

Abstract
2,6-Helic[6]arene and its derivatives were synthesized, and their complexation with 1,1′-dimethyl-4,4′-bipyridinium and protonated 4,4'-bipyridinium salts were investigated in detail. It was found that the helic[6]arene and its derivatives could all form 1:1 complexes with both 1,1′-dimethyl-4,4'-bipyridinium salts and protonated 4,4'-bipyridinium salts in solution and in the solid state. Especially, the helic[6]arene and its derivatives containing 2-hydroxyethoxy or 2-methoxyethoxy groups exhibited stronger complexation with the guests than the other helic[6]arene derivatives for the additional multiple hydrogen bonding interactions between the hosts and the guests, which were evidenced by 1H NMR titrations, X-ray crystal structures and DFT calculations. Moreover, it was also found that the association constants (Ka) of the complexes could be significantly enhanced with larger counteranions of the guests and in less polar solvents. Furthermore, the switchable complexation between the helic[6]arene and protonated 4,4'-bipyridinium salt could be efficiently controlled by acids and bases.
Graphical Abstract
Introduction
Macrocyclic host molecules [1,2] play a significant role in host–guest chemistry. Compared with noncyclic molecules, the structures of macrocyclic hosts can greatly enhance the host–guest complexation ability through preorganization. Moreover, cyclic structures are also the epitome of complex-binding pockets of enzymes [3]. Macrocyclic arenes including calixarenes [4,5], resorcinarenes [6], cyclotriveratrylenes [7,8], pillararenes [9], biphen[n]arenes [10] and others [11,12] are all composed of hydroxy-substituted aromatic rings bridged by methylene or methenyl groups. They have been a kind of important macrocyclic host molecules during the last decades due to their unique structures and a wide range of applications in host–guest chemistry [13-18], self-assembly [19], biomedicine [20] and materials science [21,22]. The derivatives of macrocyclic arenes with diverse functional groups are also important for the development of various new host–guest supramolecular systems [23-29].
Helic[6]arenes [30], a new kind of macrocyclic arenes, are composed of 2,6-dihydroxy-substituted triptycene subunits bridged by methylene groups. They have exhibited wide potential applications in supramolecular chemistry [31-36] for their unique structures and electron-rich cavities. In this paper, we report the complexation between 2,6-helic[6]arene and its four derivatives with 1,1′-dimethyl-4,4′-bipyridinium and protonated 4,4'-bipyridinium salts (Figure 1) in both solution and in the solid state. We found that the helic[6]arene and its derivatives containing 2-hydroxyethoxy or 2-methoxyethoxy groups showed stronger complexation with the guests than the other helic[6]arene derivatives. This result can be explained by the additional multiple hydrogen-bonding interactions between the hosts and the guests, which were evidenced by 1H NMR titration, X-ray crystal structures and DFT calculations. Moreover, we also found that the Ka values of the complexes could be significantly enhanced with larger counteranions of the guests and in less polar solvent. Furthermore, the controllable complexation between (O-methyl)6-2,6-helic[6]arene and protonated 4,4'-bipyridinium salt could be efficiently controlled by acids and bases.
![[1860-5397-15-173-1]](/bjoc/content/figures/1860-5397-15-173-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures and proton designations of hosts H1–5 and guests G1–4.
Figure 1: Structures and proton designations of hosts H1–5 and guests G1–4.
Results and Discussion
Synthesis of the hosts and the guests
2,6-Helic[6]arene H1 and its methyl-substituted derivative H2 were prepared according to previously reported methods [30]. Starting from helic[6]arene H1, helic[6]arene derivatives H3 and H4 were conveniently synthesized by etherification of H1 with bromobutane or 2-bromoethyl methyl ether, respectively, in tetrahydrofuran in the presence of sodium hydride. Helic[6]arene derivative H5 was synthesized by treatment of H1 with methyl bromoacetate followed by reduction with lithium aluminium hydride (Scheme 1). The guests G1–3 were prepared according to previously reported procedures [37-39]. Guest G4 was synthesized through reaction of 4,4′-bipyridine with concentrated HCl in acetonitrile followed by counteranion exchange with sodium tetrakis[3,5-di(trifluoromethyl)phenyl]borate (NaBArF) in dichloromethane. The new compounds were confirmed by NMR spectroscopy and high-resolution mass spectrometry (Supporting Information File 1, Figures S1–S8).
![[1860-5397-15-173-i1]](/bjoc/content/inline/1860-5397-15-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Host–guest complexation in solution
Firstly, we tested the complexation between hosts H1 and H4 with guest G1 in solution by 1H NMR spectroscopy. As shown in Figure 2, when mixing equivalent amounts of host and guest in CDCl3/acetone-d6 1:2 (v/v), the 1H NMR spectrum showed a new set of proton signals, which was different from the free host or guest, indicating the formation of new complexes H1·G1 and H4·G1, respectively, and the complexation was a fast exchange process on the NMR time scale. The proton signals of a and c of the bipyridinium ring showed upfield shifts, while the signal for protons b completely disappeared due to the shielding effect of the aromatic rings in hosts H1 or H4. The signals for the protons 2, 3, and 4 of H1 and 2, 3, 4, and 13 of H4 all showed downfield shifts, which might be attributed to the deshielding effect of guest G1. Other helic[6]arene derivatives (H2, H3, H5) with guests G1 and G2 showed similar complexation as described above (Supporting Information File 1, Figures S9–S14).
![[1860-5397-15-173-2]](/bjoc/content/figures/1860-5397-15-173-2.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 2: Partial 1H NMR spectra (400 MHz CDCl3/acetone-d6 1:2 (v/v), 298 K) of (a) free H1, (b) H1 with 1.0 equiv G1, (c) free G1, (d) H4 with 1.0 equiv G1, (e) free H4. [H1]0 = [H4]0 = [G1]0 = 2.0 mM.
Figure 2: Partial 1H NMR spectra (400 MHz CDCl3/acetone-d6 1:2 (v/v), 298 K) of (a) free H1, (b) H1 with 1.0 ...
We also investigated the complexation between hosts H1 and H4 with guest G4 in solution by 1H NMR spectroscopy. As shown in Figure 3, upon mixing equal equivalents of host and guest in CD2Cl2, the 1H NMR spectrum also showed a new set of proton signals, which was different from the free host or guest. These results indicated that the new complexes H1·G4 and H4·G4 were formed, and the complexation between the host and the guest was a fast exchange process on the NMR time scale as well. The signal for protons d of the 4,4'-bipyridinium ring showed an upfield shift and that for protons e completely disappeared possibly due to the shielding effect of the aromatic rings in H1 or H4. The proton signals of 2 and 3 of H1 and 3 of H4 all showed upfield shifts with broadened peaks, which indicated that π–π stacking interactions between the bipyridinium unit of G4 and the benzene ring of the hosts might exist. The signals for protons 2 and 13 of H4 showed a downfield shift with broadened signals due to deshielding effect, while the signals for protons 11 and 12 showed upfield shifts, possibly due to hydrogen bonding between the hydrogen of the bipyridinium unit of G4 and the oxygen atoms of the host. Similarly, the complexation between other helic[6]arene derivatives (H2, H3, H5) with guests G3 and G4 could also be observed (Supporting Information File 1, Figures S15–S22). Furthermore, job plots showed that throughout 1:1 host–guest complexes are formed (Supporting Information File 1, Figures S56–S88).
![[1860-5397-15-173-3]](/bjoc/content/figures/1860-5397-15-173-3.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 3: Partial 1H NMR spectra (400 MHz, CD2Cl2, 298 K) of (a) free H1, (b) H1 with 1.0 equiv G4, (c) free G4, (d) H4 with 1.0 equiv G4, (e) free H4. [H1]0 = [H4]0 = [G4]0 = 2.0 mM.
Figure 3: Partial 1H NMR spectra (400 MHz, CD2Cl2, 298 K) of (a) free H1, (b) H1 with 1.0 equiv G4, (c) free ...
To gain quantitative insight into the complexation between the hosts and the guests, we carried out 1H NMR titrations and calculated the association constants Ka by the nonlinear curve-fitting method [40]. As shown in Table 1, compared with its derivatives, the unsubstituted host helic[6]arene H1 showed the strongest complexation with all guests tested. The association constant (Ka) of complex H1·G1 was calculated to be (6.76 ± 1.02) × 103 M−1, while the Ka of H2·G1 was much lower (1.03 ± 0.15) × 102 M−1). For (O-2-methoxyethoxy)6-2,6-helic[6]arene H4 and (O-2-hydroxyethoxy)6-2,6-helic[6]arene H5, the association constants of their complexes with G1 were found to be (1.36 ± 0.17) × 103 M−1 and (3.10 ± 0.30) × 103 M−1, respectively, which are only slightly smaller than that of H1·G1, but much higher than that of H2·G1. In the case of H3 containing n-butoxy groups, almost no binding affinity toward G1 was observed under these conditions.
Table 1: Association constants (Ka) for 1:1 host–guest complexes in CDCl3/acetone-d6 1:2 (v/v) at 298 K.
| Complexes | Ka [M−1] | Complexes | Ka [M−1] |
| H1·G1 | (6.76 ± 1.02) × 103 | H1·G3 | (1.28 ± 0.17) × 102 |
| H2·G1 | (1.03 ± 0.15) × 102 | H2·G3 | –a |
| H3·G1 | –a | H3·G3 | –a |
| H4·G1 | (1.36 ± 0.17) × 103 | H4·G3 | (73.33 ± 8.09) |
| H5·G1 | (3.10 ± 0.30) × 103 | H5·G3 | (88.72 ± 0.96) |
| H1·G2 | (1.22 ± 0.17) × 104 | H1·G4 | (7.26 ± 0.93) × 103 |
| H2·G2 | (1.26 ± 0.16) × 102 | H2·G4 | –a |
| H3·G2 | –a | H3·G4 | –a |
| H4·G2 | (2.72 ± 0.39) × 103 | H4·G4 | (2.27 ± 0.31) × 103 |
| H5·G2 | (3.50 ± 0.48) × 103 | H5·G4 | (3.04 ± 0.02) × 103 |
aKa values not calculated due to too small binding.
Compared with G1, the protonated 4,4'-bipyridinium salt G3 showed similar complexation behavior but significantly lower binding abilities with helic[6]arene H1 and its derivatives H2–5.
It is known that ion-pairing effects can hamper the complexation of charged species [41-43], and thus affect the host–guest complexation [10,44,45]. Consequently, we also prepared the 4,4'-bipyridinium salts G2 and G4 with BArF− as the counteranion. As shown in Table 1, compared with guests G1 and G3 with PF6− as the counteranion, G2 and G4 exhibited higher binding abilities with the hosts probably due to a weakened ion-pairing effect. Especially, for complex H1·G2, the Ka value was high (1.22 ± 0.17) × 104 M−1.
Solvents with different polarity can also affect the complexation between the hosts and the guests. As shown in Table 2, we found that performing the 1H NMR titrations of the host–guest complexation in CDCl3/acetone-d6 1:2 (v/v), the Ka values of the 1:1 host–guest complexes were about 103 M−1 except for H2 that showed very low complexation ability with G4. When the 1H NMR titrations were carried out in CD2Cl2, the Ka values of complexes H1·G4, H3·G4 and H4·G4 were all higher than 104 M−1, while the Ka value of complex H2·G4 was found to be (6.07 ± 0.08) × 102 M−1. These results suggest that, compared with the non-polar solvent, acetone hampers or competes the intermolecular non-covalent interactions between the hosts and the guests, and thus resulted in a decrease of the host–guest complexation.
Table 2: Association constants (Ka) for the 1:1 host–guest complexes in different solvents at 298 K.
| Complexes | Ka [M−1] | |
|
in CDCl3/acetone-d6
1:2 (v/v) |
in CD2Cl2 | |
| H1·G4 | (7.26 ± 0.93) × 103 | (2.11 ± 0.28) × 104 |
| H2·G4 | –a | (6.07 ± 0.08) × 102 |
| H4·G4 | (2.27 ± 0.31) × 103 | (1.13 ± 0.15) × 104 |
| H5·G4 | (3.04 ± 0.02) × 103 | (1.92 ± 0.21) × 104 |
aKa value not calculated due to too small binding.
ESIMS studies of the formation of host–guest complexes
The electrospray ionization (ESI) mass spectra also confirmed the formation of 1:1 complexes between the hosts and the guests. By using a solution of H1 and G1 in chloroform/acetone 1:2 (v/v), the strongest peak at m/z 540.2056 corresponding to [H1·G1−2PF6]2+ was found, which was in accordance with the 1:1 complex formed in solution. Similarly, the strongest peaks at m/z 582.2526, 714.8325, 672.2842, 540.2061, 582.2522, 714.8319, 672.2836, 526.1895, 700.3148, 658.2689, 526.1898, 568.2365, 700.3143, 658.2681 corresponding to [H2·G1−2PF6]2+, [H4·G1−2PF6]2+, [H5·G1−2PF6]2+, [H1·G2−2BArF]2+, [H2·G2−2BArF]2+, [H4·G2−2BArF]2+, [H5·G2−2BArF]2+, [H1·G3−2PF6]2+, [H4·G3−2PF6]2+, [H5·G3−2PF6]2+, [H1·G4−2BArF]2+, [H2·G4−2BArF]2+, [H4·G4−2BArF]2+, [H5·G4−2BArF]2+ were observed, which further confirmed the formation of the 1:1 host–guest complexes (Supporting Information File 1, Figures S41–S55).
Host–guest complexation in the solid state
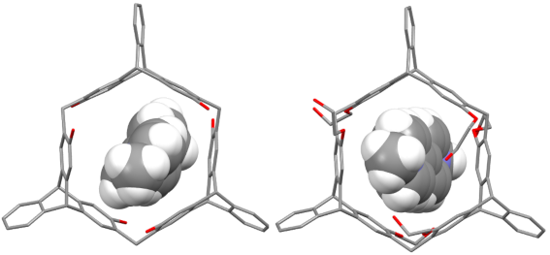
The single crystal of complex H1·G1 was obtained by vapor diffusion of isopropyl ether into acetone. As shown in Figure 4, G1 was encapsulated in the cavity of H1 to form a 1:1 complex, in which G1 is distorted by the dihedral angle between the pyridinium rings of 33.19°. There exist multiple CH···π interactions between the protons of G1 and the aromatic rings of H1 with distances of 2.683 for A, 2.845 for B, 2.788 for C, 2.802 for D, and 2.868 Å for E, respectively. There also exist π–π stacking interactions between the pyridinium of G1 and the aromatic ring of H1 with the distance of 3.854 Å for F, a CH···O hydrogen bond between the proton of G1 and oxygen of H1 in the distance of 2.683 Å for G. Moreover, C-H···F hydrogen bonds between the two adjacent guests with the distances of 2.670 (H), 2.570 (I), 2.594 (J) and 1.981 Å (K), respectively, were observed. These multiple interactions play an important role in the formation of the host–guest complex. Furthermore, it was found that adjacent complexes were nearly perpendicular to each other, which self-assembled into rhombuses with hollows along the c-axis (Figure 4c) and curved ribbons along the a- and b-axes (Supporting Information File 1, Figure S89).
![[1860-5397-15-173-4]](/bjoc/content/figures/1860-5397-15-173-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Crystal structure of complex H1·G1. (a) Top view, (b) side view, and (c) packing viewed along c-axis. Blue lines denote the non-covalent interactions between H1 and G1. Solvent molecules and hydrogen atoms not involved in the non-covalent interactions were omitted for clarity.
Figure 4: Crystal structure of complex H1·G1. (a) Top view, (b) side view, and (c) packing viewed along c-axi...
By vapor diffusion of isopropyl ether into a chloroform/acetone 1:1 (v/v) solution of the 1:1 mixture of H3 and G1, we only obtained a single crystal of H3 instead of the host–guest complex. The steric hindrance of the n-butoxy groups in H3 (Supporting Information File 1, Figure S90) might lead to weak complexation of H3 with the tested guests in solution. However, we obtained a single crystal of complex H5·G1 by vapor diffusion of isopropyl ether into an acetone solution. As shown in Figure 5, we found that G1 was encapsulated in the cavity of H5 to form a 1:1 complex, and the complex molecules are stacked into infinite channels along the a-axis (Figure 5c), which is different from that of H1·G1. There exist multiple CH···π interactions between the proton of G1 and the aromatic ring of H5 with distances of 2.892 (A), 2.844 (B), 2.893 (C) and 2.853 Å (D), respectively. A CH···π interaction between the proton of H5 and the aromatic ring of G1 with a distance of 2.860 Å, and the CH···π interaction between the proton of H5 and the aromatic ring of adjacent H5 in the distance of 2.801 (F), 2.714 (G) and 2.887 Å (H), respectively, are also observed. Moreover, there are multiple CH···O hydrogen-bonding interactions between the protons of G1 and the oxygen of H5 with the distances of 2.600 (I), 2.456 (J), 2.556 (K), 2.296 (L), 2.464 (M), 2.401 (N), 2.176 (O), 2.511 (P) and 2.547 Å (Q), respectively, and OH···O hydrogen bonding between the proton of the side chain of H5 and oxygen of the side chain of adjacent H5 in the distance of 1.989 Å (R). In addition, C-H···F hydrogen bonds between the two adjacent guests with the distance of 2.420 (S), 2.474 (T) and 2.187 Å (U), respectively, are observed as well. These multiple intermolecular hydrogen-bonding interactions between the host and the guest might be the main reason for the formation of the stable complex H5·G1.
![[1860-5397-15-173-5]](/bjoc/content/figures/1860-5397-15-173-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Crystal structure of complex H5·G1. (a) Top view, (b) side view, and (c) packing viewed along the a-axis. Blue lines denote the non-covalent interactions between H5 and G1. Solvent molecules, PF6− counteranions and hydrogen atoms not involved in the non-covalent interactions were omitted for clarity.
Figure 5: Crystal structure of complex H5·G1. (a) Top view, (b) side view, and (c) packing viewed along the a-...
DFT calculation of host–guest complexes
To further investigate the complexation mode and structural characteristics of the host–guest complexes, DFT calculations were carried out at the B3LYP/6-31G level of theory for complex H4·G1 (Supporting Information File 1, Figure S92). The calculation results revealed the C–H···π interactions between the protons on the pyridinium ring of G1 and the benzene ring units of the host H4 and C–H···O hydrogen bonds between the protons of the methyl group and pyridinium rings of G1 and the oxygen atom of H4 with distances ranging from 2.052 to 2.769 Å. Likewise, DFT calculations at the B3LYP/6-31G level of theory for the complexes H4·G3 and H5·G3 were also performed. As shown in Figure 6, in the optimized structure, the pyridinium ring of the guest is surrounded by the cavity of the host. There are C–H···π interactions between the protons on the pyridinium ring of G3 and the benzene rings encompassing the cavity of H4, and C–H···O hydrogen bonding between the protons of the pyridinium ring of G3 and the oxygen atom of H4 with distances ranging from 2.052 to 2.769 Å. Similar to H4·G3, complex H5·G3 also shows the multiple intermolecular non-covalent interactions with distances ranging from 1.651 to 2.575 Å.
![[1860-5397-15-173-6]](/bjoc/content/figures/1860-5397-15-173-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Calculated structures of the complexes at the B3LYP/6-31G level of theory. (a) Top view and (b) side view of H4·G3, and (c) top view and (d) side view of H5·G3.
Figure 6: Calculated structures of the complexes at the B3LYP/6-31G level of theory. (a) Top view and (b) sid...
Compared with hosts H2 and H3, helic[6]arene H1 and its derivatives H4 and H5 all show multiple hydrogen-bonding interactions with the examined guests, which were confirmed by not only X-ray crystal structures of the complexes but also by DFT calculations. These additional multiple hydrogen-bonding interactions might be responsible that H1 and its derivatives H4 and H5 show stronger host–guest complexation with the tested guests than those of H2 and H3. This is consistent with the results obtained in solution.
Acid–base controlled complexation between H2 and G4
4,4′-Bipyridine easily forms protonated 4,4′-pyridinium salts and vice versa. Hence we could conveniently control the association and dissociation of the host–guest complexes based on protonated 4,4′-pyridinium guests by use of acid and base. As shown in Figure 7, when 2.2 equiv of DBU were added into the solution of complex H2·G4 in CD2Cl2, the signals for protons 3 and 6 of complex H2·G4 disappeared while the proton signals of free H2 and 4,4'-bipyridine were observed, which indicated that the complex dissociated. On the other hand, when 2.2 equiv of TFA were added into the above solution, the proton signals of the free 4,4'-bipyridine and the signals for protons 3 and 6 of free H2 disappeared, while the proton signals of complex H2·G4 appeared again, thus indicating the regeneration of the host–guest complex. Therefore, the switchable complexation between H2 and G4 could be efficiently controlled by addition and removal of acid and base.
![[1860-5397-15-173-7]](/bjoc/content/figures/1860-5397-15-173-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Schematic representation of the acid–base controlled complexation process and partial 1H NMR spectra (300 MHz, CD2Cl2, 298 K) of (a) free H2, (b) to the solution of complex H2·G4 were added 2.2 equiv of DBU, (c) to the solution of b were added 2.2 equiv of TFA, and (d) free 4,4'-bipyridine. [H2]0 = 2.0 mM.
Figure 7: Schematic representation of the acid–base controlled complexation process and partial 1H NMR spectr...
Conclusion
In conclusion, we have demonstrated that 2,6-helic[6]arene and its derivatives could form 1:1 complexes with 1,1′-dimethyl-4,4′-bipyridinium and protonated 4,4'-bipyridinium salts in both solution and in the solid state. Compared with H2 and H3, hydroxylated 2,6-helic[6]arene H1 and its derivatives containing 2-hydroxyethoxy (H5) or 2-methoxyethoxy (H4) groups exhibited stronger complexation with the tested guests probably due to the additional multiple hydrogen-bonding interactions between the hosts and the guests, which were confirmed by X-ray single crystal structures and DFT calculations. Moreover, we also found that the association constants of the complexes could be significantly increased for the guests with a large counteranion (BArF−) and in non-polar solvents. Furthermore, the switchable complexation between 2,6-helic[6]arene and protonated 4,4'-bipyridinium salt could be efficiently controlled by acid and base.
Supporting Information
| Supporting Information File 1: Experimental, NMR spectra, mass spectra, determination of association constants, X-ray single crystal data and DFT calculation data. | ||
| Format: PDF | Size: 4.0 MB | Download |
| Supporting Information File 2: CIF file for H1·G1. | ||
| Format: CIF | Size: 3.6 MB | Download |
| Supporting Information File 3: CIF file for H3. | ||
| Format: CIF | Size: 2.9 MB | Download |
| Supporting Information File 4: CIF file for H5·G1. | ||
| Format: CIF | Size: 7.8 MB | Download |
References
-
Steed, J. W.; Atwood, J. L. Supramolecular Chemistry, 2nd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009. doi:10.1002/9780470740880
Return to citation in text: [1] -
Han, Y.; Meng, Z.; Ma, Y.-X.; Chen, C.-F. Acc. Chem. Res. 2014, 47, 2026–2040. doi:10.1021/ar5000677
Return to citation in text: [1] -
Le Poul, N.; Le Mest, Y.; Jabin, I.; Reinaud, O. Acc. Chem. Res. 2015, 48, 2097–2106. doi:10.1021/acs.accounts.5b00152
Return to citation in text: [1] -
Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J.; Saadioui, M., Eds. Calixarenes; Kluwer Academic Publishers: Dordrecht, Netherlands, 2001. doi:10.1007/0-306-47522-7
Return to citation in text: [1] -
Guo, D.-S.; Liu, Y. Acc. Chem. Res. 2014, 47, 1925–1934. doi:10.1021/ar500009g
Return to citation in text: [1] -
Biros, S. M.; Rebek, J., Jr. Chem. Soc. Rev. 2007, 36, 93–104. doi:10.1039/b508530f
Return to citation in text: [1] -
Hardie, M. J. Chem. Soc. Rev. 2010, 39, 516–527. doi:10.1039/b821019p
Return to citation in text: [1] -
Yu, J.-T.; Huang, Z.-T.; Zheng, Q.-Y. Org. Biomol. Chem. 2012, 10, 1359–1364. doi:10.1039/c1ob06465g
Return to citation in text: [1] -
Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765
Return to citation in text: [1] -
Chen, H.; Fan, J.; Hu, X.; Ma, J.; Wang, S.; Li, J.; Yu, Y.; Jia, X.; Li, C. Chem. Sci. 2015, 6, 197–202. doi:10.1039/c4sc02422b
Return to citation in text: [1] [2] -
Li, B.; Wang, B.; Huang, X.; Dai, L.; Cui, L.; Li, J.; Jia, X.; Li, C. Angew. Chem., Int. Ed. 2019, 58, 3885–3889. doi:10.1002/anie.201813972
Return to citation in text: [1] -
Chen, C.-F.; Han, Y. Acc. Chem. Res. 2018, 51, 2093–2106. doi:10.1021/acs.accounts.8b00268
Return to citation in text: [1] -
Yang, K.; Pei, Y.; Wen, J.; Pei, Z. Chem. Commun. 2016, 52, 9316–9326. doi:10.1039/c6cc03641d
Return to citation in text: [1] -
Ogoshi, T.; Kayama, H.; Yamafuji, D.; Aoki, T.; Yamagishi, T.-a. Chem. Sci. 2012, 3, 3221–3226. doi:10.1039/c2sc20982a
Return to citation in text: [1] -
Strutt, N. L.; Forgan, R. S.; Spruell, J. M.; Botros, Y. Y.; Stoddart, J. F. J. Am. Chem. Soc. 2011, 133, 5668–5671. doi:10.1021/ja111418j
Return to citation in text: [1] -
Shu, X.; Chen, S.; Li, J.; Chen, Z.; Weng, L.; Jia, X.; Li, C. Chem. Commun. 2012, 48, 2967–2969. doi:10.1039/c2cc00153e
Return to citation in text: [1] -
Chi, X.; Xue, M.; Yao, Y.; Huang, F. Org. Lett. 2013, 15, 4722–4725. doi:10.1021/ol402048n
Return to citation in text: [1] -
Xia, W.; Hu, X.-Y.; Chen, Y.; Lin, C.; Wang, L. Chem. Commun. 2013, 49, 5085–5087. doi:10.1039/c3cc41903g
Return to citation in text: [1] -
Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Chem. Soc. Rev. 2015, 44, 3568–3587. doi:10.1039/c4cs00390j
Return to citation in text: [1] -
Dondoni, A.; Marra, A. Chem. Rev. 2010, 110, 4949–4977. doi:10.1021/cr100027b
Return to citation in text: [1] -
Descalzo, A. B.; Martínez-Máñez, R.; Sancenón, F.; Hoffmann, K.; Rurack, K. Angew. Chem., Int. Ed. 2006, 45, 5924–5948. doi:10.1002/anie.200600734
Return to citation in text: [1] -
Xiao, T.; Zhou, L.; Xu, L.; Zhong, W.; Zhao, W.; Sun, X.-Q.; Elmes, R. B. P. Chin. Chem. Lett. 2019, 30, 271–276. doi:10.1016/j.cclet.2018.05.039
Return to citation in text: [1] -
Gattuso, G.; Notti, A.; Pappalardo, A.; Parisi, M. F.; Pisagatti, I.; Pappalardo, S.; Garozzo, D.; Messina, A.; Cohen, Y.; Slovak, S. J. Org. Chem. 2008, 73, 7280–7289. doi:10.1021/jo801202h
Return to citation in text: [1] -
Talotta, C.; De Simone, N. A.; Gaeta, C.; Neri, P. Org. Lett. 2015, 17, 1006–1009. doi:10.1021/acs.orglett.5b00115
Return to citation in text: [1] -
Fan, J.; Deng, H.; Li, J.; Jia, X.; Li, C. Chem. Commun. 2013, 49, 6343–6345. doi:10.1039/c3cc42506a
Return to citation in text: [1] -
Ogoshi, T.; Kitajima, K.; Aoki, T.; Fujinami, S.; Yamagishi, T.-a.; Nakamoto, Y. J. Org. Chem. 2010, 75, 3268–3273. doi:10.1021/jo100273n
Return to citation in text: [1] -
Wang, S.; Xu, Z.; Wang, T.; Xiao, T.; Hu, X.-Y.; Shen, Y.-Z.; Wang, L. Nat. Commun. 2018, 9, 1737. doi:10.1038/s41467-018-03827-3
Return to citation in text: [1] -
Kobayashi, K.; Yamanaka, M. Chem. Soc. Rev. 2015, 44, 449–466. doi:10.1039/c4cs00153b
Return to citation in text: [1] -
Si, W.; Chen, L.; Hu, X.-B.; Tang, G.; Chen, Z.; Hou, J.-L.; Li, Z.-T. Angew. Chem., Int. Ed. 2011, 50, 12564–12568. doi:10.1002/anie.201106857
Return to citation in text: [1] -
Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Angew. Chem., Int. Ed. 2016, 55, 5304–5308. doi:10.1002/anie.201600911
Return to citation in text: [1] [2] -
Zhang, G.-W.; Li, P.-F.; Wang, H.-X.; Han, Y.; Chen, C.-F. Chem. – Eur. J. 2017, 23, 3735–3742. doi:10.1002/chem.201605394
Return to citation in text: [1] -
Wang, J.-Q.; Li, J.; Zhang, G.-W.; Chen, C.-F. J. Org. Chem. 2018, 83, 11532–11540. doi:10.1021/acs.joc.8b01437
Return to citation in text: [1] -
Shi, Q.; Chen, C.-F. Org. Lett. 2017, 19, 3175–3178. doi:10.1021/acs.orglett.7b01296
Return to citation in text: [1] -
Shi, Q.; Han, Y.; Chen, C.-F. Chem. – Asian J. 2017, 12, 2576–2582. doi:10.1002/asia.201700857
Return to citation in text: [1] -
Zhang, G.-W.; Shi, Q.; Chen, C.-F. Chem. Commun. 2017, 53, 2582–2585. doi:10.1039/c7cc00600d
Return to citation in text: [1] -
Zhang, G.-W.; Han, Y.; Han, Y.; Wang, Y.; Chen, C.-F. Chem. Commun. 2017, 53, 10433–10436. doi:10.1039/c7cc05489k
Return to citation in text: [1] -
Han, Y.; Gu, Y.-K.; Guo, J.-B.; Chen, C.-F. Eur. J. Org. Chem. 2015, 1257–1263. doi:10.1002/ejoc.201403390
Return to citation in text: [1] -
Nagamura, T.; Sakai, K. J. Chem. Soc., Faraday Trans. 1 1988, 84, 3529–3537. doi:10.1039/f19888403529
Return to citation in text: [1] -
Han, Y.; Meng, Z.; Chen, C.-F. Chem. Commun. 2016, 52, 590–593. doi:10.1039/c5cc08166a
Return to citation in text: [1] -
Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k
Return to citation in text: [1] -
Pappalardo, S.; Villari, V.; Slovak, S.; Cohen, Y.; Gattuso, G.; Notti, A.; Pappalardo, A.; Pisagatti, I.; Parisi, M. F. Chem. – Eur. J. 2007, 13, 8164–8173. doi:10.1002/chem.200601785
Return to citation in text: [1] -
Roelens, S.; Vacca, A.; Venturi, C. Chem. – Eur. J. 2009, 15, 2635–2644. doi:10.1002/chem.200802298
Return to citation in text: [1] -
Huang, F.; Jones, J. W.; Slebodnick, C.; Gibson, H. W. J. Am. Chem. Soc. 2003, 125, 14458–14464. doi:10.1021/ja036606f
Return to citation in text: [1] -
Li, L.; Clarkson, G. J. Org. Lett. 2007, 9, 497–500. doi:10.1021/ol062912x
Return to citation in text: [1] -
Li, C.; Shu, X.; Li, J.; Fan, J.; Chen, Z.; Weng, L.; Jia, X. Org. Lett. 2012, 14, 4126–4129. doi:10.1021/ol301757q
Return to citation in text: [1]
| 41. | Pappalardo, S.; Villari, V.; Slovak, S.; Cohen, Y.; Gattuso, G.; Notti, A.; Pappalardo, A.; Pisagatti, I.; Parisi, M. F. Chem. – Eur. J. 2007, 13, 8164–8173. doi:10.1002/chem.200601785 |
| 42. | Roelens, S.; Vacca, A.; Venturi, C. Chem. – Eur. J. 2009, 15, 2635–2644. doi:10.1002/chem.200802298 |
| 43. | Huang, F.; Jones, J. W.; Slebodnick, C.; Gibson, H. W. J. Am. Chem. Soc. 2003, 125, 14458–14464. doi:10.1021/ja036606f |
| 37. | Han, Y.; Gu, Y.-K.; Guo, J.-B.; Chen, C.-F. Eur. J. Org. Chem. 2015, 1257–1263. doi:10.1002/ejoc.201403390 |
| 38. | Nagamura, T.; Sakai, K. J. Chem. Soc., Faraday Trans. 1 1988, 84, 3529–3537. doi:10.1039/f19888403529 |
| 39. | Han, Y.; Meng, Z.; Chen, C.-F. Chem. Commun. 2016, 52, 590–593. doi:10.1039/c5cc08166a |
| 1. | Steed, J. W.; Atwood, J. L. Supramolecular Chemistry, 2nd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009. doi:10.1002/9780470740880 |
| 2. | Han, Y.; Meng, Z.; Ma, Y.-X.; Chen, C.-F. Acc. Chem. Res. 2014, 47, 2026–2040. doi:10.1021/ar5000677 |
| 7. | Hardie, M. J. Chem. Soc. Rev. 2010, 39, 516–527. doi:10.1039/b821019p |
| 8. | Yu, J.-T.; Huang, Z.-T.; Zheng, Q.-Y. Org. Biomol. Chem. 2012, 10, 1359–1364. doi:10.1039/c1ob06465g |
| 31. | Zhang, G.-W.; Li, P.-F.; Wang, H.-X.; Han, Y.; Chen, C.-F. Chem. – Eur. J. 2017, 23, 3735–3742. doi:10.1002/chem.201605394 |
| 32. | Wang, J.-Q.; Li, J.; Zhang, G.-W.; Chen, C.-F. J. Org. Chem. 2018, 83, 11532–11540. doi:10.1021/acs.joc.8b01437 |
| 33. | Shi, Q.; Chen, C.-F. Org. Lett. 2017, 19, 3175–3178. doi:10.1021/acs.orglett.7b01296 |
| 34. | Shi, Q.; Han, Y.; Chen, C.-F. Chem. – Asian J. 2017, 12, 2576–2582. doi:10.1002/asia.201700857 |
| 35. | Zhang, G.-W.; Shi, Q.; Chen, C.-F. Chem. Commun. 2017, 53, 2582–2585. doi:10.1039/c7cc00600d |
| 36. | Zhang, G.-W.; Han, Y.; Han, Y.; Wang, Y.; Chen, C.-F. Chem. Commun. 2017, 53, 10433–10436. doi:10.1039/c7cc05489k |
| 6. | Biros, S. M.; Rebek, J., Jr. Chem. Soc. Rev. 2007, 36, 93–104. doi:10.1039/b508530f |
| 30. | Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Angew. Chem., Int. Ed. 2016, 55, 5304–5308. doi:10.1002/anie.201600911 |
| 4. | Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J.; Saadioui, M., Eds. Calixarenes; Kluwer Academic Publishers: Dordrecht, Netherlands, 2001. doi:10.1007/0-306-47522-7 |
| 5. | Guo, D.-S.; Liu, Y. Acc. Chem. Res. 2014, 47, 1925–1934. doi:10.1021/ar500009g |
| 23. | Gattuso, G.; Notti, A.; Pappalardo, A.; Parisi, M. F.; Pisagatti, I.; Pappalardo, S.; Garozzo, D.; Messina, A.; Cohen, Y.; Slovak, S. J. Org. Chem. 2008, 73, 7280–7289. doi:10.1021/jo801202h |
| 24. | Talotta, C.; De Simone, N. A.; Gaeta, C.; Neri, P. Org. Lett. 2015, 17, 1006–1009. doi:10.1021/acs.orglett.5b00115 |
| 25. | Fan, J.; Deng, H.; Li, J.; Jia, X.; Li, C. Chem. Commun. 2013, 49, 6343–6345. doi:10.1039/c3cc42506a |
| 26. | Ogoshi, T.; Kitajima, K.; Aoki, T.; Fujinami, S.; Yamagishi, T.-a.; Nakamoto, Y. J. Org. Chem. 2010, 75, 3268–3273. doi:10.1021/jo100273n |
| 27. | Wang, S.; Xu, Z.; Wang, T.; Xiao, T.; Hu, X.-Y.; Shen, Y.-Z.; Wang, L. Nat. Commun. 2018, 9, 1737. doi:10.1038/s41467-018-03827-3 |
| 28. | Kobayashi, K.; Yamanaka, M. Chem. Soc. Rev. 2015, 44, 449–466. doi:10.1039/c4cs00153b |
| 29. | Si, W.; Chen, L.; Hu, X.-B.; Tang, G.; Chen, Z.; Hou, J.-L.; Li, Z.-T. Angew. Chem., Int. Ed. 2011, 50, 12564–12568. doi:10.1002/anie.201106857 |
| 3. | Le Poul, N.; Le Mest, Y.; Jabin, I.; Reinaud, O. Acc. Chem. Res. 2015, 48, 2097–2106. doi:10.1021/acs.accounts.5b00152 |
| 30. | Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Angew. Chem., Int. Ed. 2016, 55, 5304–5308. doi:10.1002/anie.201600911 |
| 13. | Yang, K.; Pei, Y.; Wen, J.; Pei, Z. Chem. Commun. 2016, 52, 9316–9326. doi:10.1039/c6cc03641d |
| 14. | Ogoshi, T.; Kayama, H.; Yamafuji, D.; Aoki, T.; Yamagishi, T.-a. Chem. Sci. 2012, 3, 3221–3226. doi:10.1039/c2sc20982a |
| 15. | Strutt, N. L.; Forgan, R. S.; Spruell, J. M.; Botros, Y. Y.; Stoddart, J. F. J. Am. Chem. Soc. 2011, 133, 5668–5671. doi:10.1021/ja111418j |
| 16. | Shu, X.; Chen, S.; Li, J.; Chen, Z.; Weng, L.; Jia, X.; Li, C. Chem. Commun. 2012, 48, 2967–2969. doi:10.1039/c2cc00153e |
| 17. | Chi, X.; Xue, M.; Yao, Y.; Huang, F. Org. Lett. 2013, 15, 4722–4725. doi:10.1021/ol402048n |
| 18. | Xia, W.; Hu, X.-Y.; Chen, Y.; Lin, C.; Wang, L. Chem. Commun. 2013, 49, 5085–5087. doi:10.1039/c3cc41903g |
| 20. | Dondoni, A.; Marra, A. Chem. Rev. 2010, 110, 4949–4977. doi:10.1021/cr100027b |
| 11. | Li, B.; Wang, B.; Huang, X.; Dai, L.; Cui, L.; Li, J.; Jia, X.; Li, C. Angew. Chem., Int. Ed. 2019, 58, 3885–3889. doi:10.1002/anie.201813972 |
| 12. | Chen, C.-F.; Han, Y. Acc. Chem. Res. 2018, 51, 2093–2106. doi:10.1021/acs.accounts.8b00268 |
| 21. | Descalzo, A. B.; Martínez-Máñez, R.; Sancenón, F.; Hoffmann, K.; Rurack, K. Angew. Chem., Int. Ed. 2006, 45, 5924–5948. doi:10.1002/anie.200600734 |
| 22. | Xiao, T.; Zhou, L.; Xu, L.; Zhong, W.; Zhao, W.; Sun, X.-Q.; Elmes, R. B. P. Chin. Chem. Lett. 2019, 30, 271–276. doi:10.1016/j.cclet.2018.05.039 |
| 10. | Chen, H.; Fan, J.; Hu, X.; Ma, J.; Wang, S.; Li, J.; Yu, Y.; Jia, X.; Li, C. Chem. Sci. 2015, 6, 197–202. doi:10.1039/c4sc02422b |
| 10. | Chen, H.; Fan, J.; Hu, X.; Ma, J.; Wang, S.; Li, J.; Yu, Y.; Jia, X.; Li, C. Chem. Sci. 2015, 6, 197–202. doi:10.1039/c4sc02422b |
| 44. | Li, L.; Clarkson, G. J. Org. Lett. 2007, 9, 497–500. doi:10.1021/ol062912x |
| 45. | Li, C.; Shu, X.; Li, J.; Fan, J.; Chen, Z.; Weng, L.; Jia, X. Org. Lett. 2012, 14, 4126–4129. doi:10.1021/ol301757q |
| 9. | Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765 |
| 19. | Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Chem. Soc. Rev. 2015, 44, 3568–3587. doi:10.1039/c4cs00390j |
© 2019 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
