Abstract
8-Methoxy-γ-humulene, (E)-8-methoxy-β-farnesene, 12-methoxy-β-sesquiphellandrene and 12-methoxyzingiberene can be synthesised in amorphadiene synthase-catalysed reactions from 8- and 12-methoxyfarnesyl diphosphates due to the highly plastic yet tightly controlled carbocationic chemistry of this sesquiterpene cyclase.

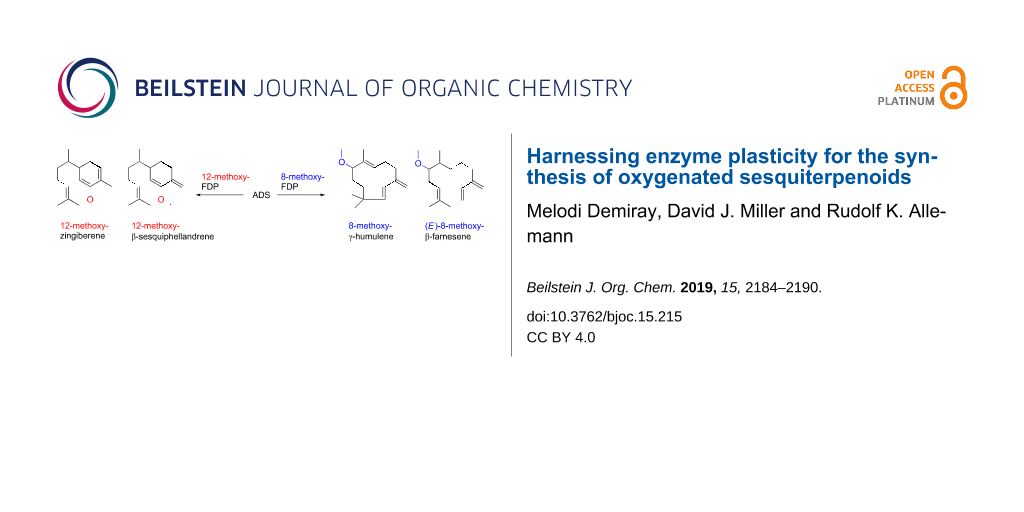
Graphical Abstract
Introduction
Amorphadiene synthase (ADS) from Artemisia annua is a key enzyme involved in the biosynthesis of the antimalarial sesquiterpene drug artemisinin (1) [1-4]. ADS catalyses the Mg2+-dependent conversion of farnesyl diphosphate (FDP, 2) to amorpha-4,11-diene (3) with high regio- and stereochemical control (Scheme 1) [5-7]. The carbocationic reaction mechanism of ADS involves an isomerisation to nerolidyl diphosphate (NDP, 4) followed by breakage of the carbon–oxygen bond to generate the allylic cation 5. This allows rotation around the C2–C3 bond and 1,6-ring closure to form the bisabolyl cation (6). A [1,3]-hydride shift to form carbocation 7 and 1,10-ring closure yield the amorphyl cation (8). Finally, deprotonation generates amorpha-4,11-diene (3) [8,9].
![[1860-5397-15-215-i1]](/bjoc/content/inline/1860-5397-15-215-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism of the ADS-catalysed conversion of FDP (2) to amorpha-4,11-diene (3), a biosynthetic precursor of artemisinin (1).
Scheme 1: Mechanism of the ADS-catalysed conversion of FDP (2) to amorpha-4,11-diene (3), a biosynthetic prec...
Several sesquiterpene synthases including ADS accept FDP analogues containing a variety of heteroatoms and functional groups to generate unnatural sesquiterpenoids that are not easily accessible by conventional organic synthesis [10-19]. Creating novel sesquiterpenoids, not normally found in nature, is of great interest due to the important applications of terpenoids in healthcare and agriculture as well as the potential to tailor their properties to specific needs. For example, fluorinated derivatives of (E)-β-farnesene, a potent alarm pheromone for aphids [20-23], are more effective as pheromones than the parent compound, and finding high potency derivatives of (E)-β-farnesene may be of significant benefit in agriculture [24]. While (S)-germacrene D is a highly volatile but unstable olfactory signal that repels invertebrate arthropod pests (insects, ticks, mites) that affect humans and livestock as well as arable crops, (S)-14,15-dimethylgermacrene D acts as an attractant of aphids [10]. β-Sesquiphellandrene and α-curcumene are both found in turmeric (Curcuma longa) and have been shown to have anticancer activity [25,26]. The oxygenated α-curcumene and β-sesquiphellandrene derivatives α- and β-turmerone are reported to possess anticonvulsant properties and are used to treat epilepsy [27,28]. This array of important compounds shows the potential of generating novel sesquiterpenoids with desirable bio-properties.
ADS is a high fidelity sesquiterpene synthase that produces almost exclusively a single product. Its active site plasticity nevertheless allows the conversion of 12-hydroxy-FDP (9) to dihydroartemisinic aldehyde (10), a biosynthetic intermediate and valuable precursor in the synthesis of artemisinin [29].
Results and Discussion
Here we report that ADS accepts the bulkier FDP analogues 8-methoxy-FDP (11) and 12-methoxy FDP (12) as substrates, thereby opening up direct and efficient synthetic routes to oxidised sesquiterpenoids. This represents in essence a reversal of the biosynthetic pathways to oxidised sesquiterpenoids since cyclisation occurs after FDP ‘oxidation’. 8-Methoxy-FDP (11) and 12-methoxy-FDP (12) were both prepared in the same manner, beginning with a tetrahydropyranyl (THP) protection of (E,E)-farnesol to form 13 [29,30]. This was followed by a selenium dioxide oxidation that produced 12-hydroxyfarnesol (14) as the major product (48%, already published in [29]) in addition to 8-hydroxyfarnesol (15, 11%) [31]. Both of these alcohols were methylated with methyl iodide to yield 16 and 17 [32]. To produce the final FDP analogues, THP was removed to generate alcohols 18 and 19, and subsequently diphosphorylated via halogenated intermediates (Scheme 2 and Supporting Information File 1) [33-35].
![[1860-5397-15-215-i2]](/bjoc/content/inline/1860-5397-15-215-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 8-methoxy-FDP (11) and 12-methoxy-FDP (12) (for full synthesis details see Supporting Information File 1).
Scheme 2: Synthesis of 8-methoxy-FDP (11) and 12-methoxy-FDP (12) (for full synthesis details see Supporting Information File 1).
GC–MS analysis (Figure 1) of the organic soluble products formed from 8-methoxy FDP (11) through ADS catalysis revealed the formation of a major sesquiterpenoid of molecular mass 234 (85%). No organic soluble products were detected when ADS was omitted from the incubation mixture. This product was identified as 8-methoxy-γ-humulene (20) by NMR spectroscopy and comparison of its 1H NMR spectrum (Figure 2) with that of 8-oxo-γ-humulene, a natural sesquiterpenoid isolated from the plant Cineraria fruticulorum [36]. The signals for H5 (δH = 5.44 ppm, d, JH,H = 16.0 Hz), H6 (δH = 6.06, d, JH,H = 16.0 Hz), H15 (δH = 5.00, bs) and H15’ (δH = 5.02, bs) of 8-oxo-γ-humulene [36] are in agreement with their corresponding equivalents in 20 (H2 (δH = 5.48, d, JH,H = 16.0 Hz), H1 (δH = 5.88, d, JH,H = 16.0 Hz), H15 (δH = 4.81, d, JH,H = 2.5 Hz) and H15’ (δH = 4.89, d, JH,H = 2.5 Hz)). The resonances for the methyl groups at C12 and C13 are also analogous, appearing as singlets at 1.03 ppm in 8-oxo-γ-humulene and 0.96 and 0.97 ppm in 20. The identity of the minor product (15%) was confirmed as (E)-8-methoxy-β-farnesene (21) by GC co-elution and comparison of its mass spectrum with an authentic sample prepared by exposing (E)-β-farnesene synthase to diphosphate 11 (Supporting Information File 1). Further support for the structure of the minor product came from the excellent agreement of the diagnostic 1H NMR signals of 21 (H1 (δH = 5.25, d, JH,H = 17.5 Hz), H1’ (δH = 5.06, d, JH,H = 11.0 Hz), H2 (δH = 6.38, dd, JH,H = 17.5 and 11.0 Hz), H15 (δH = 5.02, s), H15’ (δH = 5.00, s)) with those reported for the parent sesquiterpene (E)-β-farnesene (Supporting Information File 1) [37]. The proton on C6 (δH = 5.34, br, t) resonates further downfield for 21 compared to the corresponding proton of (E)-β-farnesene (δH = 5.17, t, JH,H = 7.0 Hz) due to the presence of the methoxy group two carbon atoms away.
![[1860-5397-15-215-1]](/bjoc/content/figures/1860-5397-15-215-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Total-ion chromatogram of the pentane extractable products formed in an incubation of ADS with 8-methoxy-FDP (11). Inset: EI+ Mass spectra of the eluted compounds 20 and 21.
Figure 1: Total-ion chromatogram of the pentane extractable products formed in an incubation of ADS with 8-me...
![[1860-5397-15-215-2]](/bjoc/content/figures/1860-5397-15-215-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectrum (500 MHz, CDCl3) of the 8-methoxy-γ-humulene (20) generated by ADS from 8-methoxy-FDP (11).
Figure 2: 1H NMR spectrum (500 MHz, CDCl3) of the 8-methoxy-γ-humulene (20) generated by ADS from 8-methoxy-F...
The ADS-catalysed 1,11-cyclisation of diphosphate 11 suggests that the 8-methoxy group prevents the formation of a conformation conducive to isomerisation to NDP (4, Scheme 1) and hence 1,6-cyclisation to generate a bisabolyl cation and the amorphane skeleton. Rather the active site conformations of 11 and cation 22 appear to enable a 1,11-cyclisation to 23.
A subsequent [1,3]-hydride shift to cation 24 and deprotonation from C15 lead to 8-methoxy-γ-humulene (20, Scheme 3A). Alternatively, the nucleophilic 8-methoxy group could react at C10 to induce a fast 1,11-cyclisation, forming cation 25, which effectively competes with the isomerization of 11 to 8-methoxy-NDP. A subsequent [1,3]-hydride shift leads to 24 (Scheme 3A). Direct deprotonation of 22 at C15 forms the minor reaction product (E)-8-methoxy-β-farnesene (21) (Scheme 3B). Due to the racemic nature of the starting diphosphate 11b it is, however, also possible that each enzymatic product arises independently from the individual enantiomers.
![[1860-5397-15-215-i3]](/bjoc/content/inline/1860-5397-15-215-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Potential mechanisms for the ADS-catalysed conversion of 8-methoxy-FDP (11) to 8-methoxy-γ-humulene (20) and (E)-8-methoxy-β-farnesene (21).
Scheme 3: Potential mechanisms for the ADS-catalysed conversion of 8-methoxy-FDP (11) to 8-methoxy-γ-humulene...
GC–MS analysis of the organic soluble products generated from an incubation of ADS with 12-methoxy-FDP (12) revealed the formation of a 1:2.4 mixture of two sesquiterpenoids of mass m/z 234 (Figure 3). Again, no organic soluble products were detected when ADS was omitted from the incubation mixture. The 1H NMR spectrum of this product mixture (Figure 4) and comparison with the 1H NMR spectra of the bisabolyl-derived sesquiterpenes β-sesquiphellandrene [38-40] and zingiberene [40], a hydrocarbon with antifertility, antiviral and anticancer activity [41], suggested that the major compound was 12-methoxy-β-sesquiphellandrene (26), while the minor product was identified as 12-methoxyzingiberene (27). The two doublets observed at 0.85 and 0.87 ppm correspond to the C14H3 groups for both compounds. The 13C-DEPT-135 spectrum (see Supporting Information File 1) showed an inverted peak at ≈109 ppm, implying the presence of an olefinic CH2 that couples to the two overlapped doublets at 4.74 ppm. The integration of these doublets as 2 protons suggested that the exocyclic alkene was present in only one of the products. In the literature, this exocyclic alkene in β-sesquiphellandrene is observed at 4.72 ppm as a multiplet [39]. A triplet at 5.39 ppm that integrates for 2 protons can be assigned to H10 in both 26 and 27, which resonates further downfield than in β-sesquiphellandrene and zingiberene due to the methoxy group positioned two carbons away. The olefinic protons H1 (δH = 5.67, d, JH,H = 10.0 Hz) and H2 (δH = 6.14, d, JH,H = 10.0 Hz) of 26 are in agreement with the equivalent protons H1 (δH = 5.66, dd, JH,H = 10.0 and 2.5 Hz) and H2 (δH = 6.13, d, JH,H = 10.0 Hz) in β-sesquiphellandrene. Similarly, the signals for the olefinic protons H1 (δH = 5.63), H2 (δH = 5.77) and H4 (δH = 5.45) in 27 correspond to the equivalent protons (δH = 5.61), H2 (δH = 5.57) and H4 (δH = 5.42) in zingiberene [40].
![[1860-5397-15-215-3]](/bjoc/content/figures/1860-5397-15-215-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Total-ion chromatogram of the pentane extractable products formed in an incubation of ADS with 12-methoxy-FDP (12). Inset: Mass spectra of 26 and 27.
Figure 3: Total-ion chromatogram of the pentane extractable products formed in an incubation of ADS with 12-m...
![[1860-5397-15-215-4]](/bjoc/content/figures/1860-5397-15-215-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR spectrum (400 MHz, CDCl3) of 12-methoxy-β-sesquiphellandrene (26) and 12-methoxyzingiberene (27) produced by ADS from 12-methoxy-FDP (12).
Figure 4: 1H NMR spectrum (400 MHz, CDCl3) of 12-methoxy-β-sesquiphellandrene (26) and 12-methoxyzingiberene (...
Mechanistically, isomerisation of 12 to the methoxy-NDP analogue 28 allows for an ADS-catalysed 1,6-cyclisation to the 12-methoxy-bisabolyl cation (29) followed by a [1,3]-hydride shift, which relocates the positive charge on C1 in 30. With FDP and 12-hydroxy-FDP, a subsequent 1,10-ring closure has been proposed previously [29]. However, the presence of monocyclic products indicates that this second ring closure does not occur with 12-methoxy-FDP (12).
It is suggested that the 12-methoxy group enforces an orientation of the distal 10,11-double bond that is not conducive to the second ring closure. Subsequent deprotonation from C15 and C4 from intermediate 30 affords 12-methoxy-β-sesquiphellandrene (26) and 12-methoxyzingiberene (27), respectively (Scheme 4).
![[1860-5397-15-215-i4]](/bjoc/content/inline/1860-5397-15-215-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Possible mechanisms for the ADS-catalysed conversion of 12-methoxy-FDP (12) to 12-methoxy-β-sesquiphellandrene (26) and 12-methoxyzingiberene (27).
Scheme 4: Possible mechanisms for the ADS-catalysed conversion of 12-methoxy-FDP (12) to 12-methoxy-β-sesquip...
Conclusion
In conclusion, the class I sesquiterpene cyclase amorphadiene synthase facilitates the efficient conversion of readily accessible synthetic methoxy-FDPs to sesquiterpenoids that may have applications in healthcare and agriculture. These results inform us of both the utility and limitations that non-natural functional groups have upon terpene cyclase-catalysed reaction cascades supporting the design of future biocatalytic syntheses. In particular, the presence of an ethereal oxygen atom containing π-acid functionality alongside its inductive withdrawal effect has a profound effect on the carbocationic reactivity of the intermediates. Of course a fully comprehensive interpretation of these results, regarding potentially interesting aspects such as anchimeric assistance is hampered by unknowns such as the conformation of binding to the enzyme and what effect the extra bulk of the substituents has upon the results observed, but nevertheless such empirical results will accumulate to inform future investigations. This reversal of the biosynthetic reaction order is expandable to other terpene synthases to generate libraries of unnatural sesquiterpenoids with a wide range of potential uses and applications across many areas of human activity.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.4 MB | Download |
Acknowledgements
We thank Dr. Veronica Gonzalez for helpful discussions. The help of Dr. Juan A. Faraldos with early parts of this work and of Dr. Rob Jenkins and Mr Thomas Williams with mass spectrometry and NMR spectroscopy is gratefully acknowledged. This work forms part of the Ph.D. project carried out by Dr. Melodi Demiray and is contained within her Ph.D. Thesis (Cardiff University, 2017) and was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC) through grants (BB/H01683X/1, BB/M022463/1, BB/N012526/1) and the Engineering and Physical Sciences Research Council (EPSRC) through grant (EP/K502819/1).
References
-
Klayman, D. Science 1985, 228, 1049–1055. doi:10.1126/science.3887571
Return to citation in text: [1] -
White, N. J. J. Clin. Invest. 2004, 113, 1084–1092. doi:10.1172/jci21682
Return to citation in text: [1] -
Krishna, S.; Bustamante, L.; Haynes, R. K.; Staines, H. M. Trends Pharmacol. Sci. 2008, 29, 520–527. doi:10.1016/j.tips.2008.07.004
Return to citation in text: [1] -
Kremsner, P. G.; Krishna, S. Lancet 2004, 364, 285–294. doi:10.1016/s0140-6736(04)16680-4
Return to citation in text: [1] -
Bouwmeester, H. J.; Wallaart, T. E.; Janssen, M. H. A.; van Loo, B.; Jansen, B. J. M.; Posthumus, M. A.; Schmidt, C. O.; De Kraker, J.-W.; König, W. A.; Franssen, M. C. R. Phytochemistry 1999, 52, 843–854. doi:10.1016/s0031-9422(99)00206-x
Return to citation in text: [1] -
Mercke, P.; Bengtsson, M.; Bouwmeester, H. J.; Posthumus, M. A.; Brodelius, P. E. Arch. Biochem. Biophys. 2000, 381, 173–180. doi:10.1006/abbi.2000.1962
Return to citation in text: [1] -
Chang, Y.-J.; Song, S.-H.; Park, S.-H.; Kim, S.-U. Arch. Biochem. Biophys. 2000, 383, 178–184. doi:10.1006/abbi.2000.2061
Return to citation in text: [1] -
Picaud, S.; Mercke, P.; He, X.; Sterner, O.; Brodelius, M.; Cane, D. E.; Brodelius, P. E. Arch. Biochem. Biophys. 2006, 448, 150–155. doi:10.1016/j.abb.2005.07.015
Return to citation in text: [1] -
Kim, S.-H.; Heo, K.; Chang, Y.-J.; Park, S.-H.; Rhee, S.-K.; Kim, S.-U. J. Nat. Prod. 2006, 69, 758–762. doi:10.1021/np050356u
Return to citation in text: [1] -
Touchet, S.; Chamberlain, K.; Woodcock, C. M.; Miller, D. J.; Birkett, M. A.; Pickett, J. A.; Allemann, R. K. Chem. Commun. 2015, 51, 7550–7553. doi:10.1039/c5cc01814e
Return to citation in text: [1] [2] -
Cascón, O.; Touchet, S.; Miller, D. J.; Gonzalez, V.; Faraldos, J. A.; Allemann, R. K. Chem. Commun. 2012, 48, 9702–9704. doi:10.1039/c2cc35542f
Return to citation in text: [1] -
Faraldos, J. A.; Miller, D. J.; González, V.; Yoosuf-Aly, Z.; Cascón, O.; Li, A.; Allemann, R. K. J. Am. Chem. Soc. 2012, 134, 5900–5908. doi:10.1021/ja211820p
Return to citation in text: [1] -
Miller, D. J.; Yu, F.; Allemann, R. K. ChemBioChem 2007, 8, 1819–1825. doi:10.1002/cbic.200700219
Return to citation in text: [1] -
Faraldos, J. A.; Zhao, Y.; O'Maille, P. E.; Noel, J. P.; Coates, R. M. ChemBioChem 2007, 8, 1826–1833. doi:10.1002/cbic.200700398
Return to citation in text: [1] -
Huynh, F.; Grundy, D. J.; Jenkins, R. L.; Miller, D. J.; Allemann, R. K. ChemBioChem 2018, 19, 1834–1838. doi:10.1002/cbic.201800218
Return to citation in text: [1] -
Huynh, F.; Miller, D. J.; Allemann, R. K. Methods Enzymol. 2018, 608, 83–95. doi:10.1016/bs.mie.2018.05.004
Return to citation in text: [1] -
Oberhauser, C.; Harms, V.; Seidel, K.; Schröder, B.; Ekramzadeh, K.; Beutel, S.; Winkler, S.; Lauterbach, L.; Dickschat, J. S.; Kirschning, A. Angew. Chem., Int. Ed. 2018, 57, 11802–11806. doi:10.1002/anie.201805526
Return to citation in text: [1] -
Rising, K. A.; Crenshaw, C. M.; Koo, H. J.; Subramanian, T.; Chehade, K. A. H.; Starks, C.; Allen, K. D.; Andres, D. A.; Spielmann, H. P.; Noel, J. P.; Chappell, J. ACS Chem. Biol. 2015, 10, 1729–1736. doi:10.1021/acschembio.5b00145
Return to citation in text: [1] -
Lauchli, R.; Rabe, K. S.; Kalbarczyk, K. Z.; Tata, A.; Heel, T.; Kitto, R. Z.; Arnold, F. H. Angew. Chem., Int. Ed. 2013, 52, 5571–5574. doi:10.1002/anie.201301362
Return to citation in text: [1] -
Beale, M. H.; Birkett, M. A.; Bruce, T. J. A.; Chamberlain, K.; Field, L. M.; Huttly, A. K.; Martin, J. L.; Parker, R.; Phillips, A. L.; Pickett, J. A.; Prosser, I. M.; Shewry, P. R.; Smart, L. E.; Wadhams, L. J.; Woodcock, C. M.; Zhang, Y. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10509–10513. doi:10.1073/pnas.0603998103
Return to citation in text: [1] -
Yu, X.-D.; Pickett, J.; Ma, Y.-Z.; Bruce, T.; Napier, J.; Jones, H. D.; Xia, L.-Q. J. Integr. Plant Biol. 2012, 54, 282–299. doi:10.1111/j.1744-7909.2012.01107.x
Return to citation in text: [1] -
Yu, X.; Zhang, Y.; Ma, Y.; Xu, Z.; Wang, G.; Xia, L. The Crop J. 2013, 1, 50–60. doi:10.1016/j.cj.2013.07.005
Return to citation in text: [1] -
Bhatia, V.; Maisnam, J.; Jain, A.; Sharma, K. K.; Bhattacharya, R. Ann. Bot. (Oxford, U. K.) 2015, 115, 581–591. doi:10.1093/aob/mcu250
Return to citation in text: [1] -
Briggs, G. G.; Cayley, G. R.; Dawson, G. W.; Griffiths, D. C.; Macaulay, E. D. M.; Pickett, J. A.; Pile, M. M.; Wadhams, L. J.; Woodcock, C. M. Pestic. Sci. 1986, 17, 441–448. doi:10.1002/ps.2780170415
Return to citation in text: [1] -
Tyagi, A. K.; Prasad, S.; Yuan, W.; Li, S.; Aggarwal, B. B. Invest. New Drugs 2015, 33, 1175–1186. doi:10.1007/s10637-015-0296-5
Return to citation in text: [1] -
Shin, Y.; Lee, Y. Toxicol. Res. (Seoul, Repub. Korea) 2013, 29, 257–261. doi:10.5487/tr.2013.29.4.257
Return to citation in text: [1] -
Orellana-Paucar, A. M.; Serruys, A.-S. K.; Afrikanova, T.; Maes, J.; De Borggraeve, W.; Alen, J.; León-Tamariz, F.; Wilches-Arizábala, I. M.; Crawford, A. D.; de Witte, P. A. M.; Esguerra, C. V. Epilepsy Behav. 2012, 24, 14–22. doi:10.1016/j.yebeh.2012.02.020
Return to citation in text: [1] -
Orellana-Paucar, A. M.; Afrikanova, T.; Thomas, J.; Aibuldinov, Y. K.; Dehaen, W.; de Witte, P. A. M.; Esguerra, C. V. PLoS One 2013, 8, e81634. doi:10.1371/journal.pone.0081634
Return to citation in text: [1] -
Demiray, M.; Tang, X.; Wirth, T.; Faraldos, J. A.; Allemann, R. K. Angew. Chem., Int. Ed. 2017, 56, 4347–4350. doi:10.1002/anie.201609557
Return to citation in text: [1] [2] [3] [4] -
Wuts, P. G. M.; Greene, T. W. Greene's Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2006. doi:10.1002/0470053488
Return to citation in text: [1] -
Rose, M. W.; Rose, N. D.; Boggs, J.; Lenevich, S.; Xu, J.; Barany, G.; Distefano, M. D. J. Pept. Res. 2005, 65, 529–537. doi:10.1111/j.1399-3011.2005.00261.x
Return to citation in text: [1] -
Marco, J. A.; García-Pla, J.; Carda, M.; Murga, J.; Falomir, E.; Trigili, C.; Notararigo, S.; Díaz, J. F.; Barasoain, I. Eur. J. Med. Chem. 2011, 46, 1630–1637. doi:10.1016/j.ejmech.2011.02.011
Return to citation in text: [1] -
Davisson, V. J.; Woodside, A. B.; Neal, T. R.; Stremler, K. E.; Muehlbacher, M.; Poulter, C. D. J. Org. Chem. 1986, 51, 4768–4779. doi:10.1021/jo00375a005
Return to citation in text: [1] -
Huang, Z.; Poulter, C. D. Org. Synth. 1988, 66, 211–215. doi:10.15227/orgsyn.066.0211
Return to citation in text: [1] -
Keller, R. K.; Thompson, R. J. Chromatogr. 1993, 645, 161–167. doi:10.1016/0021-9673(93)80630-q
Return to citation in text: [1] -
Bohlmann, F.; Singh, P.; Jakupovic, J. Phytochemistry 1982, 21, 2531–2535. doi:10.1016/0031-9422(82)85251-5
Return to citation in text: [1] [2] -
Arkoudis, E.; Stratakis, M. J. Org. Chem. 2008, 73, 4484–4490. doi:10.1021/jo800355y
Return to citation in text: [1] -
Tori, M.; Aoki, M.; Asakawa, Y. Phytochemistry 1994, 36, 73–76. doi:10.1016/s0031-9422(00)97015-8
Return to citation in text: [1] -
Kreiser, W.; Körner, F. Helv. Chim. Acta 1999, 82, 1427–1433. doi:10.1002/(sici)1522-2675(19990908)82:9<1427::aid-hlca1427>3.0.co;2-s
Return to citation in text: [1] [2] -
Sy, L.-K.; Brown, G. D. Magn. Reson. Chem. 1997, 35, 424–425. doi:10.1002/(sici)1097-458x(199706)35:6<424::aid-omr84>3.0.co;2-c
Return to citation in text: [1] [2] [3] -
Togar, B.; Turkez, H.; Tatar, A.; Hacimuftuoglu, A.; Geyikoglu, F. Cytotechnology 2015, 67, 939–946. doi:10.1007/s10616-014-9729-9
Return to citation in text: [1]
| 38. | Tori, M.; Aoki, M.; Asakawa, Y. Phytochemistry 1994, 36, 73–76. doi:10.1016/s0031-9422(00)97015-8 |
| 39. | Kreiser, W.; Körner, F. Helv. Chim. Acta 1999, 82, 1427–1433. doi:10.1002/(sici)1522-2675(19990908)82:9<1427::aid-hlca1427>3.0.co;2-s |
| 40. | Sy, L.-K.; Brown, G. D. Magn. Reson. Chem. 1997, 35, 424–425. doi:10.1002/(sici)1097-458x(199706)35:6<424::aid-omr84>3.0.co;2-c |
| 36. | Bohlmann, F.; Singh, P.; Jakupovic, J. Phytochemistry 1982, 21, 2531–2535. doi:10.1016/0031-9422(82)85251-5 |
| 37. | Arkoudis, E.; Stratakis, M. J. Org. Chem. 2008, 73, 4484–4490. doi:10.1021/jo800355y |
| 1. | Klayman, D. Science 1985, 228, 1049–1055. doi:10.1126/science.3887571 |
| 2. | White, N. J. J. Clin. Invest. 2004, 113, 1084–1092. doi:10.1172/jci21682 |
| 3. | Krishna, S.; Bustamante, L.; Haynes, R. K.; Staines, H. M. Trends Pharmacol. Sci. 2008, 29, 520–527. doi:10.1016/j.tips.2008.07.004 |
| 4. | Kremsner, P. G.; Krishna, S. Lancet 2004, 364, 285–294. doi:10.1016/s0140-6736(04)16680-4 |
| 20. | Beale, M. H.; Birkett, M. A.; Bruce, T. J. A.; Chamberlain, K.; Field, L. M.; Huttly, A. K.; Martin, J. L.; Parker, R.; Phillips, A. L.; Pickett, J. A.; Prosser, I. M.; Shewry, P. R.; Smart, L. E.; Wadhams, L. J.; Woodcock, C. M.; Zhang, Y. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10509–10513. doi:10.1073/pnas.0603998103 |
| 21. | Yu, X.-D.; Pickett, J.; Ma, Y.-Z.; Bruce, T.; Napier, J.; Jones, H. D.; Xia, L.-Q. J. Integr. Plant Biol. 2012, 54, 282–299. doi:10.1111/j.1744-7909.2012.01107.x |
| 22. | Yu, X.; Zhang, Y.; Ma, Y.; Xu, Z.; Wang, G.; Xia, L. The Crop J. 2013, 1, 50–60. doi:10.1016/j.cj.2013.07.005 |
| 23. | Bhatia, V.; Maisnam, J.; Jain, A.; Sharma, K. K.; Bhattacharya, R. Ann. Bot. (Oxford, U. K.) 2015, 115, 581–591. doi:10.1093/aob/mcu250 |
| 33. | Davisson, V. J.; Woodside, A. B.; Neal, T. R.; Stremler, K. E.; Muehlbacher, M.; Poulter, C. D. J. Org. Chem. 1986, 51, 4768–4779. doi:10.1021/jo00375a005 |
| 34. | Huang, Z.; Poulter, C. D. Org. Synth. 1988, 66, 211–215. doi:10.15227/orgsyn.066.0211 |
| 35. | Keller, R. K.; Thompson, R. J. Chromatogr. 1993, 645, 161–167. doi:10.1016/0021-9673(93)80630-q |
| 10. | Touchet, S.; Chamberlain, K.; Woodcock, C. M.; Miller, D. J.; Birkett, M. A.; Pickett, J. A.; Allemann, R. K. Chem. Commun. 2015, 51, 7550–7553. doi:10.1039/c5cc01814e |
| 11. | Cascón, O.; Touchet, S.; Miller, D. J.; Gonzalez, V.; Faraldos, J. A.; Allemann, R. K. Chem. Commun. 2012, 48, 9702–9704. doi:10.1039/c2cc35542f |
| 12. | Faraldos, J. A.; Miller, D. J.; González, V.; Yoosuf-Aly, Z.; Cascón, O.; Li, A.; Allemann, R. K. J. Am. Chem. Soc. 2012, 134, 5900–5908. doi:10.1021/ja211820p |
| 13. | Miller, D. J.; Yu, F.; Allemann, R. K. ChemBioChem 2007, 8, 1819–1825. doi:10.1002/cbic.200700219 |
| 14. | Faraldos, J. A.; Zhao, Y.; O'Maille, P. E.; Noel, J. P.; Coates, R. M. ChemBioChem 2007, 8, 1826–1833. doi:10.1002/cbic.200700398 |
| 15. | Huynh, F.; Grundy, D. J.; Jenkins, R. L.; Miller, D. J.; Allemann, R. K. ChemBioChem 2018, 19, 1834–1838. doi:10.1002/cbic.201800218 |
| 16. | Huynh, F.; Miller, D. J.; Allemann, R. K. Methods Enzymol. 2018, 608, 83–95. doi:10.1016/bs.mie.2018.05.004 |
| 17. | Oberhauser, C.; Harms, V.; Seidel, K.; Schröder, B.; Ekramzadeh, K.; Beutel, S.; Winkler, S.; Lauterbach, L.; Dickschat, J. S.; Kirschning, A. Angew. Chem., Int. Ed. 2018, 57, 11802–11806. doi:10.1002/anie.201805526 |
| 18. | Rising, K. A.; Crenshaw, C. M.; Koo, H. J.; Subramanian, T.; Chehade, K. A. H.; Starks, C.; Allen, K. D.; Andres, D. A.; Spielmann, H. P.; Noel, J. P.; Chappell, J. ACS Chem. Biol. 2015, 10, 1729–1736. doi:10.1021/acschembio.5b00145 |
| 19. | Lauchli, R.; Rabe, K. S.; Kalbarczyk, K. Z.; Tata, A.; Heel, T.; Kitto, R. Z.; Arnold, F. H. Angew. Chem., Int. Ed. 2013, 52, 5571–5574. doi:10.1002/anie.201301362 |
| 36. | Bohlmann, F.; Singh, P.; Jakupovic, J. Phytochemistry 1982, 21, 2531–2535. doi:10.1016/0031-9422(82)85251-5 |
| 8. | Picaud, S.; Mercke, P.; He, X.; Sterner, O.; Brodelius, M.; Cane, D. E.; Brodelius, P. E. Arch. Biochem. Biophys. 2006, 448, 150–155. doi:10.1016/j.abb.2005.07.015 |
| 9. | Kim, S.-H.; Heo, K.; Chang, Y.-J.; Park, S.-H.; Rhee, S.-K.; Kim, S.-U. J. Nat. Prod. 2006, 69, 758–762. doi:10.1021/np050356u |
| 31. | Rose, M. W.; Rose, N. D.; Boggs, J.; Lenevich, S.; Xu, J.; Barany, G.; Distefano, M. D. J. Pept. Res. 2005, 65, 529–537. doi:10.1111/j.1399-3011.2005.00261.x |
| 29. | Demiray, M.; Tang, X.; Wirth, T.; Faraldos, J. A.; Allemann, R. K. Angew. Chem., Int. Ed. 2017, 56, 4347–4350. doi:10.1002/anie.201609557 |
| 5. | Bouwmeester, H. J.; Wallaart, T. E.; Janssen, M. H. A.; van Loo, B.; Jansen, B. J. M.; Posthumus, M. A.; Schmidt, C. O.; De Kraker, J.-W.; König, W. A.; Franssen, M. C. R. Phytochemistry 1999, 52, 843–854. doi:10.1016/s0031-9422(99)00206-x |
| 6. | Mercke, P.; Bengtsson, M.; Bouwmeester, H. J.; Posthumus, M. A.; Brodelius, P. E. Arch. Biochem. Biophys. 2000, 381, 173–180. doi:10.1006/abbi.2000.1962 |
| 7. | Chang, Y.-J.; Song, S.-H.; Park, S.-H.; Kim, S.-U. Arch. Biochem. Biophys. 2000, 383, 178–184. doi:10.1006/abbi.2000.2061 |
| 32. | Marco, J. A.; García-Pla, J.; Carda, M.; Murga, J.; Falomir, E.; Trigili, C.; Notararigo, S.; Díaz, J. F.; Barasoain, I. Eur. J. Med. Chem. 2011, 46, 1630–1637. doi:10.1016/j.ejmech.2011.02.011 |
| 27. | Orellana-Paucar, A. M.; Serruys, A.-S. K.; Afrikanova, T.; Maes, J.; De Borggraeve, W.; Alen, J.; León-Tamariz, F.; Wilches-Arizábala, I. M.; Crawford, A. D.; de Witte, P. A. M.; Esguerra, C. V. Epilepsy Behav. 2012, 24, 14–22. doi:10.1016/j.yebeh.2012.02.020 |
| 28. | Orellana-Paucar, A. M.; Afrikanova, T.; Thomas, J.; Aibuldinov, Y. K.; Dehaen, W.; de Witte, P. A. M.; Esguerra, C. V. PLoS One 2013, 8, e81634. doi:10.1371/journal.pone.0081634 |
| 29. | Demiray, M.; Tang, X.; Wirth, T.; Faraldos, J. A.; Allemann, R. K. Angew. Chem., Int. Ed. 2017, 56, 4347–4350. doi:10.1002/anie.201609557 |
| 30. | Wuts, P. G. M.; Greene, T. W. Greene's Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2006. doi:10.1002/0470053488 |
| 39. | Kreiser, W.; Körner, F. Helv. Chim. Acta 1999, 82, 1427–1433. doi:10.1002/(sici)1522-2675(19990908)82:9<1427::aid-hlca1427>3.0.co;2-s |
| 25. | Tyagi, A. K.; Prasad, S.; Yuan, W.; Li, S.; Aggarwal, B. B. Invest. New Drugs 2015, 33, 1175–1186. doi:10.1007/s10637-015-0296-5 |
| 26. | Shin, Y.; Lee, Y. Toxicol. Res. (Seoul, Repub. Korea) 2013, 29, 257–261. doi:10.5487/tr.2013.29.4.257 |
| 29. | Demiray, M.; Tang, X.; Wirth, T.; Faraldos, J. A.; Allemann, R. K. Angew. Chem., Int. Ed. 2017, 56, 4347–4350. doi:10.1002/anie.201609557 |
| 40. | Sy, L.-K.; Brown, G. D. Magn. Reson. Chem. 1997, 35, 424–425. doi:10.1002/(sici)1097-458x(199706)35:6<424::aid-omr84>3.0.co;2-c |
| 10. | Touchet, S.; Chamberlain, K.; Woodcock, C. M.; Miller, D. J.; Birkett, M. A.; Pickett, J. A.; Allemann, R. K. Chem. Commun. 2015, 51, 7550–7553. doi:10.1039/c5cc01814e |
| 40. | Sy, L.-K.; Brown, G. D. Magn. Reson. Chem. 1997, 35, 424–425. doi:10.1002/(sici)1097-458x(199706)35:6<424::aid-omr84>3.0.co;2-c |
| 24. | Briggs, G. G.; Cayley, G. R.; Dawson, G. W.; Griffiths, D. C.; Macaulay, E. D. M.; Pickett, J. A.; Pile, M. M.; Wadhams, L. J.; Woodcock, C. M. Pestic. Sci. 1986, 17, 441–448. doi:10.1002/ps.2780170415 |
| 29. | Demiray, M.; Tang, X.; Wirth, T.; Faraldos, J. A.; Allemann, R. K. Angew. Chem., Int. Ed. 2017, 56, 4347–4350. doi:10.1002/anie.201609557 |
| 41. | Togar, B.; Turkez, H.; Tatar, A.; Hacimuftuoglu, A.; Geyikoglu, F. Cytotechnology 2015, 67, 939–946. doi:10.1007/s10616-014-9729-9 |
© 2019 Demiray et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)