.; Li, Y.; Zhai, H. RSC Adv. 2017, 7, 54087–54090. doi:10.1039/c7ra12117b
Return to citation in text: [1] -
Shirtcliff, L. D.; Hayes, A. G.; Haley, M. M.; Köhler, F.; Hess, K.; Herges, R. J. Am. Chem. Soc. 2006, 128, 9711–9721. doi:10.1021/ja054547v
Return to citation in text: [1] -
APEX3, SAINT and SADABS, Bruker 2016; Bruker AXS Inc.: Madison, Wisconsin, USA, 2016.
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Macrae, C. F.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Shields, G. P.; Taylor, R.; Towler, M.; van de Streek, J. J. Appl. Crystallogr. 2006, 39, 453–457. doi:10.1107/s002188980600731x
Return to citation in text: [1]
| 34. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 35. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 36. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 1. | Brouwer, A. J.; Jonker, A.; Werkhoven, P.; Kuo, E.; Li, N.; Gallastegui, N.; Kemmink, J.; Florea, B. I.; Groll, M.; Overkleeft, H. S.; Liskamp, R. M. J. J. Med. Chem. 2012, 55, 10995–11003. doi:10.1021/jm301443r |
| 2. | Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650–2659. doi:10.1039/c5sc00408j |
| 3. | Grimster, N. P.; Connelly, S.; Baranczak, A.; Dong, J.; Krasnova, L. B.; Sharpless, K. B.; Powers, E. T.; Wilson, I. A.; Kelly, J. W. J. Am. Chem. Soc. 2013, 135, 5656–5668. doi:10.1021/ja311729d |
| 5. | Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z |
| 5. | Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z |
| 18. | Crabtree, R. H. Science 1998, 282, 2000–2001. doi:10.1126/science.282.5396.2000 |
| 19. | Custelcean, R.; Jackson, J. E. Chem. Rev. 2001, 101, 1963–1980. doi:10.1021/cr000021b |
| 5. | Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z |
| 15. | Bhosekar, G.; Murali, C.; Gonnade, R. G.; Shashidhar, M. S.; Bhadbhade, M. M. Cryst. Growth Des. 2005, 5, 1977–1982. doi:10.1021/cg050272j |
| 4. | Inkster, J. A. H.; Liu, K.; Ait-Mohand, S.; Schaffer, P.; Guérin, B.; Ruth, T. J.; Storr, T. Chem. – Eur. J. 2012, 18, 11079–11087. doi:10.1002/chem.201103450 |
| 5. | Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z |
| 11. | Meiries, S.; Speck, K.; Cordes, D. B.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2013, 32, 330–339. doi:10.1021/om3011867 |
| 13. | Zhou, J.; Liu, L. L.; Cao, L. L.; Stephan, D. W. Angew. Chem., Int. Ed. 2018, 57, 3322–3326. doi:10.1002/anie.201713118 |
| 7. | Emmett, E. J.; Willis, M. C. Asian J. Org. Chem. 2015, 4, 602–611. doi:10.1002/ajoc.201500103 |
| 8. | Woolven, H.; González-Rodríguez, C.; Marco, I.; Thompson, A. L.; Willis, M. C. Org. Lett. 2011, 13, 4876–4878. doi:10.1021/ol201957n |
| 9. | Deeming, A. S.; Russell, C. J.; Hennessy, A. J.; Willis, M. C. Org. Lett. 2014, 16, 150–153. doi:10.1021/ol403122a |
| 10. | Hu, T.; Baxendale, I. R.; Baumann, M. Molecules 2016, 21, 918. doi:10.3390/molecules21070918 |
| 14. | Desiraju, G. R.; Steiner, T. The Weak Hydrogen Bond: In Structure Chemistry and Biology; Oxford University Press: Oxford, United Kingdom, 1999. |
| 5. | Matesic, L.; Wyatt, N. A.; Fraser, B. H.; Roberts, M. P.; Pham, T. Q.; Greguric, I. J. Org. Chem. 2013, 78, 11262–11270. doi:10.1021/jo401759z |
| 37. | Macrae, C. F.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Shields, G. P.; Taylor, R.; Towler, M.; van de Streek, J. J. Appl. Crystallogr. 2006, 39, 453–457. doi:10.1107/s002188980600731x |
| 6. | Smith, T. A. D. J. Labelled Compd. Radiopharm. 2012, 55, 281–288. doi:10.1002/jlcr.2940 |
| 12. | Malet-Sanz, L.; Madrzak, J.; Ley, S. V.; Baxendale, I. R. Org. Biomol. Chem. 2010, 8, 5324–5332. doi:10.1039/c0ob00450b |
| 22. | Echeverría, J.; Aullón, G.; Danovich, D.; Shaik, S.; Alvarez, S. Nat. Chem. 2011, 3, 323–330. doi:10.1038/nchem.1004 |
| 23. | Danovich, D.; Shaik, S.; Neese, F.; Echeverría, J.; Aullón, G.; Alvarez, S. J. Chem. Theory Comput. 2013, 9, 1977–1991. doi:10.1021/ct400070j |
| 20. | Matta, C. F.; Hernández-Trujillo, J.; Tang, T.-H.; Bader, R. F. W. Chem. – Eur. J. 2003, 9, 1940–1951. doi:10.1002/chem.200204626 |
| 21. | Wang, C.-C.; Tang, T.-H.; Wu, L.-C.; Wang, Y. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004, 60, 488–493. doi:10.1107/s0108767304015375 |
| 22. | Echeverría, J.; Aullón, G.; Danovich, D.; Shaik, S.; Alvarez, S. Nat. Chem. 2011, 3, 323–330. doi:10.1038/nchem.1004 |
| 23. | Danovich, D.; Shaik, S.; Neese, F.; Echeverría, J.; Aullón, G.; Alvarez, S. J. Chem. Theory Comput. 2013, 9, 1977–1991. doi:10.1021/ct400070j |
| 33. | APEX3, SAINT and SADABS, Bruker 2016; Bruker AXS Inc.: Madison, Wisconsin, USA, 2016. |
| 33. | APEX3, SAINT and SADABS, Bruker 2016; Bruker AXS Inc.: Madison, Wisconsin, USA, 2016. |
| 29. | Jin, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2007, 46, 3323–3325. doi:10.1002/anie.200700101 |
| 30. | Liu, Z.; Shi, F.; Martinez, P. D. G.; Raminelli, C.; Larock, R. C. J. Org. Chem. 2008, 73, 219–226. doi:10.1021/jo702062n |
| 31. | Cheng, B.; Bao, B.; Zu, B.; Duan, X.; Duan, S.; Li, Y.; Zhai, H. RSC Adv. 2017, 7, 54087–54090. doi:10.1039/c7ra12117b |
| 32. | Shirtcliff, L. D.; Hayes, A. G.; Haley, M. M.; Köhler, F.; Hess, K.; Herges, R. J. Am. Chem. Soc. 2006, 128, 9711–9721. doi:10.1021/ja054547v |
| 25. | Stadlbauer, W. Indazoles. In Science of Synthesis; Neier, R., Ed.; Georg-Thieme Verlag: Stuttgart, New York, 2002; Vol. 12, pp 227–324. |
| 26. | Jacobson, P.; Huber, L. Ber. Dtsch. Chem. Ges. 1908, 41, 660–671. doi:10.1002/cber.190804101127 |
| 27. | Rüchardt, C.; Hassmann, V. Liebigs Ann. Chem. 1980, 908–927. doi:10.1002/jlac.198019800611 |
| 28. | Yoshida, T.; Matsuura, N.; Yamamoto, K.; Doi, M.; Shimada, K.; Morie, T.; Kato, S. Heterocycles 1996, 43, 2701–2712. doi:10.3987/com-96-7614 |
| 21. | Wang, C.-C.; Tang, T.-H.; Wu, L.-C.; Wang, Y. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004, 60, 488–493. doi:10.1107/s0108767304015375 |
| 23. | Danovich, D.; Shaik, S.; Neese, F.; Echeverría, J.; Aullón, G.; Alvarez, S. J. Chem. Theory Comput. 2013, 9, 1977–1991. doi:10.1021/ct400070j |
| 24. | Gaikwad, D. D.; Chapolikar, A. D.; Devkate, C. G.; Warad, K. D.; Tayade, A. P.; Pawar, R. P.; Domb, A. J. Eur. J. Med. Chem. 2015, 90, 707–731. doi:10.1016/j.ejmech.2014.11.029 |
© 2019 King et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)