Abstract
We propose a two-step ligand exchange for the selective end-functionalization of gold nanorods (AuNR) by thiolated cyclodextrin (CD) host molecules. As a result of the complete removal of the precursor capping agent cetyltrimethylammonium bromide (CTAB) by a tetraethylene glycol derivative, competitive binding to the host cavity was prevented, and reversible, light-responsive assembly and disassembly of the AuNR could be induced by host–guest interaction of CD on the nanorods and a photoswitchable arylazopyrazole cross-linker in aqueous solution. The end-to-end assembly of AuNR could be effectively controlled by irradiation with UV and visible light, respectively, over four cycles. By the introduction of AAP, previous disassembly limitations based on the photostationary states of azobenzenes could be solved. The combination photoresponsive interaction and selectively end-functionalized nanoparticles shows significant potential in the reversible self-assembly of inorganic–organic hybrid nanomaterials.
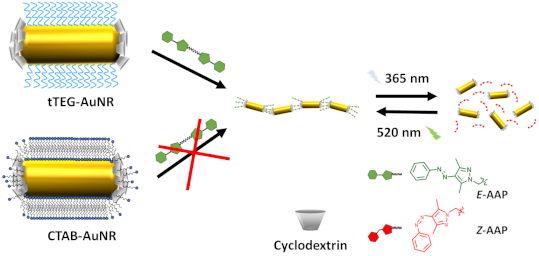
Graphical Abstract
Introduction
Metallic nanomaterials have received intense and interdisciplinary interest due to their unique optical [1], electronic [2,3] and sensing properties [4,5]. In particular, noble metal nanoparticles of sizes smaller than the wavelength of the incident light show interesting optical behavior as a result of collective oscillations of the valence electrons. This leads to surface plasmon resonance (SPR), which is highly dependent on size, shape and chemical environment giving rise to different SPR band wavelengths [6-8]. Especially gold nanorods (AuNR) are of interest because of their good synthetic availability and their unique optical properties. Due to their anisotropy AuNR possess a transversal SPR (TSPR) band in the visible and a longitudinal SPR (LSPR) band that can reach up to the near infrared (NIR) region [9]. The LSPR band can be tuned over a wider wavelength range than for isotropic nanoparticles by varying the aspect ratio of the AuNR or by the assembly of multiple AuNR into linear clusters [10]. These linear aggregates can be realized more or less efficiently through various approaches based on supramolecular interactions like metal–metal and π–π interactions [11], DNA mediated [12] or by host–guest chemistry [13]. Most of these approaches require selective functionalization of the ends of the AuNR and take advantage of the different ligand exchange kinetics of CTAB on the ends and the side of the particles [14,15]. CTAB serves as a capping agent in AuNR synthesis by the seed-mediated growth process and is essential for the anisotropic growth. Due to the different crystallographic environments on the particle surface, weaker packing and binding forces enable the preferential exchange of ligands on the facets at the ends [16]. Removal of CTAB on the side facets is relatively slow and requires a higher ligand concentration. However, for biotechnological applications it might be advantageous to remove CTAB from the complete surface because of its cell toxicity [17], hence strategies for replacing this coating are desirable.
Host–guest chemistry is a supramolecular interaction that is tailor-made for self-assembly due to its lock–key mechanism and has been applied in our and other groups to various nanoparticle systems [18-24]. The host–guest interaction can be responsive to external stimuli such as redox, pH or light, with the latter being the most desirable for assembling nanoparticles by virtue of its noninvasive nature [25]. Prominent light-responsive guest molecules are azobenzenes that form inclusion complexes with α- or β-CD exclusively in the trans configuration, not in the cis configuration [26]. This light-responsive interaction has been recently applied by Ma et al. for the end-to-end assembly of AuNR [27]. However, the system showed some limitations as the assembly was only achieved when the CD ligand and the divalent azobenzene linker were premixed to generate the host–guest complex. This solution was then added to the AuNR leading to a ligand exchange preferentially at the ends and therefore to the linear arrangement of AuNR. If the AuNR were first end-functionalized with the CD, no assembly could be observed after addition of the divalent azobenzene linker. Moreover, the assemblies could only once be disassembled by the combination of UV irradiation and physical forces by sonication. The light-induced back-isomerization of azobenzenes did not form similar end-to-end aggregates. Ma et al. suggested that the host–guest interaction is not strong enough to assemble the particles if not preformed. Yet, this is not consistent with publications that report photoswitchable azobenzene–CD interaction on surfaces [28], in hydrogels [29], the assembly of CD vesicles [30] or the assembly of Au nanoparticles [31].
It is our hypothesis that CTAB, which serves as surfactant and is essential in the synthesis of AuNR in seed-mediated growth processes, acts as a competitive binder to the CD cavity and prevents the reversible self-assembly. In fact, CTAB has a more than 20-fold higher binding constant to β-CD (6.5 × 104 M−1) [32] than unmodified azobenzenes (2.5 × 103 M−1) [33]. Therefore, the competitive binding of CTAB to β-CD has been applied previously, e.g., in sensing applications [34] or to enhance the water solubility of a cyclodextrin polymer [35]. On account of the experimental conditions that lead to a CTAB double layer formation around the AuNR, a significant amount of unbound CTAB may remain in solution and interact with host molecules. Furthermore, the light-responsive dispersion of the AuNR was only possible by the combination of irradiation and sonication, whilst some dimers could not be fully dispersed. Possibly this observation can be explained from the limited photostationary states (PSS) of azobenzenes (E – Z: 80%, Z – E: 70%) [36].
Arylazopyrazoles (AAP) are a new class of molecular switches introduced by Fuchter et al. with excellent photophysical properties like nearly quantitative isomerization, straight forward synthesis in excellent yields and very long Z-isomer half-life times up to 1000 days due to less steric repulsion [37]. In previous reports we could show that the AAP guest inclusion properties to β-CD are comparable to azobenzenes leading due to the superior photophysical properties to fully reversible supramolecular systems, which showed limited feasibility with azobenzenes [20,38,39].
Herein, we present the application of a light-responsive CD–AAP host–guest system for the reversible end-to-end assembly of AuNR. The AuNR are functionalized selectively in a two-step ligand exchange reaction. The ends are capped with per-6-thio-β-cyclodextrin (tCD) and CTAB is removed from the remaining surface by monothiolated tetraethylene glycol (tTEG) enabling reversible host–guest chemistry. The addition of a divalent AAP linker molecule (dAAP) led to light-responsive reversible self-assembly of AuNR in an end-to-end manner as depicted in Scheme 1 to open up a novel strategy for the design of hybrid nanomaterials by supramolecular chemistry.
![[1860-5397-15-140-i1]](/bjoc/content/inline/1860-5397-15-140-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Light-responsive end-to-end assembly of host-functionalized gold nanorods (AuNR) by cyclodextrin–AAP interaction.
Scheme 1: Light-responsive end-to-end assembly of host-functionalized gold nanorods (AuNR) by cyclodextrin–AA...
Results and Discussion
The AuNR (length: 58 nm, width: 25 nm, aspect-ratio: 2.3) were synthesized according to the seed-mediated growth procedure developed by Murphy [40] and El-Sayed [41]. CD-end functionalized AuNR were obtained in a two-step ligand exchange procedure as depicted in Scheme 2. For an efficient ligand exchange the primary alcohol groups of β-CD were replaced with thiols by a known two-step synthesis [42,43] to give a multivalent ligand (tCD) that is tightly bound to the gold surface and is not replaced by an excess of monothiolated tetraethylene glycol (tTEG). It is important to note the importance of the addition of ethanol during the ligand exchange to increase the solubility of CTAB and destabilize its double layer. Furthermore, ethanol weakens the hydrophobic effect of the solvent so that the inclusion of CTAB into β-CD is suppressed.
![[1860-5397-15-140-i2]](/bjoc/content/inline/1860-5397-15-140-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Two-step ligand exchange reaction for the synthesis of water-soluble cyclodextrin end-functionalized gold nanorods.
Scheme 2: Two-step ligand exchange reaction for the synthesis of water-soluble cyclodextrin end-functionalize...
The ligand exchange was followed by ζ-potential measurements giving direct information about the surface charge potential and therefore about the capping agent (Figure 1a). CTAB-functionalized AuNR have a highly positive surface charge (+55 mV) due to the formation of CTAB double layers. The intermediate state [tCD]AuNR shows a decreased surface charge (+42 mV) indicating a reduction of the CTAB surface coverage. It is assumed by preliminary studies that the ligand exchange mainly emerges at the AuNR ends due to its high ligand exchange kinetics [10,15,16]. The addition of an excess of tTEG resulted in a negative surface charge (−32 mV) indicating a complete removal of CTAB from the gold surface. Furthermore, the high negative ζ-potential evidences electrostatic repulsion between the particles and thus good colloidal stability against uncontrolled aggregation. It is well known that PEGylated surfaces and particles show a negative ζ-potential in water due to the preferential absorption of hydroxide anions [44]. Furthermore, the ζ-potential is known to be highest in deionized water and highly pH-dependent [45].
![[1860-5397-15-140-1]](/bjoc/content/figures/1860-5397-15-140-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) ζ-Potential measurement of different stages of the ligand exchange. b) UV–vis spectroscopy before (black) and after complete ligand exchange (red).
Figure 1: a) ζ-Potential measurement of different stages of the ligand exchange. b) UV–vis spectroscopy befor...
The ligand exchange reaction was also analyzed by UV–vis spectroscopy (Figure 1b). The synthesized [CTAB]AuNR show the two expected absorption maxima at λ = 525 nm for the TSPR band and at λ = 690 nm for the LSPR band. After the ligand exchange the bands shift to λ = 528 nm TSPR and to λ = 721 nm LSPR accompanied by a general broadening of the SPR bands. This broadening can be explained by different chemical environments for diverse ligands and is a known characteristic for dual surfactant nanoparticle systems [46]. This interpretation is also supported by TEM images (Figure 4a, see below) indicating significant interparticle repulsion and no aggregation-induced absorption maxima broadening.
For further experiments the [tCD+tTEG]AuNR stock solution was diluted with ddH2O. For the photoresponsive aggregation of isotropic nanoparticles, the 1,4-bis(2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)piperazine-based AAP linker molecule (dAAP) has been successfully applied before and it was synthesized as described in detail in the literature [38]. This divalent linker molecule can form a light-responsive 1:2 complex with host-functionalized AuNR as shown in Scheme 1. The light-responsive AAP moiety isomerizes upon irradiation with λ = 365 nm to the Z-state and upon irradiation with λ = 520 nm back to the E-state. A non-ionic linker design was chosen to eliminate the possibility that the assembly is affected by changes of the ionic strength.
Shortly after the addition of dAAP to the AuNR solution, UV–vis spectra were recorded (Figure 2a) indicating a strong LSPR shift of λ = 36 nm to λ = 757 nm while the TSPR band change is negligible. This indicates selective end-to-end assembly, whereas side-to-side or uncontrolled aggregation would result in an additional shift of the TSPR band [15]. In the course of repeated photoswitching experiments, the spectra show a small but significant decrease of the overall nanoparticle absorbance which is most likely due to sedimentation of irreversible aggregates or size-dependent nanoparticle settling and is in agreement with other studies on linear assemblies of AuNR [12]. To visualize the AuNR end-to-end assemblies, TEM measurements have been conducted (Figure 4b and c, see below) showing chains of AuNR. Hardly any unselective side-by-side or random assemblies can be observed which are often seen in various publications and are due to unspecific drying effects during the TEM grid preparation [11,27]. Adding the dAAP to the intermediate state of the ligand exchange ([tCD]AuNR, with CTAB surface coating on the sides of the particles) showed no significant shift of the SPR maxima (Figure 2b). This observation is consistent with the results of Ma et al. and reinforces the assumption that CTAB behaves as a competitive guest molecule towards β-CD. Therefore, reversible host–guest chemistry was not possible unless CTAB is completely removed.
![[1860-5397-15-140-2]](/bjoc/content/figures/1860-5397-15-140-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectroscopy of a) [tCD+tTEG]AuNR with different amount of dAAP (0-35 µM). b) [tCD]AuNR with dAAP (0–25 µM). c) [tCD+tTEG]AuNR with dAAP (15 µM). Assembly and disassembly by irradiation (5 min, 3 W lamp, λ = 360 nm and λ = 520 nm) over four cycles. d) Zoomed-in region of the LSPR.
Figure 2: UV–vis spectroscopy of a) [tCD+tTEG]AuNR with different amount of dAAP (0-35 µM). b) [tCD]AuNR with...
The reversibility of the light-responsive assembly was further investigated via UV–vis spectroscopy (Figure 2c and d). The spectrum measured after injection of dAAP shows a broadened LSPR maximum with a red shift of λ = 7 nm while the TSPR band remains unchanged. This indicates longitudinal coupling of AuNR as a result of the formation of end-to-end assemblies. Irradiation with UV light (λ = 365 nm, 5 min, 3 W) led to a sharpening of the LSPR band in addition to a blue-shifted maximum to λ = 721 nm, which is the same value recorded before the addition of dAAP. The LSPR and TSPR maxima have been plotted over four cycles showing only minor changes of the TSPR over the assembly (Figure 3). For reversible end-to-end assembly of AuNR, a shift λ < 10 nm is the expected range as it has been reported previously [18,38]. Longer irradiation times did not lead to stronger SPR band shifts and therefore all samples were irradiated for 5 min of the respective wavelength.
![[1860-5397-15-140-3]](/bjoc/content/figures/1860-5397-15-140-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: SPR maxima of [tCD+tTEG]AuNR with dAAP during four cycles of irradiation. a) Longitudinal SPR. b) Transversal SPR.
Figure 3: SPR maxima of [tCD+tTEG]AuNR with dAAP during four cycles of irradiation. a) Longitudinal SPR. b) T...
Via irradiation with visible light (λ = 520 nm, 5 min, 3 W), the LSPR band is again broadened and shifted to a higher wavelength indicating the assembly of AuNR. This procedure could be repeated over four cycles without any fatigue effect and shows the good reversibility of the switching behavior. The dispersion of AuNR assemblies after visible light irradiation can also be verified by TEM bright field images (Figure 4d) showing that the disassembly is possible without the combination of irradiation and sonication. This can be attributed to the favorable photostationary states of the AAP moiety in comparison to azobenzenes used in previous studies. Furthermore, dynamic light scattering was conducted to analyze the assembly in solution (Figure 5). The mean diameter increases upon dAAP addition from ca. 100 nm to ca. 230 nm. Upon UV-light irradiation, the mean diameter decreases to 120 nm. We note that for anisotropic particles, the hydrodynamic diameter values give only a rough overestimation of the nanoparticle diameter and are not suitable to characterize end-to-end assembly. However dynamic light scattering clearly confirms the light-responsive aggregation of the AuNR [47].
![[1860-5397-15-140-4]](/bjoc/content/figures/1860-5397-15-140-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: TEM-BF images of a) [tCD+tTEG]AuNR. b and c) AuNR end-to-end assemblies by dAAP (15 µM). d) Dissolved AuNR by UV irradiation (λ = 360 nm).
Figure 4: TEM-BF images of a) [tCD+tTEG]AuNR. b and c) AuNR end-to-end assemblies by dAAP (15 µM). d) Dissolv...
![[1860-5397-15-140-5]](/bjoc/content/figures/1860-5397-15-140-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Reversible aggregation of [tCD+tTEG]AuNR by addition of dAAP (15 µM) monitored by dynamic light scattering.
Figure 5: Reversible aggregation of [tCD+tTEG]AuNR by addition of dAAP (15 µM) monitored by dynamic light sca...
Finally, to confirm that end-to-end assembly of AuNR occurs through the formation of a 2:1 complex of the [tCD+tTEG]AuNR and dAAP, a control experiment was performed by the addition of a monovalent AAP (mAAP) molecule (see Scheme S1 and Figure S3 in Supporting Information File 1). Even upon addition of rather high concentrations of mAAP (100 µM) no strong shift can be observed by UV–vis spectroscopy, indicating that supramolecular cross-linking is essential for end-to-end assembly of the AuNR.
Conclusion
In summary, we reported a supramolecular system based on the reversible light-responsive interaction between arylazopyrazoles and β-CD for the end-to-end assembly of AuNR. The substitution of azobenzenes by AAP as guest molecules, which feature nearly quantitative photostationary states, allows the control over assembled and disassembled states solely by irradiation. The divalent AAP linker forms a 1:2 complex with per-6-thiolated β-CD which is selectively located at the ends of the AuNR. This was achieved through a novel two-step ligand exchange reaction including the complete removal of CTAB by the addition of thiolated TEG. The removal of CTAB is crucial due to its competitive interaction with β-CD since it has a higher binding constant than the AAP moieties. The success of the consecutive ligand exchange strategy was verified by ζ-potential measurements. The selective end-to-end assembly of AuNR was evidenced by UV–vis and TEM measurements. The LSPR shift could reversibly be shifted back and forth ending at the original wavelength. Switching of the end-to-end assembly was possible over four cycles without fatigue appearance. Previously reported limited feasibilities of azobenzenes could be resolved by the implementation of arylazopyrazoles. This report shows a successful method for the fabrication of new hybrid nanomaterials by the application of light-responsive host–guest chemistry allowing for effective control over assembled and disassembled states of anisotropic nanoparticles over several cycles.
Experimental
Instrumentation and materials
All chemicals were purchased from Sigma-Aldrich, Acros Organics, Merck, VWR or TCI and used without further purification if not stated otherwise. UV–vis absorbance measurements were performed with a V-770 double beam spectrophotometer (JASCO) at 25 °C. Samples for spectroscopic measurements were prepared in disposable 1 mL semi-micro PMMA cuvettes (BRAND). Transmission electron microscopy was performed using a Titan Themis G3 300 TEM (FEI) operating at 300 kV or a Libra 200 FE electron microscope (Zeiss) operating at 200 kV. The ζ-potential measurements were carried out on a Nano ZS Zetasizer (Malvern Instruments) at 25 °C and samples were prepared in disposable DTS 1060 capillary cells (Malvern Instruments). The syntheses of the tCD and tTEG can be found in Supporting Information File 1.
AuNR seed-mediated growth
The CTAB-stabilized AuNR were synthesized according to a literature procedure [38,39]. All glassware used were rinsed with aqua regia before use. A 0.1 M aqueous solution of CTAB was prepared by gentle heating and sonication. Au seeds were prepared as follows: An aqueous solution of HAuCl4·3H2O (250 μL, 0.01 M) was added to an aqueous solution of CTAB (9.75 mL, 0.1 M) in a round-bottomed flask. The solution was stirred at 25 °C for 10 min. Then, a freshly prepared ice-cold aqueous solution of NaBH4 (600 μL, 0.01 M, prepared by diluting an 0.1 M solution) was added in one portion under vigorous stirring. After 10 min, stirring was slowed down to 200 rpm and continued at 25 °C. The seeds were kept at this temperature until their further use.
Growth of AuNR: Aqueous solutions of CTAB (95 mL, 0.1 M), HAuCl4·3H2O (5 mL, 0.01 M), AgNO3 (800 μL, 0.01 M) and ascorbic acid (800 μL, 0.1 M) were added in the given order to a 100 mL Erlenmeyer flask and were mixed after every addition by stirring. The addition of ascorbic acid turned the yellow solution colourless due to Au3+ to Au+ reduction. The flask was placed into a water bath of 25 °C. After 10 min, seed solution (120 μL) was added and mixed by gentle stirring. Stirring was continued for 18 h. The obtained AuNR solution was separated into two falcon tubes (50 mL) and centrifuged (6000 rpm, 20 min). The AuNR pellet was redispersed in H2O (50 mL each tube) and centrifuged again (6000 rpm, 20 min). The AuNR were dissolved in H2O stock solution of a total amount of 10 mL and stored in the fridge.
[tCD+tTEG]AuNR ligand exchange
The isolated AuNR (1.9 mL of the stock solution) were diluted with H2O (8.1 mL) and tCD (3 mL, 0.5 mM, freshly prepared) was added dropwise under sonication. The solution was further sonicated for 10 min and then stirred for 18 h at 30 °C. A fresh aqueous solution of tTEG (2 mL, 10 mM) was added under sonication. The solution was stirred for 1 h at 30 °C. After this, EtOH abs. (3 mL) was added and stirring was continued at 30 °C for 1.5 h. DMSO (10 mL) was added and the resulting solution was centrifuged (6000 rpm, 10 min). The AuNR pellet was redispersed in H2O/EtOH 1:1 (10 mL) and centrifuged again (6000 rpm, 10 min). For final removal of excess ligands, the AuNR were purified by centrifugal filtration (2 × 3 mL H2O, 6000 rpm, 10 min, 10 kDa MWCO).
Supporting Information
| Supporting Information File 1: Experimental details and additional characterization data. | ||
| Format: PDF | Size: 940.1 KB | Download |
References
-
Motl, N. E.; Smith, A. F.; DeSantis, C. J.; Skrabalak, S. E. Chem. Soc. Rev. 2014, 43, 3823–3834. doi:10.1039/c3cs60347d
Return to citation in text: [1] -
Gschneidtner, T. A.; Diaz Fernandez, Y. A.; Moth-Poulsen, K. J. Mater. Chem. C 2013, 1, 7127–7133. doi:10.1039/c3tc31483a
Return to citation in text: [1] -
Makarenko, K. S.; Liu, Z.; de Jong, M. P.; Zwanenburg, F. A.; Huskens, J.; van der Wiel, W. G. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1702920. doi:10.1002/adma.201702920
Return to citation in text: [1] -
Howes, P. D.; Chandrawati, R.; Stevens, M. M. Science 2014, 346, 1247390. doi:10.1126/science.1247390
Return to citation in text: [1] -
Polavarapu, L.; Pérez-Juste, J.; Xu, Q.-H.; Liz-Marzán, L. M. J. Mater. Chem. C 2014, 2, 7460–7467. doi:10.1039/c4tc01142b
Return to citation in text: [1] -
Willets, K. A.; Van Duyne, R. P. Annu. Rev. Phys. Chem. 2007, 58, 267–297. doi:10.1146/annurev.physchem.58.032806.104607
Return to citation in text: [1] -
Eustis, S.; El-Sayed, M. A. Chem. Soc. Rev. 2006, 35, 209–217. doi:10.1039/b514191e
Return to citation in text: [1] -
Jensen, T. R.; Malinsky, M. D.; Haynes, C. L.; Van Duyne, R. P. J. Phys. Chem. B 2000, 104, 10549–10556. doi:10.1021/jp002435e
Return to citation in text: [1] -
Chen, H.; Shao, L.; Li, Q.; Wang, J. Chem. Soc. Rev. 2013, 42, 2679–2724. doi:10.1039/c2cs35367a
Return to citation in text: [1] -
Liu, K.; Nie, Z.; Zhao, N.; Li, W.; Rubinstein, M.; Kumacheva, E. Science 2010, 329, 197–200. doi:10.1126/science.1189457
Return to citation in text: [1] [2] -
Leung, F. C.-M.; Leung, S. Y.-L.; Chung, C. Y.-S.; Yam, V. W.-W. J. Am. Chem. Soc. 2016, 138, 2989–2992. doi:10.1021/jacs.6b01382
Return to citation in text: [1] [2] -
Ma, W.; Kuang, H.; Xu, L.; Ding, L.; Xu, C.; Wang, L.; Kotov, N. A. Nat. Commun. 2013, 4, No. 2689. doi:10.1038/ncomms3689
Return to citation in text: [1] [2] -
Jones, S. T.; Zayed, J. M.; Scherman, O. A. Nanoscale 2013, 5, 5299–5302. doi:10.1039/c3nr01454a
Return to citation in text: [1] -
Carbó-Argibay, E.; Rodríguez-González, B.; Gómez-Graña, S.; Guerrero-Martínez, A.; Pastoriza-Santos, I.; Pérez-Juste, J.; Liz-Marzán, L. M. Angew. Chem., Int. Ed. 2010, 49, 9397–9400. doi:10.1002/anie.201004910
Return to citation in text: [1] -
Nepal, D.; Park, K.; Vaia, R. A. Small 2012, 8, 1013–1020. doi:10.1002/smll.201102152
Return to citation in text: [1] [2] [3] -
Katz-Boon, H.; Rossouw, C. J.; Weyland, M.; Funston, A. M.; Mulvaney, P.; Etheridge, J. Nano Lett. 2011, 11, 273–278. doi:10.1021/nl103726k
Return to citation in text: [1] [2] -
Indrasekara, A. S. D. S.; Wadams, R. C.; Fabris, L. Part. Part. Syst. Charact. 2014, 31, 819–838. doi:10.1002/ppsc.201400006
Return to citation in text: [1] -
Engel, S.; Möller, N.; Stricker, L.; Peterlechner, M.; Ravoo, B. J. Small 2018, 14, 1704287. doi:10.1002/smll.201704287
Return to citation in text: [1] [2] -
Möller, N.; Hellwig, T.; Stricker, L.; Engel, S.; Fallnich, C.; Ravoo, B. J. Chem. Commun. 2017, 53, 240–243. doi:10.1039/c6cc08321h
Return to citation in text: [1] -
Sagebiel, S.; Stricker, L.; Engel, S.; Ravoo, B. J. Chem. Commun. 2017, 53, 9296–9299. doi:10.1039/c7cc04594h
Return to citation in text: [1] [2] -
Krings, J. A.; Vonhören, B.; Tegeder, P.; Siozios, V.; Peterlechner, M.; Ravoo, B. J. J. Mater. Chem. A 2014, 2, 9587–9593. doi:10.1039/c4ta01359j
Return to citation in text: [1] -
Han, K.; Go, D.; Tigges, T.; Rahimi, K.; Kuehne, A. J. C.; Walther, A. Angew. Chem., Int. Ed. 2017, 56, 2176–2182. doi:10.1002/anie.201612196
Angew. Chem., 2017, 129, 2208–2214. doi:10.1002/ange.201612196
Return to citation in text: [1] -
Liu, J.; Mendoza, S.; Román, E.; Lynn, M. J.; Xu, R.; Kaifer, A. E. J. Am. Chem. Soc. 1999, 121, 4304–4305. doi:10.1021/ja990330n
Return to citation in text: [1] -
de laRica, R. M.; Fratila, R. M.; Szarpak, A.; Huskens, J.; Velders, A. H. Angew. Chem., Int. Ed. 2011, 50, 5704–5707. doi:10.1002/anie.201008189
Angew. Chem. 2011, 123, 5822–5825. doi:10.1002/ange.201008189
Return to citation in text: [1] -
Engel, S.; Möller, N.; Ravoo, B. J. Chem. – Eur. J. 2018, 24, 4741–4748. doi:10.1002/chem.201705540
Return to citation in text: [1] -
Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g
Return to citation in text: [1] -
Wu, J.; Xu, Y.; Li, D.; Ma, X.; Tian, H. Chem. Commun. 2017, 53, 4577–4580. doi:10.1039/c7cc01678f
Return to citation in text: [1] [2] -
Klajn, R. Pure Appl. Chem. 2010, 82, 2247–2279. doi:10.1351/pac-con-10-09-04
Return to citation in text: [1] -
Zhao, Y.-L.; Stoddart, J. F. Langmuir 2009, 25, 8442–8446. doi:10.1021/la804316u
Return to citation in text: [1] -
Schenkel, J. H.; Samanta, A.; Ravoo, B. J. Adv. Mater. (Weinheim, Ger.) 2014, 26, 1076–1080. doi:10.1002/adma.201304689
Return to citation in text: [1] -
Liu, Z.; Jiang, M. J. Mater. Chem. 2007, 17, 4249–4254. doi:10.1039/b707910a
Return to citation in text: [1] -
Samanta, A.; Ravoo, B. J. Chem. – Eur. J. 2014, 20, 4966–4973. doi:10.1002/chem.201304658
Return to citation in text: [1] -
Dharmawardana, U. R.; Christian, S. D.; Tucker, E. E.; Taylor, R. W.; Scamehorn, J. F. Langmuir 1993, 9, 2258–2263. doi:10.1021/la00033a003
Return to citation in text: [1] -
Afkhami, A.; Khalafi, L. J. Chin. Chem. Soc. 2007, 54, 431–436. doi:10.1002/jccs.200700060
Return to citation in text: [1] -
Dwivedi, A. K.; Singh, R.; Singh, A.; Wei, K.-H.; Wu, C.-Y.; Lyu, P.-C.; Lin, H.-C. Macromolecules 2016, 49, 5587–5598. doi:10.1021/acs.macromol.6b00789
Return to citation in text: [1] -
Nalluri, S. K. M.; Voskuhl, J.; Bultema, J. B.; Boekema, E. J.; Ravoo, B. J. Angew. Chem., Int. Ed. 2011, 50, 9747–9751. doi:10.1002/anie.201103707
Angew. Chem. 2011, 123 9921–9925. doi:10.1002/ange.201103707
Return to citation in text: [1] -
Weston, C. E.; Richardson, R. D.; Haycock, P. R.; White, A. J. P.; Fuchter, M. J. J. Am. Chem. Soc. 2014, 136, 11878–11881. doi:10.1021/ja505444d
Return to citation in text: [1] -
Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484
Return to citation in text: [1] [2] [3] [4] -
Chu, C.-W.; Ravoo, B. J. Chem. Commun. 2017, 53, 12450–12453. doi:10.1039/c7cc07859e
Return to citation in text: [1] [2] -
Jana, N. R.; Gearheart, L.; Murphy, C. J. J. Phys. Chem. B 2001, 105, 4065–4067. doi:10.1021/jp0107964
Return to citation in text: [1] -
Nikoobakht, B.; El-Sayed, M. A. Chem. Mater. 2003, 15, 1957–1962. doi:10.1021/cm020732l
Return to citation in text: [1] -
Gadelle, A.; Defaye, J. Angew. Chem., Int. Ed. Engl. 1991, 30, 78–80. doi:10.1002/anie.199100781
Angew. Chem. 1991, 103, 94–95. doi:10.1002/ange.19911030121
Return to citation in text: [1] -
Rojas, M. T.; Koeniger, R.; Stoddart, J. F.; Kaifer, A. E. J. Am. Chem. Soc. 1995, 117, 336–343. doi:10.1021/ja00106a036
Return to citation in text: [1] -
Chan, Y.-H. M.; Schweiss, R.; Werner, C.; Grunze, M. Langmuir 2003, 19, 7380–7385. doi:10.1021/la0343977
Return to citation in text: [1] -
Jokerst, J. V.; Lobovkina, T.; Zare, R. N.; Gambhir, S. S. Nanomedicine (London, U. K.) 2011, 6, 715–728. doi:10.2217/nnm.11.19
Return to citation in text: [1] -
Henson, C. B.; Harris, J. T.; Homan, K. A. Mater. Matters 2017, 12, 26–30.
Return to citation in text: [1] -
Wang, Y.; Li, Y. F.; Wang, J.; Sang, Y.; Huang, C. Z. Chem. Commun. 2010, 46, 1332–1334. doi:10.1039/b921464j
Return to citation in text: [1]
| 40. | Jana, N. R.; Gearheart, L.; Murphy, C. J. J. Phys. Chem. B 2001, 105, 4065–4067. doi:10.1021/jp0107964 |
| 41. | Nikoobakht, B.; El-Sayed, M. A. Chem. Mater. 2003, 15, 1957–1962. doi:10.1021/cm020732l |
| 42. |
Gadelle, A.; Defaye, J. Angew. Chem., Int. Ed. Engl. 1991, 30, 78–80. doi:10.1002/anie.199100781
Angew. Chem. 1991, 103, 94–95. doi:10.1002/ange.19911030121 |
| 43. | Rojas, M. T.; Koeniger, R.; Stoddart, J. F.; Kaifer, A. E. J. Am. Chem. Soc. 1995, 117, 336–343. doi:10.1021/ja00106a036 |
| 1. | Motl, N. E.; Smith, A. F.; DeSantis, C. J.; Skrabalak, S. E. Chem. Soc. Rev. 2014, 43, 3823–3834. doi:10.1039/c3cs60347d |
| 9. | Chen, H.; Shao, L.; Li, Q.; Wang, J. Chem. Soc. Rev. 2013, 42, 2679–2724. doi:10.1039/c2cs35367a |
| 26. | Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g |
| 12. | Ma, W.; Kuang, H.; Xu, L.; Ding, L.; Xu, C.; Wang, L.; Kotov, N. A. Nat. Commun. 2013, 4, No. 2689. doi:10.1038/ncomms3689 |
| 6. | Willets, K. A.; Van Duyne, R. P. Annu. Rev. Phys. Chem. 2007, 58, 267–297. doi:10.1146/annurev.physchem.58.032806.104607 |
| 7. | Eustis, S.; El-Sayed, M. A. Chem. Soc. Rev. 2006, 35, 209–217. doi:10.1039/b514191e |
| 8. | Jensen, T. R.; Malinsky, M. D.; Haynes, C. L.; Van Duyne, R. P. J. Phys. Chem. B 2000, 104, 10549–10556. doi:10.1021/jp002435e |
| 27. | Wu, J.; Xu, Y.; Li, D.; Ma, X.; Tian, H. Chem. Commun. 2017, 53, 4577–4580. doi:10.1039/c7cc01678f |
| 11. | Leung, F. C.-M.; Leung, S. Y.-L.; Chung, C. Y.-S.; Yam, V. W.-W. J. Am. Chem. Soc. 2016, 138, 2989–2992. doi:10.1021/jacs.6b01382 |
| 27. | Wu, J.; Xu, Y.; Li, D.; Ma, X.; Tian, H. Chem. Commun. 2017, 53, 4577–4580. doi:10.1039/c7cc01678f |
| 4. | Howes, P. D.; Chandrawati, R.; Stevens, M. M. Science 2014, 346, 1247390. doi:10.1126/science.1247390 |
| 5. | Polavarapu, L.; Pérez-Juste, J.; Xu, Q.-H.; Liz-Marzán, L. M. J. Mater. Chem. C 2014, 2, 7460–7467. doi:10.1039/c4tc01142b |
| 18. | Engel, S.; Möller, N.; Stricker, L.; Peterlechner, M.; Ravoo, B. J. Small 2018, 14, 1704287. doi:10.1002/smll.201704287 |
| 19. | Möller, N.; Hellwig, T.; Stricker, L.; Engel, S.; Fallnich, C.; Ravoo, B. J. Chem. Commun. 2017, 53, 240–243. doi:10.1039/c6cc08321h |
| 20. | Sagebiel, S.; Stricker, L.; Engel, S.; Ravoo, B. J. Chem. Commun. 2017, 53, 9296–9299. doi:10.1039/c7cc04594h |
| 21. | Krings, J. A.; Vonhören, B.; Tegeder, P.; Siozios, V.; Peterlechner, M.; Ravoo, B. J. J. Mater. Chem. A 2014, 2, 9587–9593. doi:10.1039/c4ta01359j |
| 22. |
Han, K.; Go, D.; Tigges, T.; Rahimi, K.; Kuehne, A. J. C.; Walther, A. Angew. Chem., Int. Ed. 2017, 56, 2176–2182. doi:10.1002/anie.201612196
Angew. Chem., 2017, 129, 2208–2214. doi:10.1002/ange.201612196 |
| 23. | Liu, J.; Mendoza, S.; Román, E.; Lynn, M. J.; Xu, R.; Kaifer, A. E. J. Am. Chem. Soc. 1999, 121, 4304–4305. doi:10.1021/ja990330n |
| 24. |
de laRica, R. M.; Fratila, R. M.; Szarpak, A.; Huskens, J.; Velders, A. H. Angew. Chem., Int. Ed. 2011, 50, 5704–5707. doi:10.1002/anie.201008189
Angew. Chem. 2011, 123, 5822–5825. doi:10.1002/ange.201008189 |
| 38. | Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484 |
| 2. | Gschneidtner, T. A.; Diaz Fernandez, Y. A.; Moth-Poulsen, K. J. Mater. Chem. C 2013, 1, 7127–7133. doi:10.1039/c3tc31483a |
| 3. | Makarenko, K. S.; Liu, Z.; de Jong, M. P.; Zwanenburg, F. A.; Huskens, J.; van der Wiel, W. G. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1702920. doi:10.1002/adma.201702920 |
| 25. | Engel, S.; Möller, N.; Ravoo, B. J. Chem. – Eur. J. 2018, 24, 4741–4748. doi:10.1002/chem.201705540 |
| 15. | Nepal, D.; Park, K.; Vaia, R. A. Small 2012, 8, 1013–1020. doi:10.1002/smll.201102152 |
| 13. | Jones, S. T.; Zayed, J. M.; Scherman, O. A. Nanoscale 2013, 5, 5299–5302. doi:10.1039/c3nr01454a |
| 16. | Katz-Boon, H.; Rossouw, C. J.; Weyland, M.; Funston, A. M.; Mulvaney, P.; Etheridge, J. Nano Lett. 2011, 11, 273–278. doi:10.1021/nl103726k |
| 45. | Jokerst, J. V.; Lobovkina, T.; Zare, R. N.; Gambhir, S. S. Nanomedicine (London, U. K.) 2011, 6, 715–728. doi:10.2217/nnm.11.19 |
| 12. | Ma, W.; Kuang, H.; Xu, L.; Ding, L.; Xu, C.; Wang, L.; Kotov, N. A. Nat. Commun. 2013, 4, No. 2689. doi:10.1038/ncomms3689 |
| 17. | Indrasekara, A. S. D. S.; Wadams, R. C.; Fabris, L. Part. Part. Syst. Charact. 2014, 31, 819–838. doi:10.1002/ppsc.201400006 |
| 11. | Leung, F. C.-M.; Leung, S. Y.-L.; Chung, C. Y.-S.; Yam, V. W.-W. J. Am. Chem. Soc. 2016, 138, 2989–2992. doi:10.1021/jacs.6b01382 |
| 10. | Liu, K.; Nie, Z.; Zhao, N.; Li, W.; Rubinstein, M.; Kumacheva, E. Science 2010, 329, 197–200. doi:10.1126/science.1189457 |
| 15. | Nepal, D.; Park, K.; Vaia, R. A. Small 2012, 8, 1013–1020. doi:10.1002/smll.201102152 |
| 16. | Katz-Boon, H.; Rossouw, C. J.; Weyland, M.; Funston, A. M.; Mulvaney, P.; Etheridge, J. Nano Lett. 2011, 11, 273–278. doi:10.1021/nl103726k |
| 10. | Liu, K.; Nie, Z.; Zhao, N.; Li, W.; Rubinstein, M.; Kumacheva, E. Science 2010, 329, 197–200. doi:10.1126/science.1189457 |
| 14. | Carbó-Argibay, E.; Rodríguez-González, B.; Gómez-Graña, S.; Guerrero-Martínez, A.; Pastoriza-Santos, I.; Pérez-Juste, J.; Liz-Marzán, L. M. Angew. Chem., Int. Ed. 2010, 49, 9397–9400. doi:10.1002/anie.201004910 |
| 15. | Nepal, D.; Park, K.; Vaia, R. A. Small 2012, 8, 1013–1020. doi:10.1002/smll.201102152 |
| 44. | Chan, Y.-H. M.; Schweiss, R.; Werner, C.; Grunze, M. Langmuir 2003, 19, 7380–7385. doi:10.1021/la0343977 |
| 30. | Schenkel, J. H.; Samanta, A.; Ravoo, B. J. Adv. Mater. (Weinheim, Ger.) 2014, 26, 1076–1080. doi:10.1002/adma.201304689 |
| 18. | Engel, S.; Möller, N.; Stricker, L.; Peterlechner, M.; Ravoo, B. J. Small 2018, 14, 1704287. doi:10.1002/smll.201704287 |
| 38. | Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484 |
| 29. | Zhao, Y.-L.; Stoddart, J. F. Langmuir 2009, 25, 8442–8446. doi:10.1021/la804316u |
| 47. | Wang, Y.; Li, Y. F.; Wang, J.; Sang, Y.; Huang, C. Z. Chem. Commun. 2010, 46, 1332–1334. doi:10.1039/b921464j |
| 38. | Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484 |
| 39. | Chu, C.-W.; Ravoo, B. J. Chem. Commun. 2017, 53, 12450–12453. doi:10.1039/c7cc07859e |
| 37. | Weston, C. E.; Richardson, R. D.; Haycock, P. R.; White, A. J. P.; Fuchter, M. J. J. Am. Chem. Soc. 2014, 136, 11878–11881. doi:10.1021/ja505444d |
| 20. | Sagebiel, S.; Stricker, L.; Engel, S.; Ravoo, B. J. Chem. Commun. 2017, 53, 9296–9299. doi:10.1039/c7cc04594h |
| 38. | Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484 |
| 39. | Chu, C.-W.; Ravoo, B. J. Chem. Commun. 2017, 53, 12450–12453. doi:10.1039/c7cc07859e |
| 35. | Dwivedi, A. K.; Singh, R.; Singh, A.; Wei, K.-H.; Wu, C.-Y.; Lyu, P.-C.; Lin, H.-C. Macromolecules 2016, 49, 5587–5598. doi:10.1021/acs.macromol.6b00789 |
| 36. |
Nalluri, S. K. M.; Voskuhl, J.; Bultema, J. B.; Boekema, E. J.; Ravoo, B. J. Angew. Chem., Int. Ed. 2011, 50, 9747–9751. doi:10.1002/anie.201103707
Angew. Chem. 2011, 123 9921–9925. doi:10.1002/ange.201103707 |
| 33. | Dharmawardana, U. R.; Christian, S. D.; Tucker, E. E.; Taylor, R. W.; Scamehorn, J. F. Langmuir 1993, 9, 2258–2263. doi:10.1021/la00033a003 |
| 34. | Afkhami, A.; Khalafi, L. J. Chin. Chem. Soc. 2007, 54, 431–436. doi:10.1002/jccs.200700060 |
| 32. | Samanta, A.; Ravoo, B. J. Chem. – Eur. J. 2014, 20, 4966–4973. doi:10.1002/chem.201304658 |
© 2019 Niehues et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)