Abstract
The Pd-catalyzed C–H bond functionalization of lilolidine was investigated. The use of a palladium-diphosphine catalyst associated to acetate bases in DMA was found to promote the regioselective arylation at α-position of the nitrogen atom of lilolidine with a wide variety of aryl bromides. From these α-arylated lilolidines, a second arylation at the β-position gives the access to α,β-diarylated lilolidines containing two different aryl groups. The one pot access to α,β-diarylated lilolidines with two identical aryl groups is also possible by using a larger amount of aryl bromide. The synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles from lilolidine via three successive direct arylations is also described. Therefore, this methodology provides a straightforward access to several lilolidine derivatives from commercially available compounds via one, two or three C–H bond functionalization steps allowing to tune their biological properties.
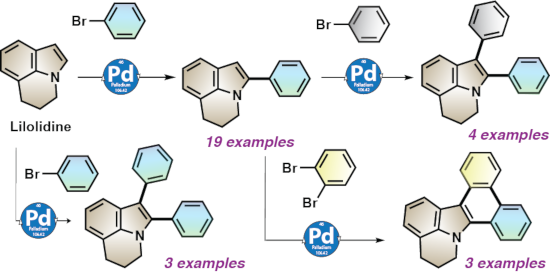
Graphical Abstract
Introduction
Lilolidine (Figure 1, left), which is a commercially available compound, contains a 5,6-dihydropyrrolo[3,2,1-ij]quinoline skeleton found in several bioactive molecules. For example, tivantinib (Figure 1, middle) exhibits MET inhibitor properties [1]; whereas tarazepide (Figure 1, right) is being investigated for the treatment of gastrointestinal diseases. Other lilolidine derivatives also exhibit properties for the treatment of cancers [2-5]. Therefore, the discovery of simple methods for the preparation of lilolidine derivatives is an important research area in pharmaceutical chemistry.
![[1860-5397-15-204-1]](/bjoc/content/figures/1860-5397-15-204-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of lilolidine, tivantinib and tarazepide.
Figure 1: Structures of lilolidine, tivantinib and tarazepide.
To the best of our knowledge, so far only a few methods allow the synthesis of lilolidines arylated at α- [6,7] or β- [8-13] positions of the nitrogen atom. In 2014, Chen and Tang et al. reported that NH4PF6 promotes the cyclodehydration of α-aminocarbonyl compounds, leading to the formation of β-arylated 5,6-dihydropyrrolo[3,2,1-ij]quinoline derivatives [6] (Scheme 1a). Three α-arylated 5,6-dihydropyrrolo[3,2,1-ij]quinoline derivatives have been prepared by Pal et al. via the cyclization of 8-arylethynyl-1,2,3,4-tetrahydroquinolines [9] (Scheme 1b). The best results were obtained using 10 mol % of CuI as catalyst in DMF at 100 °C.
![[1860-5397-15-204-i1]](/bjoc/content/inline/1860-5397-15-204-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Access to α- and β-arylated lilolidine derivatives.
Scheme 1: Access to α- and β-arylated lilolidine derivatives.
The late stage C–H bond functionalization of molecules represents a powerful method for the easy screening of the biological properties of compounds containing a bioactive unit. Since the seminal work by Ohta et al. on the Pd-catalyzed C–H bond functionalization of heteroarenes such as thiophenes, furans, pyrroles and indoles [14,15], this methodology has been widely applied for the preparation of new aryl-substituted heteroarenes [16-21]. Several results dealing with the C–H bond functionalization of indoles have been reported allowing to prepare either α- [22-31] or β-arylated [32-37] indoles, depending on the reaction conditions. However, to the best of our knowledge, no example of regioselective α- or β-arylations via the C–H bond functionalization of lilolidine has been reported so far (Scheme 1c).
Herein, we report i) on the simple access to α-arylated 5,6-dihydropyrrolo[3,2,1-ij]quinolines using an air-stable Pd catalyst associated to an inexpensive base, ii) on the sequential access to α,β-diarylated 5,6-dihydropyrrolo[3,2,1-ij]quinolines containing identical or different aryl groups at α- and β-positions via two-fold Pd-catalyzed C–H bond functionalizations, iii) on the synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles via three successive C–H bond functionalization steps (Scheme 1c).
Results and Discussion
Based on our previous results on the arylation of heteroaromatics [38], we initially employed 1 mol % Pd(OAc)2 catalyst associated to KOAc as the base in DMA at 150 °C as the reaction conditions to promote the coupling of lilolidine with 3-bromobenzonitrile (Table 1, entry 1). Under these conditions a mixture of the α- and β-arylated lilolidines 1a and 1b was obtained in a 64:36 ratio. Then, the influence of some bases on the regioselectivity with Pd(OAc)2 catalyst was examined. With CsOAc a similar regioselectivity than with KOAc was obtained, whereas the use of NaOAc afforded the products 1a and 1b in an 85:15 ratio, but with a moderate conversion of 3-bromobenzonitrile (Table 1, entries 2 and 3). High regioselectivities in favor of isomer 1a (85–90%) were also obtained using K2CO3 and Na2CO3, but partial conversions of 3-bromobenzonitrile were observed (Table 1, entries 4–6). In order to improve the conversions of the aryl bromide, the thermally more stable PdCl(C3H5)(dppb) catalyst [39] was employed. With K2CO3 and Na2CO3, the conversion of 3-bromobenzonitrile was not improved, whereas using NaOAc, a complete conversion of the aryl bromide was observed (Table 1, entries 8–10). Moreover, the regioselectivity in favor of α-arylated lilolidine was improved to 93% affording 1a in 83% yield. The use of KOAc associated to PdCl(C3H5)(dppb) catalyst also afforded the regioisomer 1a in a quite good regioselectivity and yield (Table 1, entry 11). The higher conversions observed in the presence of acetate bases compared to carbonate bases (Table 1, entries 4–6 and 8–11) might be due to an easier coordination of acetates to palladium which favors the concerted metallation deprotonation (CMD) mechanism [40]. The regioselectivities observed using acetate bases are consistent with a CMD mechanism.
Table 1: Influence of the reaction conditions on the palladium-catalyzed direct coupling of lilolidine with 3-bromobenzonitrile.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-204-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst (mol %) | Base | Conv. (%) | Ratio 1a:1b |
Yield in
1a (%) |
| 1 | Pd(OAc)2 (1) | KOAc | 100 | 64:36 | 63 |
| 2 | Pd(OAc)2 (1) | CsOAc | 100 | 69:31 | 58 |
| 3 | Pd(OAc)2 (1) | NaOAc | 65 | 85:15 | 43 |
| 4 | Pd(OAc)2 (1) | K2CO3 | 40 | 90:10 | 37 |
| 5 | Pd(OAc)2 (1) | Cs2CO3 | 0 | – | – |
| 6 | Pd(OAc)2 (1) | Na2CO3 | 33 | 85:15 | n.d. |
| 7 | Pd(OAc)2 (1) | KOAc | 0b | – | – |
| 8 | PdCl(C3H5)(dppb) (2) | K2CO3 | 42 | 90:10 | n.d. |
| 9 | PdCl(C3H5)(dppb) (2) | Na2CO3 | 35 | 91:9 | n.d. |
| 10 | PdCl(C3H5)(dppb) (2) | NaOAc | 100 | 93:7 | 83 |
| 11 | PdCl(C3H5)(dppb) (2) | KOAc | 100 | 82:18 | 68 |
aConditions: lilolidine (1.5 mmol), 3-bromobenzonitrile (1 mmol), base (2 mmol), DMA, under argon, 16 h, 150 °C, isolated yields. bIn xylene.
Then, a set of aryl bromides was reacted with lilolidine using 2 mol % of PdCl(C3H5)(dppb) catalyst, NaOAc or KOAc as bases in DMA at 150 °C (Scheme 2). We initially studied the reactivity of electron-deficient aryl bromides. Acetyl, propionyl, benzoyl and ester as para-substituents on the aryl bromides were tolerated affording the target products 2–6 in 64–77% yields. The structure of 2 was confirmed by X-ray analysis. A lower yield of 7 was obtained for the reaction of 4-bromobenzaldehyde with lilolidine due to the formation of degradation products. Good yields in 8 and 9 were obtained from 4-chloro- and 4-carbonitrile-substituted aryl bromides. In all cases, with these para-substituted aryl bromides, high regioselectivities in favor of the α-arylations were observed. The meta-substituted 3-bromoacetophenone and methyl 3-bromobenzoate also afforded the α-arylated lilolidines 10 and 11 with high regioselectivities. Conversely, with the more sterically hindered aryl source 2-bromobenzonitrile, a mixture of α- and β-arylated lilolidine derivatives was obtained (ratio α:β 69:31). The reactivity of two electron-rich aryl bromides was also examined. With 4-tert-butylbromobenzene and 4-bromoanisole, the target products 13 and 14 were obtained with high regioselectivities, but in low yields due to a partial conversion of these aryl bromides. It should be mentioned that the use of aryl chlorides instead of aryl bromides did not allow to improve the regioselectivities or the reaction yields. With 4-chlorobenzonitrile, product 2 was obtained in 80% regioselectivity and in 38% yield; whereas the use of 2-chlorobenzonitrile afforded 12 in 62% regioselectivity and in 34% yield. In both cases, partial conversions of the aryl chlorides were observed.
![[1860-5397-15-204-i2]](/bjoc/content/inline/1860-5397-15-204-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of α-arylated lilolidine derivatives.
Scheme 2: Synthesis of α-arylated lilolidine derivatives.
Pyridines and quinoline heterocycles are very important structures in pharmaceutical chemistry as more than 100 currently marketed drugs contain these units. Therefore, the reactivity of 3- and 4-bromo-substituted pyridines, 3-bromoquinoline and 4-bromoisoquinoline for the α-arylation of lilolidine was also studied. In all cases, the desired N-containing coupling products 16–19 were obtained in high regioselectivities and in 58–74% yields.
Then, the one-pot synthesis of α,β-di(hetero)arylated 5,6-dihydropyrrolo[3,2,1-ij]quinolines was attempted (Scheme 3). The use of a larger amount of aryl bromides (3 equiv) provided the target diarylated lilolidines 20–22 in good yields. Under these conditions, the mono-arylated lilolidines were detected in very low yields by GC–MS analysis of the crude mixtures. The structure of 20 was confirmed by X-ray diffraction.
![[1860-5397-15-204-i3]](/bjoc/content/inline/1860-5397-15-204-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of α,β-di(hetero)arylated lilolidine derivatives.
Scheme 3: Synthesis of α,β-di(hetero)arylated lilolidine derivatives.
As α-arylated lilolidines can be easily obtained under the reaction conditions shown in Scheme 2, the synthesis of α,β-diarylated 5,6-dihydropyrrolo[3,2,1-ij]quinolines containing two different aryl groups at α- and β-positions via sequential Pd-catalyzed C–H bond functionalization steps was studied (Scheme 4). The reaction of 1 equiv of 4-(5,6-dihydropyrrolo[3,2,1-ij]quinolin-2-yl)benzonitrile 2 and 1.5 equiv of a set of aryl bromides using again 2 mol % PdCl(C3H5)(dppb) catalyst associated to KOAc provided the desired diarylated lilolidines 23–26 in 55–87% yields. The structure of 23 was confirmed by X-ray diffraction.
![[1860-5397-15-204-i4]](/bjoc/content/inline/1860-5397-15-204-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of α,β-diarylated lilolidine derivatives via successive direct arylations.
Scheme 4: Synthesis of α,β-diarylated lilolidine derivatives via successive direct arylations.
Finally, the synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles via β-arylation of the previously obtained α-arylated lilolidines followed by an intramolecular Pd-catalyzed direct arylation was examined (Scheme 5). The reaction of compound 2 with 1,2-dibromobenzene in the presence of 2 mol % PdCl(C3H5)(dppb) catalyst and KOAc as base afforded the desired carbazole 27 in moderate yield after 16 h due to a partial conversion of 2. However, the use of a longer reaction time (48 h) allowed to reach an almost complete conversion of 2, and the carbazole 27 was isolated in 62% yield. A slightly lower yield in the carbazole 28 was obtained from (4-(5,6-dihydropyrrolo[3,2,1-ij]quinolin-2-yl)phenyl)(phenyl)methanone 5 and 1,2-dibromobenzene. This sequential C–H bond arylation strategy was also effective for the synthesis of the carbazole 29 from the pyridine α-substituted lilolidine 17.
![[1860-5397-15-204-i5]](/bjoc/content/inline/1860-5397-15-204-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles via successive direct arylations.
Scheme 5: Synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles via successive direct arylations.
Conclusion
The late stage Pd-catalyzed C–H bond functionalization allows to prepare (di)arylated lilolidine derivatives in only one or two steps. The α-arylated lilolidines were generally obtained in high regioselectivities and in good yields using aryl bromides as easily available aryl sources, acetates as inexpensive bases and PdCl(C3H5)(dppb) as air-stable catalyst. The reaction tolerated a wide variety of useful functional groups such as nitrile, formyl, acetyl, propionyl, benzoyl, esters, chloro, or acetonitrile on the aryl bromide and the heteroaryl bromides 3- or 4-bromopyridines and 3-bromoquinoline. From these α-arylated lilolidines, a second Pd-catalyzed direct arylation at β-position gave rise to α,β-diarylated lilolidines with two different aryl units. The one pot access to α,β-diarylated lilolidines with two identical aryl groups was also possible by using a larger amount of aryl bromide. The synthesis of 5,6-dihydrodibenzo[a,c]pyrido[3,2,1-jk]carbazoles from lilolidine via three successive direct C–H bond arylations also proceed nicely. Therefore, this methodology provides a straightforward access to a wide variety of α- and β-(hetero)aryl-substituted lilolidines allowing to tune or modify their biological properties.
Supporting Information
Supporting information features experimental procedures, products characterizations, copies of 1H and 13C NMR spectra of all products and CCDC numbers of products 2, 20 and 23.
| Supporting Information File 1: Experimental procedures and NMR spectra of compounds 1–29. | ||
| Format: PDF | Size: 4.4 MB | Download |
References
-
Remsing Rix, L. L.; Kuenzi, B. M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J. M.; Haura, E. B.; Lawrence, H. R.; Rix, U. ACS Chem. Biol. 2014, 9, 353–358. doi:10.1021/cb400660a
Return to citation in text: [1] -
Huang, W.-S.; Li, F.; Dalgarno, D. C.; Gong, Y.; Ishchenko, A. V.; Kohlmann, A.; Shakespeare, W. C.; West, A. V.; Xu, Y.; Youngsaye, W.; Zhang, Y.; Zhou, T.; Zhu, X. Preparation of 2-anilino-4-heteroarylpyrimidine compounds for inhibition of mutant EGFR and mutant HER2 kinase. PCT Int. Appl. WO2015175632A1, Nov 19, 2015.
Return to citation in text: [1] -
Wei, Y.; Li, Y.; Zhang, G.; Qui, G.; Hu, S.; Chen, L.; Li, S.; Zhang, Q. Preparation of (substituted phenyl) (substituted pyrimidine) amino deriv. as anticancer drugs. PCT Int. Appl. WO2016029839A1, March 3, 2016.
Return to citation in text: [1] -
Zhu, Y.; Liu, Z.; Feng, C.; Hu, S.; Chen, H.; Bai, E.; Wang, J.; Shi, J. Deuterated 3-(4,5-substituted pyrimidinamine) phenyl derivative useful in treatment of cancer and its preparation. PCT Int. Appl. WO2018050108A1, March 22, 2018.
Return to citation in text: [1] -
Zhang, H.; Wu, W.; Feng, C.; Liu, Z.; Bai, E.; Wang, X.; Lei, M.; Cheng, H.; Feng, H.; Shi, J.; Wang, J.; Zhang, Z.; Jin, T.; Chen, S.; Hu, S.; Zhu, Y. Eur. J. Med. Chem. 2017, 135, 12–23. doi:10.1016/j.ejmech.2017.04.036
Return to citation in text: [1] -
Ji, X.-M.; Zhou, S.-J.; Deng, C.-L.; Chen, F.; Tang, R.-Y. RSC Adv. 2014, 4, 53837–53841. doi:10.1039/c4ra11168k
Return to citation in text: [1] [2] -
Ghorai, J.; Reddy, A. C. S.; Anbarasan, P. Chem. – Eur. J. 2016, 22, 16042–16046. doi:10.1002/chem.201604111
Return to citation in text: [1] -
Cao, C.; Shi, Y.; Odom, A. L. Org. Lett. 2002, 4, 2853–2856. doi:10.1021/ol0201052
Return to citation in text: [1] -
Layek, M.; Dhanunjaya Rao, A. V.; Gajare, V.; Kalita, D.; Barange, D. K.; Islam, A.; Mukkanti, K.; Pal, M. Tetrahedron Lett. 2009, 50, 4878–4881. doi:10.1016/j.tetlet.2009.06.041
Return to citation in text: [1] [2] -
Wang, C.; Huang, Y. Org. Lett. 2013, 15, 5294–5297. doi:10.1021/ol402523x
Return to citation in text: [1] -
Wang, J.; Wang, M.; Chen, K.; Zha, S.; Song, C.; Zhu, J. Org. Lett. 2016, 18, 1178–1181. doi:10.1021/acs.orglett.6b00310
Return to citation in text: [1] -
Panferova, L. I.; Smirnov, V. O.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 745–753. doi:10.1021/acs.joc.6b02344
Return to citation in text: [1] -
Xu, C.; Xu, J. J. Org. Chem. 2018, 83, 14733–14742. doi:10.1021/acs.joc.8b02203
Return to citation in text: [1] -
Akita, Y.; Inoue, A.; Yamamoto, K.; Ohta, A.; Kurihara, T.; Shimizu, M. Heterocycles 1985, 23, 2327–2333. doi:10.3987/r-1985-09-2327
Return to citation in text: [1] -
Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951–1958. doi:10.3987/com-90-5467
Return to citation in text: [1] -
Satoh, T.; Miura, M. Chem. Lett. 2007, 36, 200–205. doi:10.1246/cl.2007.200
Return to citation in text: [1] -
Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015
Return to citation in text: [1] -
Ackermann, L.; Vicente, R.; Kapdi, A. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. doi:10.1002/anie.200902996
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Adv. Synth. Catal. 2014, 356, 17–117. doi:10.1002/adsc.201300922
Return to citation in text: [1] -
Gensch, T.; James, M. J.; Dalton, T.; Glorius, F. Angew. Chem., Int. Ed. 2018, 57, 2296–2306. doi:10.1002/anie.201710377
Return to citation in text: [1] -
Mao, S.; Li, H.; Shi, X.; Soulé, J.-F.; Doucet, H. ChemCatChem 2019, 11, 269–286. doi:10.1002/cctc.201801448
Return to citation in text: [1] -
Wang, X.; Gribkov, D. V.; Sames, D. J. Org. Chem. 2007, 72, 1476–1479. doi:10.1021/jo061979v
Return to citation in text: [1] -
Bellina, F.; Calandri, C.; Cauteruccio, S.; Rossi, R. Tetrahedron 2007, 63, 1970–1980. doi:10.1016/j.tet.2006.12.068
Return to citation in text: [1] -
Yang, S.-D.; Sun, C.-L.; Fang, Z.; Li, B.-J.; Li, Y.-Z.; Shi, Z.-J. Angew. Chem., Int. Ed. 2008, 47, 1473–1476. doi:10.1002/anie.200704619
Return to citation in text: [1] -
Zhao, J.; Zhang, Y.; Cheng, K. J. Org. Chem. 2008, 73, 7428–7431. doi:10.1021/jo801371w
Return to citation in text: [1] -
Joucla, L.; Batail, N.; Djakovitch, L. Adv. Synth. Catal. 2010, 352, 2929–2936. doi:10.1002/adsc.201000512
Return to citation in text: [1] -
Liang, Z.; Yao, B.; Zhang, Y. Org. Lett. 2010, 12, 3185–3187. doi:10.1021/ol101147b
Return to citation in text: [1] -
Zhou, J.; Hu, P.; Zhang, M.; Huang, S.; Wang, M.; Su, W. Chem. – Eur. J. 2010, 16, 5876–5881. doi:10.1002/chem.201000529
Return to citation in text: [1] -
Wang, L.; Yi, W.-b.; Cai, C. Chem. Commun. 2011, 47, 806–808. doi:10.1039/c0cc01666g
Return to citation in text: [1] -
Lu, G.-p.; Cai, C. Synlett 2012, 23, 2992–2996. doi:10.1055/s-0032-1317702
Return to citation in text: [1] -
Wu, M.; Luo, J.; Xiao, F.; Zhang, S.; Deng, G.-J.; Luo, H.-A. Adv. Synth. Catal. 2012, 354, 335–340. doi:10.1002/adsc.201100603
Return to citation in text: [1] -
Akita, Y.; Itagaki, Y.; Takizawa, S.; Ohta, A. Chem. Pharm. Bull. 1989, 37, 1477–1480. doi:10.1248/cpb.37.1477
Return to citation in text: [1] -
Zhang, Z.; Hu, Z.; Yu, Z.; Lei, P.; Chi, H.; Wang, Y.; He, R. Tetrahedron Lett. 2007, 48, 2415–2419. doi:10.1016/j.tetlet.2007.01.173
Return to citation in text: [1] -
Cusati, G.; Djakovitch, L. Tetrahedron Lett. 2008, 49, 2499–2502. doi:10.1016/j.tetlet.2008.02.130
Return to citation in text: [1] -
Bellina, F.; Benelli, F.; Rossi, R. J. Org. Chem. 2008, 73, 5529–5535. doi:10.1021/jo8007572
Return to citation in text: [1] -
Ackermann, L.; Barfüßer, S. Synlett 2009, 808–812. doi:10.1055/s-0028-1087951
Return to citation in text: [1] -
Cornella, J.; Lu, P.; Larrosa, I. Org. Lett. 2009, 11, 5506–5509. doi:10.1021/ol902304n
Return to citation in text: [1] -
Zhao, L.; Bruneau, C.; Doucet, H. ChemCatChem 2013, 5, 255–262. doi:10.1002/cctc.201200521
Return to citation in text: [1] -
Cantat, T.; Génin, E.; Giroud, C.; Meyer, G.; Jutand, A. J. Organomet. Chem. 2003, 687, 365–376. doi:10.1016/s0022-328x(03)00791-5
Return to citation in text: [1] -
Potavathri, S.; Pereira, K. C.; Gorelsky, S. I.; Pike, A.; LeBris, A. P.; DeBoef, B. J. Am. Chem. Soc. 2010, 132, 14676–14681. doi:10.1021/ja107159b
Return to citation in text: [1]
| 1. | Remsing Rix, L. L.; Kuenzi, B. M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J. M.; Haura, E. B.; Lawrence, H. R.; Rix, U. ACS Chem. Biol. 2014, 9, 353–358. doi:10.1021/cb400660a |
| 6. | Ji, X.-M.; Zhou, S.-J.; Deng, C.-L.; Chen, F.; Tang, R.-Y. RSC Adv. 2014, 4, 53837–53841. doi:10.1039/c4ra11168k |
| 8. | Cao, C.; Shi, Y.; Odom, A. L. Org. Lett. 2002, 4, 2853–2856. doi:10.1021/ol0201052 |
| 9. | Layek, M.; Dhanunjaya Rao, A. V.; Gajare, V.; Kalita, D.; Barange, D. K.; Islam, A.; Mukkanti, K.; Pal, M. Tetrahedron Lett. 2009, 50, 4878–4881. doi:10.1016/j.tetlet.2009.06.041 |
| 10. | Wang, C.; Huang, Y. Org. Lett. 2013, 15, 5294–5297. doi:10.1021/ol402523x |
| 11. | Wang, J.; Wang, M.; Chen, K.; Zha, S.; Song, C.; Zhu, J. Org. Lett. 2016, 18, 1178–1181. doi:10.1021/acs.orglett.6b00310 |
| 12. | Panferova, L. I.; Smirnov, V. O.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2017, 82, 745–753. doi:10.1021/acs.joc.6b02344 |
| 13. | Xu, C.; Xu, J. J. Org. Chem. 2018, 83, 14733–14742. doi:10.1021/acs.joc.8b02203 |
| 6. | Ji, X.-M.; Zhou, S.-J.; Deng, C.-L.; Chen, F.; Tang, R.-Y. RSC Adv. 2014, 4, 53837–53841. doi:10.1039/c4ra11168k |
| 7. | Ghorai, J.; Reddy, A. C. S.; Anbarasan, P. Chem. – Eur. J. 2016, 22, 16042–16046. doi:10.1002/chem.201604111 |
| 40. | Potavathri, S.; Pereira, K. C.; Gorelsky, S. I.; Pike, A.; LeBris, A. P.; DeBoef, B. J. Am. Chem. Soc. 2010, 132, 14676–14681. doi:10.1021/ja107159b |
| 2. | Huang, W.-S.; Li, F.; Dalgarno, D. C.; Gong, Y.; Ishchenko, A. V.; Kohlmann, A.; Shakespeare, W. C.; West, A. V.; Xu, Y.; Youngsaye, W.; Zhang, Y.; Zhou, T.; Zhu, X. Preparation of 2-anilino-4-heteroarylpyrimidine compounds for inhibition of mutant EGFR and mutant HER2 kinase. PCT Int. Appl. WO2015175632A1, Nov 19, 2015. |
| 3. | Wei, Y.; Li, Y.; Zhang, G.; Qui, G.; Hu, S.; Chen, L.; Li, S.; Zhang, Q. Preparation of (substituted phenyl) (substituted pyrimidine) amino deriv. as anticancer drugs. PCT Int. Appl. WO2016029839A1, March 3, 2016. |
| 4. | Zhu, Y.; Liu, Z.; Feng, C.; Hu, S.; Chen, H.; Bai, E.; Wang, J.; Shi, J. Deuterated 3-(4,5-substituted pyrimidinamine) phenyl derivative useful in treatment of cancer and its preparation. PCT Int. Appl. WO2018050108A1, March 22, 2018. |
| 5. | Zhang, H.; Wu, W.; Feng, C.; Liu, Z.; Bai, E.; Wang, X.; Lei, M.; Cheng, H.; Feng, H.; Shi, J.; Wang, J.; Zhang, Z.; Jin, T.; Chen, S.; Hu, S.; Zhu, Y. Eur. J. Med. Chem. 2017, 135, 12–23. doi:10.1016/j.ejmech.2017.04.036 |
| 22. | Wang, X.; Gribkov, D. V.; Sames, D. J. Org. Chem. 2007, 72, 1476–1479. doi:10.1021/jo061979v |
| 23. | Bellina, F.; Calandri, C.; Cauteruccio, S.; Rossi, R. Tetrahedron 2007, 63, 1970–1980. doi:10.1016/j.tet.2006.12.068 |
| 24. | Yang, S.-D.; Sun, C.-L.; Fang, Z.; Li, B.-J.; Li, Y.-Z.; Shi, Z.-J. Angew. Chem., Int. Ed. 2008, 47, 1473–1476. doi:10.1002/anie.200704619 |
| 25. | Zhao, J.; Zhang, Y.; Cheng, K. J. Org. Chem. 2008, 73, 7428–7431. doi:10.1021/jo801371w |
| 26. | Joucla, L.; Batail, N.; Djakovitch, L. Adv. Synth. Catal. 2010, 352, 2929–2936. doi:10.1002/adsc.201000512 |
| 27. | Liang, Z.; Yao, B.; Zhang, Y. Org. Lett. 2010, 12, 3185–3187. doi:10.1021/ol101147b |
| 28. | Zhou, J.; Hu, P.; Zhang, M.; Huang, S.; Wang, M.; Su, W. Chem. – Eur. J. 2010, 16, 5876–5881. doi:10.1002/chem.201000529 |
| 29. | Wang, L.; Yi, W.-b.; Cai, C. Chem. Commun. 2011, 47, 806–808. doi:10.1039/c0cc01666g |
| 30. | Lu, G.-p.; Cai, C. Synlett 2012, 23, 2992–2996. doi:10.1055/s-0032-1317702 |
| 31. | Wu, M.; Luo, J.; Xiao, F.; Zhang, S.; Deng, G.-J.; Luo, H.-A. Adv. Synth. Catal. 2012, 354, 335–340. doi:10.1002/adsc.201100603 |
| 38. | Zhao, L.; Bruneau, C.; Doucet, H. ChemCatChem 2013, 5, 255–262. doi:10.1002/cctc.201200521 |
| 16. | Satoh, T.; Miura, M. Chem. Lett. 2007, 36, 200–205. doi:10.1246/cl.2007.200 |
| 17. | Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015 |
| 18. | Ackermann, L.; Vicente, R.; Kapdi, A. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. doi:10.1002/anie.200902996 |
| 19. | Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Adv. Synth. Catal. 2014, 356, 17–117. doi:10.1002/adsc.201300922 |
| 20. | Gensch, T.; James, M. J.; Dalton, T.; Glorius, F. Angew. Chem., Int. Ed. 2018, 57, 2296–2306. doi:10.1002/anie.201710377 |
| 21. | Mao, S.; Li, H.; Shi, X.; Soulé, J.-F.; Doucet, H. ChemCatChem 2019, 11, 269–286. doi:10.1002/cctc.201801448 |
| 39. | Cantat, T.; Génin, E.; Giroud, C.; Meyer, G.; Jutand, A. J. Organomet. Chem. 2003, 687, 365–376. doi:10.1016/s0022-328x(03)00791-5 |
| 14. | Akita, Y.; Inoue, A.; Yamamoto, K.; Ohta, A.; Kurihara, T.; Shimizu, M. Heterocycles 1985, 23, 2327–2333. doi:10.3987/r-1985-09-2327 |
| 15. | Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951–1958. doi:10.3987/com-90-5467 |
| 9. | Layek, M.; Dhanunjaya Rao, A. V.; Gajare, V.; Kalita, D.; Barange, D. K.; Islam, A.; Mukkanti, K.; Pal, M. Tetrahedron Lett. 2009, 50, 4878–4881. doi:10.1016/j.tetlet.2009.06.041 |
| 32. | Akita, Y.; Itagaki, Y.; Takizawa, S.; Ohta, A. Chem. Pharm. Bull. 1989, 37, 1477–1480. doi:10.1248/cpb.37.1477 |
| 33. | Zhang, Z.; Hu, Z.; Yu, Z.; Lei, P.; Chi, H.; Wang, Y.; He, R. Tetrahedron Lett. 2007, 48, 2415–2419. doi:10.1016/j.tetlet.2007.01.173 |
| 34. | Cusati, G.; Djakovitch, L. Tetrahedron Lett. 2008, 49, 2499–2502. doi:10.1016/j.tetlet.2008.02.130 |
| 35. | Bellina, F.; Benelli, F.; Rossi, R. J. Org. Chem. 2008, 73, 5529–5535. doi:10.1021/jo8007572 |
| 36. | Ackermann, L.; Barfüßer, S. Synlett 2009, 808–812. doi:10.1055/s-0028-1087951 |
| 37. | Cornella, J.; Lu, P.; Larrosa, I. Org. Lett. 2009, 11, 5506–5509. doi:10.1021/ol902304n |
© 2019 Huang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)