Abstract
Functionalized O6-corona[3]arene[3]tetrazines were synthesized efficiently and conveniently by means of a macrocyclic condensation reaction between N-functionalized 3,6-dihydroxyphthalimides and 3,6-dichlorotetrazine under mild conditions in a one-pot reaction manner. The novel macrocycles exist as a mixture of rapidly interconvertible conformers in solution while in the solid state they adopt the conformation in which three phthalimide units are cis,trans-orientated. Acting as electron-deficient macrocyclic hosts, the synthesized O6-corona[3]arene[3]tetrazines self-regulated conformational structures to complex anions in the gas phase and in the solid state owing to the anion–π noncovalent interactions between anions and the tetrazine rings.

Graphical Abstract
Introduction
Synthetic macrocycles [1,2] are always attractive and important because they are unique molecular systems to study molecular recognition and the nature of noncovalent interactions. Functional macrocycles also provide essential components for the fabrication or assembly of sophisticated (supra)molecular structures [1-4], advanced materials [1,2,5-10] and machinery systems [1,2,11,12]. Moreover, the designed macrocycles are useful molecular tools in the investigation of supramolecular catalysis and reaction mechanisms [1,2,13-16].
Heteracalixaromatics or heteroatom-bridged calix(het)arenes [17-21] are synthetic macrocycles composed of heteroatoms and meta-(het)arenes in an alternative manner. Because of the interplay between heteroatoms and aromatic rings, heteracalixaromatics possess versatile molecular recognition properties and have found wide supramolecular applications. Very recently, we have devised coronarenes [22] simply through editing or varying the meta-substitution of arylenes within heteracalixaromatics into the para-substitution. In contrast to heteracalixaromatics which adopt generally 1,3-alternate conformations giving V-shaped cleft structures [17-21], the resulting coronarenes form, however, cylindroid cavities [22]. We have also shown that the combination of and the interplay between heteroatoms and para-(het)arylenes produce diverse macrocycles with coarse- and fine-tunable cavity shapes and sizes. The resulting coronarenes exhibit interesting molecular recognition properties towards anions, cations and electron-neutral organic guests [22-31].
In our previous study we have developed an efficient protocol to synthesize oxygen and sulfur-linked corona[m]arene[n]tetrazines from aromatic diol and dithiol derivatives and 3,6-dichlorotetrazine, respectively, in a simply operational one-pot reaction fashion on the basis of a nucleophilic aromatic substitution reaction [23-31]. To prepare functionalized corona[6]arenes using diethyl terephthalate as a starting material, we observed, however, the formation of a mixture of macrocyclic isomers because of a high energy barrier for the rotation of diethyl terephthalate units through the corona[6]arene macrocyclic annulus [23,24]. To circumvent the formation of structural isomers arising from the restricted rotation of aromatic fragments though the macrocycle annulus, we selected in the current study 3,6-dihydroxyphthalimide derivatives as aromatic diols to construct functionalized O6-corona[3]arene[3]tetrazins. Being different from terephthalate in terms of substitution pattern, we envisioned that the phthalimide unit would flip freely owing to the less steric hindrance. In addition, N-substituted 3,6-dihydroxyphthalimide derivatives are accessible conveniently from the reaction between various commercially available functional primary amines and 3,6-dihydoxyphthalic anhydride. The ready availability of N-functionalized 3,6-dihydroxyphthalimides would therefore enable the construction of functionalized coronarene macrocycles. Moreover, the electronic feature of the phthalimide would render the resulting O6-corona[3]arene[3]tetrazines the electron-deficient hosts to form anion-π complexes [32-47]. We report herein the synthesis, structure and anion recognition of phthalimide-containing corona[3]arene[3]tetrazines.
Results and Discussion
In the presence of diisopropylethylamine (DIPEA) as an acid scavenger, N-phenyl (1a), N-cyclohexyl (1b) and N-n-hexyl (1c)-substituted 3,6-dihydroxyphthalimides were able to react efficiently with 3,6-dichlorotetrazine [48] (2) under very mild conditions. The one-pot macrocyclic condensation reaction went to completion within 0.5 h to afford the corresponding O6-corona[3]arene[3]tetrazines 3a–c as the only macrocyclic ring products in yields of 38% to 63%. We then prepared N-allyl (1d), N-(2-(2-hydroxyethoxy)ethyl (1e) and N-(2-(3-(4-(trifluoromethyl)phenyl)ureido)ethyl) (1f) substituted 3,6-dihydroxyphthalimides to synthesize functionalized O6-coronarene macrocycles. Pleasingly, the 3,6-dihydroxyphthalimide substrates 1d and 1e underwent the same reaction to produce corona[6]arene macrocycles 3d and 3e in 54% and 35% yields, respectively. Corona[3]arene[3]tetrazine bearing even a urea group (3f) was synthesized analogously from the reaction of 1f with 2 when DABCO was employed as a base instead of DIPEA (Scheme 1). It was worth addressing that a chemical yield of 52% implied roughly a 90% yield for each step in these six-bond-forming synthesis. The high efficiency for the construction of a macrocyclic ring involving the formation of six new C–O bonds between two reactants was noteworthy. The successful synthesis was most probably due to both the nature of dynamic chemical bonding between tetrazine and phenolic oxygen and the high stability of corona[6]arene macrocycle under the reaction conditions.
![[1860-5397-15-193-i1]](/bjoc/content/inline/1860-5397-15-193-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of phthalimide-containing O6-corona[3]arene[3]tetrazines.
Scheme 1: Synthesis of phthalimide-containing O6-corona[3]arene[3]tetrazines.
The spectroscopic data supported the macrocyclic structure of all products. To determine the structure beyond any doubt, and also to shed light on the conformation of phthalimide-containing O6-corona[6]arenes in the solid state, high quality single crystals of 3a were grown at room temperature from diffusion of diethyl ether vapor into the solution of 3a in acetonitrile. X-ray diffraction analysis revealed that the macrocycle 3a adopted an interesting conformation. As depicted in Figure 1, it was evident that six bridging oxygen atoms formed roughly a plane. Interestingly, three tetrazine rings are procumbent on the plane while three phthalimide units were almost perpendicular to the plane. Judging from the bond lengths, all oxygen atoms in the linking positions tended to form conjugation with their adjacent tetrazine rings rather than with the phthalimide units. Most noticeably, the three phthalimide moieties are not cis-configured. The orientation of one phthalimide was just opposite to that of the other two phthalimide segments.
![[1860-5397-15-193-1]](/bjoc/content/figures/1860-5397-15-193-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray molecular structure of 3a (CCDC 1913907) with side (left) and top (right) views. All solvent molecules are omitted for clarity.
Figure 1: X-ray molecular structure of 3a (CCDC 1913907) with side (left) and top (right) views. All solvent ...
It is important to note that corona[3]arene[3]tetrazines 3 displayed only one set of proton and carbon signals in the 1H and 13C NMR spectra, respectively, in acetone-d6 at room temperature (Figure 2). This is in sharp contradiction to O6-corona[3]arene[3]tetrazines composed of diethyl terephthalate which give several sets of resonance peaks [23,24]. The observation of the single set of proton and carbon resonance signals of 3 indicated the presence of the conformer with high symmetry or most likely a mixture of conformers which underwent very fast interconversion relative to the NMR time scale. In comparison to diethyl terephthalate, a phthalimide segment even containing a large substituent on the imide nitrogen atom is able to flip readily between two sides of the plane defined by bridging oxygen atoms. The conformational fluxionality of the macrocyclic ring would be beneficial to molecular recognition.
![[1860-5397-15-193-2]](/bjoc/content/figures/1860-5397-15-193-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H (top) and 13C (bottom) NMR spectra of 3a in acetone-d6 at 25 °C.
Figure 2: 1H (top) and 13C (bottom) NMR spectra of 3a in acetone-d6 at 25 °C.
To gain a deep insight into the redox properties of phthalimide-bearing O6-corona[3]arene[3]tetrazines, a cyclic voltammogram (CV) and a differential pulse voltammogram of 3a were recorded. As depicted in Figure 3, macrocycle 3a undergoes a reversible sequential one-electron redox process at −811, −883, −1871, and −2367 mV. The electrochemical result indicated the occurrence of electronic communication between aromatic rings. The potential for the first redox process (−811 mV) obtained from CV and DPV revealed the electron deficiency of the macrocycle.
![[1860-5397-15-193-3]](/bjoc/content/figures/1860-5397-15-193-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Normalized cyclic voltammograms (left) and differential pulse voltammograms (right) of 3a. CV and DPV were recorded (scan rate 100 mV·s−1) using a glassy carbon electrode. All of the experiments were performed at 298 K in argon-purged MeCN solutions 0.5 mM) with 0.01 M of [Bu4N]+[PF6]− as the supporting electrolyte. Potentials were recorded versus Fc+/Fc.
Figure 3: Normalized cyclic voltammograms (left) and differential pulse voltammograms (right) of 3a. CV and D...
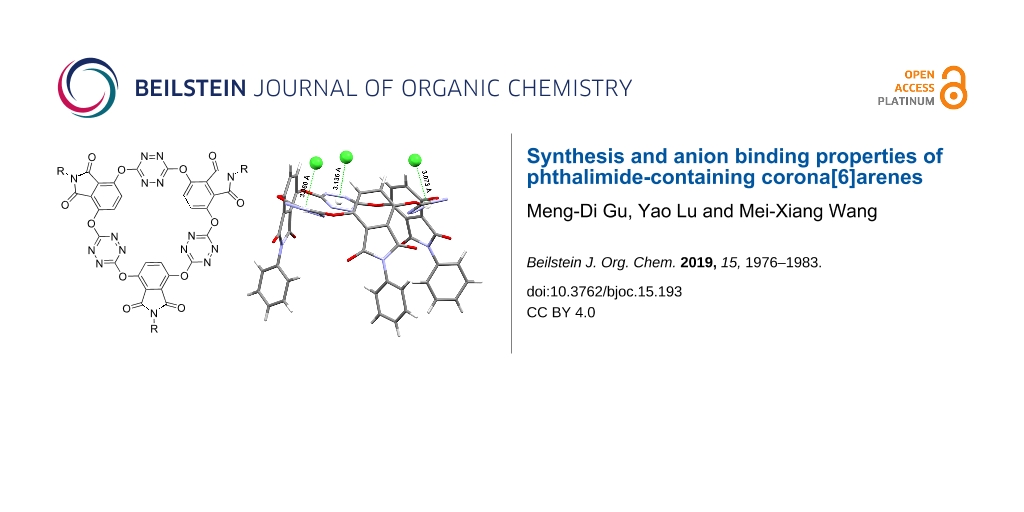
The resulting electron-deficient O6-corona[3]arene[3]tetrazines prompted us to investigate their anion binding behaviour. Taking compound 3a as a representative, we examined the interaction of macrocycles 3 with anions of tetra-n-butylammonium salts by means of electron spray ionization (ESI) mass spectrometry. It was found that the mass spectra of mixed samples of 3a with n-Bu4NX gave ion peaks corresponding to [3a − X]−, [3a-n-Bu4N − 2X]− and [3a-n-2Bu4N − 3X]− complexes in which anions X− included spherical Cl−, Br−, I−, linear SCN−, planar triangle NO3−, tetrahedral BF4− and octahedral PF6− (see Supporting Information File 1). These results demonstrated clearly the outstanding ability of 3a to bind various anion species in gas phase. To our delight, host molecule 3a co-crystalized with n-Bu4NX (X = Cl, Br) from diffusion of diethyl ether vapor into ethyl acetate solution at ambient temperature to give single crystals of the host–guest complexes (n-Bu4NX)3-3a (X = Cl, Br). X-ray crystallography then allowed us to understand the host–guest interactions at the molecular level. As illustrated in the molecular structures in Figure 4, above the centroid of each tetrazine ring there resided a chloride or a bromide. The distance of the anion to the centroid of tetrazine ranged from 3.060 Å to 3.136 Å (Cl−) or from 3.194 Å to 3.280 Å (Br−). The location of anion above the tetrazine centroid with the shorter distance than the sum of van der Waals radius indicated explicitly the typical noncovalent anion–π attraction. It is interesting to point out that in the host–guest complexes, all phthalimide units or their N-phenyl substituents became parallelly aligned (Figure 4). It seems that macrocyclic host 3a changed from its cis,trans-conformation (Figure 2) to the cis,cis one in order to best complex guest species. Another noteworthy structure feature was the deformation of all tetrazine rings. Upon complexation with a halide, the planar aromatic ring adopted a heavily pinched boat conformation, a result consistent with theoretical prediction [49]. We also examined the host–guest interaction in solution phase employing NMR and UV–vis spectroscopy and fluorescence technology. Unfortunately, titration of the host with the guest species did not cause appreciable spectral changes. Isotherm titration calorimetry (ITC) did not give satisfactory results either.
![[1860-5397-15-193-4]](/bjoc/content/figures/1860-5397-15-193-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray molecular structures of complexes (n-Bu4NCl)3-3a (1913908) (top) and (n-Bu4NBr)3-3a (1913909) (bottom) with side (left) and top (right) views. Cations and solvent molecules are omitted for clarity.
Figure 4: X-ray molecular structures of complexes (n-Bu4NCl)3-3a (1913908) (top) and (n-Bu4NBr)3-3a (1913909)...
Conclusion
In summary, we have synthesized phthalimide-containing functionalized O6-corona[3]arene[3]tetrazines by means of a one-pot macrocyclic condensation reaction between N-functionalized 3,6-dihydroxyphthalimides and 3,6-dichlorotetrazine. The unprecedented macrocycles exist as a mixture of rapidly inter-convertible conformers in solution relative to the NMR time scale. The novel O6-corona[3]arene[3]tetrazines self-regulated conformational structures to complex anions in the gas phase and in the solid state owing to the anion–π noncovalent interactions between anions and the tetrazine rings. The easy accessibility, cylindroid cavity and diverse functionality would engender phthalimide-containing functionalized O6-corona[3]arene[3]tetrazines a useful macrocyclic platform for the study of supramolecular chemistry. Applications of the phthalimide-containing functionalized O6-corona[3]arene[3]tetrazines are being actively perused and results will be reported in due course.
Experimental
General procedure for the synthesis of 3a–e. To a solution of DIPEA (2.1 mmol) in acetonitrile (150 mL) which was pre-heated to 70 °C was added dropwise a solution of 3,6-dihydroxyphthalimide derivatives 1a–e (1 mmol) and 3,6-dichlorotetrazine (2, 1 mmol) in acetonitrile (25 mL) during 25 min. After being stirred for another 5 min, the reaction was quenched by cooling down the mixture to ambient temperature and adding water (200 mL). The resulting mixture was neutralized with dilute hydrochloric acid (1 M) and extracted with EtOAc (3 × 150 mL). The combined organic solution was washed with brine (3 × 200 mL) and then dried over anhydrous Na2SO4. After filtration and concentration, the residue was chromatographed on a silica gel column using a mixture of DCM and EtOAc (v/v = 100:1) as an eluent to give product 3a–e.
3a: 140 mg, yield 42%, red solid, mp 275 °C (decomp.); 1H NMR (400 MHz, acetone-d6, 25 °C) δ 8.00 (s, 6H), 7.20–7.34 (m, 15H); 13C NMR (101 MHz, acetone-d6, 25 °C) δ 168.8, 164.4, 147.1, 132.2, 132.1, 129.6, 128.8, 127.5, 125.6; IR (KBr, cm−1) ν: 3078, 1777, 1724, 1494, 1384, 1230, 1115, 955; HRMS-APCI: [M + H]+ calcd for C48H22N15O12, 1000.1567; found, 1000.1553; anal. calcd for C48H21N15O12: C, 57.66; H, 2.12; N, 21.01; found: C, 57.71; H, 2.08; N, 20.70.
3b: 129 mg, yield 38%, red solid, mp 275 °C (decomp.); 1H NMR (400 MHz, MeCN-d3, 25 °C) δ 7.72 (s, 6H), 3.77–3.70 (m, 3H), 1.83–1.76 (m,6H), 1.71–1.68 (m, 6H), 1.25–1.04 (m, 9H); 13C NMR (101 MHz, MeCN-d3, 25 °C) δ 168.6, 165.5, 146.4, 131.5, 125.5, 52.0, 30.0, 26.5, 25.8; IR (KBr, cm−1) ν: 2934, 2858, 1772, 1715, 1382, 1228, 955; HRMS-APCI: [M + H]+ calcd for C48H40N15O12, 1018.2975; found, 1018.2954; anal. calcd for C48H39N15O12: C, 56.64; H, 3.86; N, 20.64; found: C, 56.51; H, 3.83; N, 20.24.
3c: 215 mg, yield 63%, red solid, mp 275 °C (decomp.); 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.63 (s, 6H), 3.38 (t, J = 6.8 Hz, 6H), 1.54–1.48 (m, 6H), 1.23–1.19 (m, 18H), 0.82 (t, J = 6.8 Hz, 9H); 13C NMR (101 MHz, CDCl3, 25 °C) δ 167.4, 164.4, 145.5, 130.1, 124.7, 38.6, 31.2, 29.7, 28.1, 26.4, 22.4, 13.9; IR (KBr, cm−1) ν: 3081, 2931, 2859, 1774, 1717, 1494, 1440, 1417, 1379, 1227, 1065, 954; HRMS-APCI: [M + H]+ calcd for C48H46N15O12, 1024.3445; found, 1024.3437; anal. calcd for C48H45N15O12·acetone: C, 56.61; H, 4.75; N, 19.42; found: C, 56.17; H, 4.66; N, 19.34.
3d: 160 mg, yield 54%, red solid, mp > 300 °C; 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.66 (s, 6H), 5.70–5.64 (m, 3H), 5.12–5.07 (m, 6H), 4.00 (d, J = 6.0 Hz, 3H); 13C NMR (101 MHz, CDCl3, 25 °C) δ 167.4, 163.8, 145.6, 130.4, 130.2, 124.6, 118.6, 40.5; IR (KBr, cm−1) ν: 3288, 1781, 1725, 1428, 1380, 1344, 1229, 1229, 1122, 954, 924; HRMS-APCI: [M + H]+ calcd for C39H22N15O12, 892.1567; found, 892.1563. anal. calcd for C39H21N15O12·0.5H2O: C, 52.01; H, 2.46; N, 23.33; found: C, 52.39; H, 2.11; N, 22.94.
3e: 121 mg, yield 35%, red solid, mp 182–186 °C; 1H NMR (400 MHz, acetone-d6, 25 °C) δ 7.95 (s, 6H), 3.50–3.55 (m, 12H), 3.34–3.41 (m, 12H), 2.83 (s, 3H); 13C NMR (101 MHz, acetone-d6, 25 °C) δ 168.7, 165.3, 146.6, 131.8, 125.7, 73.2, 67.8, 61.8, 38.6; IR (KBr, cm−1) ν: 3472, 1776, 1716, 1382, 1228, 956; HRMS-APCI: [M + Na]+ calcd for C42H33N15O18, 1058.2020; found, 1058.2029; anal. calcd for C42H33N15O18·H2O: C, 48.87; H, 3.35; N, 19.94; found: C, 48.12; H, 3.01; N, 19.90.
Synthesis of 3f: To a solution of DABCO (2.1 mmol) in acetonitrile (150 mL) which was pre-heated to 70 °C was added dropwise a solution of 1f (1 mmol) and 3,6-dichlorotetrazine (2, mmol) in acetonitrile (25 mL) during 25 min. After being stirred for another 5 min, the reaction was quenched by cooling the mixture down to ambient temperature and adding water (200 mL). The resulting mixture was neutralized with dilute hydrochloric acid (1 M) and extracted with EtOAc (3 × 150 mL). The combined organic solution was washed with brine (3 × 200 mL) and then dried over anhydrous Na2SO4. After filtration and concentration, the residue was chromatographed on a silica gel column using a mixture of DCM and EtOAc (v/v = 25:1) as an eluent to give product 3f (261 mg, yield 52%) as a red solid, mp 207–210 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C) δ 8.98 (s, 3H), 7.88 (s, 5H), 7.53 (s, 12H), 6.23 (t, J = 5.7 Hz, 3H), 3.36 (m, 6H), 3.01 (m, 6H), 1.60 (t, J = 6.4 Hz, 6H); 19F NMR (376 MHz, DMSO-d6, 25 °C) δ 59.8; 13C NMR (101 MHz, MeCN-d3, 25 °C) δ 167.1, 164.5, 154.8, 145.0, 144.2, 131.2, 125.9, 124.7 (q,1J(C, F) = 271.6 Hz), 124.5, 120.9 (q, 2J(C, F) = 31.8 Hz), 117.1, 36.5, 35.5, 28.2; IR (KBr, cm−1) ν: 3372, 3080, 2937, 1775, 1717, 1603, 1545, 1382, 1325, 1228, 1113, 1066. HRMS-APCI: [M − H]− calcd for C63H41N21O15F9, 1502.2953; found, 1502.2972; anal. calcd for C63H42F9N21O15·H2O: C, 49.71; H, 2.91; N, 19.32. found: C, 49.42; H, 3.04; N, 19.01.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization of products and copies of mass and NMR spectra. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017.
Return to citation in text: [1] [2] [3] [4] [5] -
Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996.
Return to citation in text: [1] [2] [3] [4] [5] -
Wang, Q.-Q.; Luo, N.; Wang, X.-D.; Ao, Y.-F.; Chen, Y.-F.; Liu, J.-M.; Su, C.-Y.; Wang, D.-X.; Wang, M.-X. J. Am. Chem. Soc. 2017, 139, 635–638. doi:10.1021/jacs.6b12386
Return to citation in text: [1] -
Jiang, B.; Wang, W.; Zhang, Y.; Lu, Y.; Zhang, C.-W.; Yin, G.-Q.; Zhao, X.-L.; Xu, L.; Tan, H.; Li, X.; Jin, G.-X.; Yang, H.-B. Angew. Chem., Int. Ed. 2017, 56, 14438–14442. doi:10.1002/anie.201707209
Return to citation in text: [1] -
Liu, Z.; Nalluri, S. K. M.; Stoddart, J. F. Chem. Soc. Rev. 2017, 46, 2459–2478. doi:10.1039/c7cs00185a
Return to citation in text: [1] -
Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765
Return to citation in text: [1] -
Shetty, D.; Khedkar, J. K.; Park, K. M.; Kim, K. Chem. Soc. Rev. 2015, 44, 8747–8761. doi:10.1039/c5cs00631g
Return to citation in text: [1] -
Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Chem. Soc. Rev. 2015, 44, 3568–3587. doi:10.1039/c4cs00390j
Return to citation in text: [1] -
Han, Y.; Meng, Z.; Ma, Y.-X.; Chen, C.-F. Acc. Chem. Res. 2014, 47, 2026–2040. doi:10.1021/ar5000677
Return to citation in text: [1] -
Wu, G.-Y.; Chen, L.-J.; Xu, L.; Zhao, X.-L.; Yang, H.-B. Coord. Chem. Rev. 2018, 369, 39–75. doi:10.1016/j.ccr.2018.05.009
Return to citation in text: [1] -
Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080–11093. doi:10.1002/anie.201702992
Return to citation in text: [1] -
Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216
Return to citation in text: [1] -
Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature 2017, 543, 637–646. doi:10.1038/nature21701
Return to citation in text: [1] -
Galan, A.; Ballester, P. Chem. Soc. Rev. 2016, 45, 1720–1737. doi:10.1039/c5cs00861a
Return to citation in text: [1] -
Zhang, H.; Yao, B.; Zhao, L.; Wang, D.-X.; Xu, B.-Q.; Wang, M.-X. J. Am. Chem. Soc. 2014, 136, 6326–6332. doi:10.1021/ja412615h
Return to citation in text: [1] -
Zhang, Q.; Liu, Y.; Wang, T.; Zhang, X.; Long, C.; Wu, Y.-D.; Wang, M.-X. J. Am. Chem. Soc. 2018, 140, 5579–5587. doi:10.1021/jacs.8b01896
Return to citation in text: [1] -
Wang, M.-X. Chem. Commun. 2008, 4541. doi:10.1039/b809287g
Return to citation in text: [1] [2] -
Wang, M.-X. Acc. Chem. Res. 2012, 45, 182–195. doi:10.1021/ar200108c
Return to citation in text: [1] [2] -
Maes, W.; Dehaen, W. Chem. Soc. Rev. 2008, 37, 2393. doi:10.1039/b718356a
Return to citation in text: [1] [2] -
Tsue, H.; Ishibashi, K.; Tamura, R. Top. Heterocycl. Chem. 2008, 17, 73. doi:10.1007/7081_2007_094
Return to citation in text: [1] [2] -
Neri, P.; Sessler, J. L.; Wang, M.-X., Eds. Calixarenes and Beyond; Springer: Berlin, Germany, 2016. doi:10.1007/978-3-319-31867-7
Chapters 14–16 and 18.
Return to citation in text: [1] [2] -
Wang, M.-X. Sci. China: Chem. 2018, 61, 993–1003. doi:10.1007/s11426-018-9328-8
Return to citation in text: [1] [2] [3] -
Guo, Q.-H.; Fu, Z.-D.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2014, 53, 13548–13552. doi:10.1002/anie.201407670
Return to citation in text: [1] [2] [3] [4] -
Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179
Return to citation in text: [1] [2] [3] [4] -
Fu, Z.-D.; Guo, Q.-H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2016, 18, 2668–2671. doi:10.1021/acs.orglett.6b01112
Return to citation in text: [1] [2] -
Wu, Z.-C.; Guo, G.-H.; Wang, M.-X. Angew. Chem., Int. Ed. 2017, 56, 7151. doi:10.1002/anie.201703008
Return to citation in text: [1] [2] -
Liu, H.-B.; Zhang, Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2018, 57, 6536. doi:10.1002/anie.201802020
Return to citation in text: [1] [2] -
Guo, Q.-H.; Zhao, L.; Wang, M.-X. Chem. – Eur. J. 2016, 22, 6947–6955. doi:10.1002/chem.201600462
Return to citation in text: [1] [2] -
Lu, Y.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Org. Lett. 2017, 19, 1590–1593. doi:10.1021/acs.orglett.7b00409
Return to citation in text: [1] [2] -
Ren, W.-S.; Zhao, L.; Wang, M.-X. Org. Lett. 2016, 18, 3126–3129. doi:10.1021/acs.orglett.6b01330
Return to citation in text: [1] [2] -
Lu, Y.; Liang, D.-D.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Chin. J. Chem. 2018, 36, 630–634. doi:10.1002/cjoc.201800131
Return to citation in text: [1] [2] -
Gamez, P. Inorg. Chem. Front. 2014, 1, 35–43. doi:10.1039/c3qi00055a
Return to citation in text: [1] -
Bauzá, A.; Mooibroek, T. J.; Frontera, A. ChemPhysChem 2015, 16, 2496–2517. doi:10.1002/cphc.201500314
Return to citation in text: [1] -
Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223
Return to citation in text: [1] -
Tuo, D.-H.; Liu, W.; Wang, X.-Y.; Wang, X.-D.; Ao, Y.-F.; Wang, Q.-Q.; Li, Z.-Y.; Wang, D.-X. J. Am. Chem. Soc. 2019, 141, 1118–1125. doi:10.1021/jacs.8b12018
Return to citation in text: [1] -
Luo, J.; Ao, Y.-F.; Wang, Q.-Q.; Wang, D.-X. Angew. Chem., Int. Ed. 2018, 57, 15827–15831. doi:10.1002/anie.201810836
Return to citation in text: [1] -
He, Q.; Ao, Y.-F.; Huang, Z.-T.; Wang, D.-X. Angew. Chem., Int. Ed. 2015, 54, 11785–11790. doi:10.1002/anie.201504710
Return to citation in text: [1] -
Wang, D.-X.; Zheng, Q.-Y.; Wang, Q.-Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2008, 47, 7485. doi:10.1002/anie.200801705
Return to citation in text: [1] -
Wang, D.-X.; Wang, M.-X. J. Am. Chem. Soc. 2013, 135, 892–897. doi:10.1021/ja310834w
Return to citation in text: [1] -
Wang, D.-X.; Wang, Q.-Q.; Han, Y.; Wang, Y.; Huang, Z.-T.; Wang, M.-X. Chem. – Eur. J. 2010, 16, 13053–13057. doi:10.1002/chem.201002307
Return to citation in text: [1] -
Chifotides, H. T.; Schottel, B. L.; Dunbar, K. R. Angew. Chem., Int. Ed. 2010, 49, 7202. doi:10.1002/anie.201001755
Return to citation in text: [1] -
Rosokha, Y. S.; Lindeman, S. V.; Rosokha, S. V.; Kochi, J. K. Angew. Chem., Int. Ed. 2004, 43, 4650–4652. doi:10.1002/anie.200460337
Return to citation in text: [1] -
Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E. C.; Ballester, P. J. Am. Chem. Soc. 2014, 136, 3208–3218. doi:10.1021/ja412098v
Return to citation in text: [1] -
Zhang, J.; Zhou, B.; Sun, Z.-R.; Wang, X.-B. Phys. Chem. Chem. Phys. 2015, 17, 3131–3141. doi:10.1039/c4cp04687k
Return to citation in text: [1] -
Anstöter, C. S.; Rogers, J. P.; Verlet, J. R. R. J. Am. Chem. Soc. 2019, 141, 6132–6135. doi:10.1021/jacs.9b01345
Return to citation in text: [1] -
Xi, J.; Xu, X. Phys. Chem. Chem. Phys. 2016, 18, 6913–6924. doi:10.1039/c5cp08065g
Return to citation in text: [1] -
Zheng, X.; Shuai, Z.; Wang, D. J. Phys. Chem. A 2013, 117, 3844–3851. doi:10.1021/jp3113478
Return to citation in text: [1] -
Clavier, G.; Audebert, P. Chem. Rev. 2010, 110, 3299–3314. doi:10.1021/cr900357e
Return to citation in text: [1] -
Garau, C.; Quiñonero, D.; Frontera, A.; Costa, A.; Ballester, P.; Deyà, P. M. Chem. Phys. Lett. 2003, 370, 7–13. doi:10.1016/s0009-2614(03)00020-4
Return to citation in text: [1]
| 1. | Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017. |
| 2. | Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996. |
| 1. | Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017. |
| 2. | Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996. |
| 13. | Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature 2017, 543, 637–646. doi:10.1038/nature21701 |
| 14. | Galan, A.; Ballester, P. Chem. Soc. Rev. 2016, 45, 1720–1737. doi:10.1039/c5cs00861a |
| 15. | Zhang, H.; Yao, B.; Zhao, L.; Wang, D.-X.; Xu, B.-Q.; Wang, M.-X. J. Am. Chem. Soc. 2014, 136, 6326–6332. doi:10.1021/ja412615h |
| 16. | Zhang, Q.; Liu, Y.; Wang, T.; Zhang, X.; Long, C.; Wu, Y.-D.; Wang, M.-X. J. Am. Chem. Soc. 2018, 140, 5579–5587. doi:10.1021/jacs.8b01896 |
| 23. | Guo, Q.-H.; Fu, Z.-D.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2014, 53, 13548–13552. doi:10.1002/anie.201407670 |
| 24. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179 |
| 1. | Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017. |
| 2. | Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996. |
| 11. | Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080–11093. doi:10.1002/anie.201702992 |
| 12. | Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216 |
| 49. | Garau, C.; Quiñonero, D.; Frontera, A.; Costa, A.; Ballester, P.; Deyà, P. M. Chem. Phys. Lett. 2003, 370, 7–13. doi:10.1016/s0009-2614(03)00020-4 |
| 1. | Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017. |
| 2. | Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996. |
| 5. | Liu, Z.; Nalluri, S. K. M.; Stoddart, J. F. Chem. Soc. Rev. 2017, 46, 2459–2478. doi:10.1039/c7cs00185a |
| 6. | Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765 |
| 7. | Shetty, D.; Khedkar, J. K.; Park, K. M.; Kim, K. Chem. Soc. Rev. 2015, 44, 8747–8761. doi:10.1039/c5cs00631g |
| 8. | Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Chem. Soc. Rev. 2015, 44, 3568–3587. doi:10.1039/c4cs00390j |
| 9. | Han, Y.; Meng, Z.; Ma, Y.-X.; Chen, C.-F. Acc. Chem. Res. 2014, 47, 2026–2040. doi:10.1021/ar5000677 |
| 10. | Wu, G.-Y.; Chen, L.-J.; Xu, L.; Zhao, X.-L.; Yang, H.-B. Coord. Chem. Rev. 2018, 369, 39–75. doi:10.1016/j.ccr.2018.05.009 |
| 32. | Gamez, P. Inorg. Chem. Front. 2014, 1, 35–43. doi:10.1039/c3qi00055a |
| 33. | Bauzá, A.; Mooibroek, T. J.; Frontera, A. ChemPhysChem 2015, 16, 2496–2517. doi:10.1002/cphc.201500314 |
| 34. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 35. | Tuo, D.-H.; Liu, W.; Wang, X.-Y.; Wang, X.-D.; Ao, Y.-F.; Wang, Q.-Q.; Li, Z.-Y.; Wang, D.-X. J. Am. Chem. Soc. 2019, 141, 1118–1125. doi:10.1021/jacs.8b12018 |
| 36. | Luo, J.; Ao, Y.-F.; Wang, Q.-Q.; Wang, D.-X. Angew. Chem., Int. Ed. 2018, 57, 15827–15831. doi:10.1002/anie.201810836 |
| 37. | He, Q.; Ao, Y.-F.; Huang, Z.-T.; Wang, D.-X. Angew. Chem., Int. Ed. 2015, 54, 11785–11790. doi:10.1002/anie.201504710 |
| 38. | Wang, D.-X.; Zheng, Q.-Y.; Wang, Q.-Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2008, 47, 7485. doi:10.1002/anie.200801705 |
| 39. | Wang, D.-X.; Wang, M.-X. J. Am. Chem. Soc. 2013, 135, 892–897. doi:10.1021/ja310834w |
| 40. | Wang, D.-X.; Wang, Q.-Q.; Han, Y.; Wang, Y.; Huang, Z.-T.; Wang, M.-X. Chem. – Eur. J. 2010, 16, 13053–13057. doi:10.1002/chem.201002307 |
| 41. | Chifotides, H. T.; Schottel, B. L.; Dunbar, K. R. Angew. Chem., Int. Ed. 2010, 49, 7202. doi:10.1002/anie.201001755 |
| 42. | Rosokha, Y. S.; Lindeman, S. V.; Rosokha, S. V.; Kochi, J. K. Angew. Chem., Int. Ed. 2004, 43, 4650–4652. doi:10.1002/anie.200460337 |
| 43. | Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E. C.; Ballester, P. J. Am. Chem. Soc. 2014, 136, 3208–3218. doi:10.1021/ja412098v |
| 44. | Zhang, J.; Zhou, B.; Sun, Z.-R.; Wang, X.-B. Phys. Chem. Chem. Phys. 2015, 17, 3131–3141. doi:10.1039/c4cp04687k |
| 45. | Anstöter, C. S.; Rogers, J. P.; Verlet, J. R. R. J. Am. Chem. Soc. 2019, 141, 6132–6135. doi:10.1021/jacs.9b01345 |
| 46. | Xi, J.; Xu, X. Phys. Chem. Chem. Phys. 2016, 18, 6913–6924. doi:10.1039/c5cp08065g |
| 47. | Zheng, X.; Shuai, Z.; Wang, D. J. Phys. Chem. A 2013, 117, 3844–3851. doi:10.1021/jp3113478 |
| 1. | Atwood, J.; Gokel, G. W.; Barbour, L., Eds. Comprehensive Supramolecular Chemistry II, 2nd ed.; Elsevier: Amsterdam, Netherlands, 2017. |
| 2. | Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vögtle, F., Eds. Comprehensive Supramolecular Chemistry; Pergamon: Oxford, United Kingdom, 1996. |
| 3. | Wang, Q.-Q.; Luo, N.; Wang, X.-D.; Ao, Y.-F.; Chen, Y.-F.; Liu, J.-M.; Su, C.-Y.; Wang, D.-X.; Wang, M.-X. J. Am. Chem. Soc. 2017, 139, 635–638. doi:10.1021/jacs.6b12386 |
| 4. | Jiang, B.; Wang, W.; Zhang, Y.; Lu, Y.; Zhang, C.-W.; Yin, G.-Q.; Zhao, X.-L.; Xu, L.; Tan, H.; Li, X.; Jin, G.-X.; Yang, H.-B. Angew. Chem., Int. Ed. 2017, 56, 14438–14442. doi:10.1002/anie.201707209 |
| 48. | Clavier, G.; Audebert, P. Chem. Rev. 2010, 110, 3299–3314. doi:10.1021/cr900357e |
| 22. | Wang, M.-X. Sci. China: Chem. 2018, 61, 993–1003. doi:10.1007/s11426-018-9328-8 |
| 23. | Guo, Q.-H.; Fu, Z.-D.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2014, 53, 13548–13552. doi:10.1002/anie.201407670 |
| 24. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179 |
| 25. | Fu, Z.-D.; Guo, Q.-H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2016, 18, 2668–2671. doi:10.1021/acs.orglett.6b01112 |
| 26. | Wu, Z.-C.; Guo, G.-H.; Wang, M.-X. Angew. Chem., Int. Ed. 2017, 56, 7151. doi:10.1002/anie.201703008 |
| 27. | Liu, H.-B.; Zhang, Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2018, 57, 6536. doi:10.1002/anie.201802020 |
| 28. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Chem. – Eur. J. 2016, 22, 6947–6955. doi:10.1002/chem.201600462 |
| 29. | Lu, Y.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Org. Lett. 2017, 19, 1590–1593. doi:10.1021/acs.orglett.7b00409 |
| 30. | Ren, W.-S.; Zhao, L.; Wang, M.-X. Org. Lett. 2016, 18, 3126–3129. doi:10.1021/acs.orglett.6b01330 |
| 31. | Lu, Y.; Liang, D.-D.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Chin. J. Chem. 2018, 36, 630–634. doi:10.1002/cjoc.201800131 |
| 17. | Wang, M.-X. Chem. Commun. 2008, 4541. doi:10.1039/b809287g |
| 18. | Wang, M.-X. Acc. Chem. Res. 2012, 45, 182–195. doi:10.1021/ar200108c |
| 19. | Maes, W.; Dehaen, W. Chem. Soc. Rev. 2008, 37, 2393. doi:10.1039/b718356a |
| 20. | Tsue, H.; Ishibashi, K.; Tamura, R. Top. Heterocycl. Chem. 2008, 17, 73. doi:10.1007/7081_2007_094 |
| 21. |
Neri, P.; Sessler, J. L.; Wang, M.-X., Eds. Calixarenes and Beyond; Springer: Berlin, Germany, 2016. doi:10.1007/978-3-319-31867-7
Chapters 14–16 and 18. |
| 23. | Guo, Q.-H.; Fu, Z.-D.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2014, 53, 13548–13552. doi:10.1002/anie.201407670 |
| 24. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179 |
| 22. | Wang, M.-X. Sci. China: Chem. 2018, 61, 993–1003. doi:10.1007/s11426-018-9328-8 |
| 17. | Wang, M.-X. Chem. Commun. 2008, 4541. doi:10.1039/b809287g |
| 18. | Wang, M.-X. Acc. Chem. Res. 2012, 45, 182–195. doi:10.1021/ar200108c |
| 19. | Maes, W.; Dehaen, W. Chem. Soc. Rev. 2008, 37, 2393. doi:10.1039/b718356a |
| 20. | Tsue, H.; Ishibashi, K.; Tamura, R. Top. Heterocycl. Chem. 2008, 17, 73. doi:10.1007/7081_2007_094 |
| 21. |
Neri, P.; Sessler, J. L.; Wang, M.-X., Eds. Calixarenes and Beyond; Springer: Berlin, Germany, 2016. doi:10.1007/978-3-319-31867-7
Chapters 14–16 and 18. |
| 22. | Wang, M.-X. Sci. China: Chem. 2018, 61, 993–1003. doi:10.1007/s11426-018-9328-8 |
| 23. | Guo, Q.-H.; Fu, Z.-D.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2014, 53, 13548–13552. doi:10.1002/anie.201407670 |
| 24. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179 |
| 25. | Fu, Z.-D.; Guo, Q.-H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2016, 18, 2668–2671. doi:10.1021/acs.orglett.6b01112 |
| 26. | Wu, Z.-C.; Guo, G.-H.; Wang, M.-X. Angew. Chem., Int. Ed. 2017, 56, 7151. doi:10.1002/anie.201703008 |
| 27. | Liu, H.-B.; Zhang, Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2018, 57, 6536. doi:10.1002/anie.201802020 |
| 28. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Chem. – Eur. J. 2016, 22, 6947–6955. doi:10.1002/chem.201600462 |
| 29. | Lu, Y.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Org. Lett. 2017, 19, 1590–1593. doi:10.1021/acs.orglett.7b00409 |
| 30. | Ren, W.-S.; Zhao, L.; Wang, M.-X. Org. Lett. 2016, 18, 3126–3129. doi:10.1021/acs.orglett.6b01330 |
| 31. | Lu, Y.; Liang, D.-D.; Fu, Z.-D.; Guo, Q.-H.; Wang, M.-X. Chin. J. Chem. 2018, 36, 630–634. doi:10.1002/cjoc.201800131 |
© 2019 Gu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)