Abstract
We report a detailed structure–activity relationship for the scaffold of VUF16216, a compound we have previously communicated as a small-molecule efficacy photoswitch for the peptidergic chemokine GPCR CXCR3. A series of photoswitchable azobenzene ligands was prepared through various synthetic strategies and multistep syntheses. Photochemical and pharmacological properties were used to guide the design iterations. Investigations of positional and substituent effects reveal that halogen substituents on the ortho-position of the outer ring are preferred for conferring partial agonism on the cis form of the ligands. This effect could be expanded by an electron-donating group on the para-position of the central ring. A variety of efficacy differences between the trans and cis forms emerges from these compounds. Tool compounds VUF15888 (4d) and VUF16620 (6e) represent more subtle efficacy switches, while VUF16216 (6f) displays the largest efficacy switch, from antagonism to full agonism. The compound class disclosed here can aid in new photopharmacology studies of CXCR3 signaling.
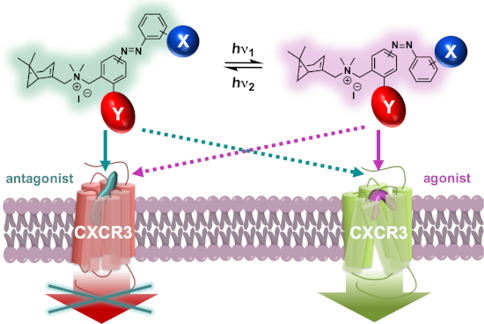
Graphical Abstract
Introduction
Photopharmacology is an emerging discipline at the interface of medicinal chemistry and photochemistry. Classical medicinal chemistry approaches make use of small-molecule ligands binding a target protein, thereby modifying its activity. Photopharmacological approaches use light-sensitive photochromic ligands that provide an advantageous and more precise pharmacological alternative, especially with respect to spatial and temporal precision [1,2]. Photochromic ligands usually contain a molecular photoswitch (photoswitchable moiety) that under certain wavelengths of illumination undergoes an isomerization event, thereby changing the properties of the molecule and the binding affinity for the target protein [3-5] or the intrinsic functional activity (efficacy) [6,7]. Despite the considerable number of photoswitches reported to date, such as spiropyrans, diarylethenes, fulgides or azobenzenes, the most widely used moiety in the photopharmacology is the latter one. One of the main reasons is that an azobenzene has a relatively simple structure that can resemble various biaryl moieties of bioactive compounds: two aromatic rings linked with a bridge (e.g., amide, ether, alkane or alkyne) [8]. In the case of azobenzene the bridge is a diazene group (also called azo group) and depending on the wavelength of illumination, a linear trans-isomer or a bent cis-isomer can be obtained [9]. If certain biaryl moieties are replaced by an azobenzene (i.e., azologization approach), there is a relatively good chance that one of the resulting photoisomers will have a spatial disposition similar to the original biaryl unit and, therefore, a similar biological activity that might change upon isomerization of the azobenzene [8]. The second reason for the success of azobenzene in the photopharmacology field is the robust photoisomerization. It provides typically high yields of photoisomerization with relatively low intensity of light and minimal photobleaching even over hundreds of cycles. A third reason is the relatively high synthetic accessibility to azobenzenes. All these properties make azobenzene compounds ideal molecular photoswitches to control protein activity and physiological events with light.
A number of protein targets have been explored with photochromic small-molecule ligands, such as ion channels, microtubules, enzymes and GPCRs (G protein-coupled receptors) [1,10]. We focus our photopharmacology research on GPCRs [3,7,11], which constitute a superfamily of membrane proteins that regulate many physiological processes [12]. Despite the high relevance of GPCRs both functionally and as a drug target [12], the first synthetic GPCR photochromic small-molecule ligands appeared only five years ago [13-15]. Since then, photopharmacology has been explored on GPCRs targeted endogenously by small molecules [3,4,11,16-20], small peptides [5,13] and larger peptides [7,21,22]. Most of the targeted GPCRs belong to the three rhodopsin-, secretin- and glutamate-like subfamilies and involve GPCRs that endogenously bind small-molecule ligands [10]. The ensuing photochromic GPCR ligands are usually orthosteric and the photoswitching generally affects the functional potency [4,11,23] and/or the binding affinity [3-5,11] of the ligand (Figure 1A). However, as mentioned, GPCRs that endogenously bind large molecules (large peptides or proteins) can be also targeted by allosteric photochromic ligands. In an initial communication [7], we recently reported a photochromic ligand class that is based on azologization of a biaryl ligand class [24] and that activates the chemokine CXCR3 receptor (CXCR3), a GPCR endogenously activated by large peptides CXCL9, CXCL10 and CXCL10 and involved in inflammatory responses. In fact, the six compounds reported in that study represent the first photochromic small-molecule class that harbors a dynamic efficacy photoswitch (from antagonism to agonism) on a peptidergic GPCR (Figure 1B). Here, we report the rationale and synthetic strategies behind this series of compounds, a detailed analysis of the molecular determinants that control the efficacy of the ligands (SAR, structure-activity relationship) and a toolbox of pharmacologically useful photoswitchable small-molecule CXCR3 agonists.
![[1860-5397-15-244-1]](/bjoc/content/figures/1860-5397-15-244-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Design of the CXCR3 efficacy photowitchable ligands. A,B) Schematic representation of a GPCR photochromic ligand that photoisomerizes and thereby photoswitches (A) binding affinity and/or (B) functional efficacy. Red represents the inactive GPCR, while green represents the active GPCR. C) General structure and exemplary functional dose-response curves of the parent biaryl family of CXCR3 ligands disclosed in Wijtmans et al. [24], in which ortho substitution on the outer aromatic ring gives partial or full agonists, while meta substitution provides antagonists. D) Azologization of the biaryl moiety provides a family of photowitchable CXCR3 small-molecule ligands.
Figure 1: Design of the CXCR3 efficacy photowitchable ligands. A,B) Schematic representation of a GPCR photoc...
Results and Discussion
Azologization design
The chemokine receptor CXCR3 is endogenously activated by the chemotactic peptides CXCL11, CXCL10 and CXCL9. Synthetic small-molecule ligands can also bind to CXCR3 [25]. Multiple small-molecule CXCR3 antagonist scaffolds have been disclosed but small-molecule CXCR3 agonists are scarce and are mostly limited to peptidomimetics [25], which makes our published biaryl series a notable exception [24]. The general scaffold of these biaryl ligands consists of a polycycloaliphatic anchor and a biaryl moiety both linked to an ammonium ion. Depending on the substitution pattern of this biaryl moiety, a broad spectrum of efficacies for CXCR3 can be obtained, i.e., from antagonists to partial agonists and full agonists (Figure 1C) [24]. Meta and para-substitution yields antagonists (exemplified by 1a,b), while ortho-substitution with halogen atoms provides agonists, exemplified by partial agonist 1c and equal full agonists 1d and 1e (VUF11418, Figure 2A). A tentative explanation for this efficacy switch includes a variation of the dihedral angle of the biaryl moiety, an increase of the electron density in the biaryl unit and/or a postulated halogen bond of the halogen substituent to the binding site of the receptor [24].
![[1860-5397-15-244-2]](/bjoc/content/figures/1860-5397-15-244-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Conformational alignment of a biaryl CXCR3 agonist with a designed azobenzene analogue. A) 2D structure of biaryl CXCR3 agonist 1e and designed ligand trans-2a and cis-2a. B,C) Alignments of 1e (grey carbon atoms) and 2a, in which the trans and cis-isomers are shown in (B) turquoise and (C) light magenta carbon atoms, respectively. The iodine atom is shown in red for clarity.
Figure 2: Conformational alignment of a biaryl CXCR3 agonist with a designed azobenzene analogue. A) 2D struc...
In order to obtain an efficacy photoswitching, we opted for replacing the biaryl moiety for an azobenzene in an azologization approach (Figure 1D) with the expectation that the isomerization of the azobenzene would provide changes in 3D shape that are similar to those observed in the biaryl series. To reinforce this hypothesis, molecular alignments were performed with Molecular Operating Environment (MOE) software [26] in which 1e was used as a model for full agonism (Figure 2). Its 3D structure was superposed with both the trans and cis-isomers of parent azobenzene compound 2a allowing flexibility of the molecules except for the conformation of the trans and cis-azobenzene moieties, which were fixed in the lowest energy conformation to ensure a shape that has also been validated by crystallographic data [27]. The results show a reasonable overall alignment between trans-2a and the agonist 1e (Figure 2B), since the planar azobenzene is partially overlapping with the biaryl moiety. However, the two aromatic rings of both compounds can evidently not be in exactly the same plane because the azobenzene moiety is planar while the tilting of the dihedral angle of the biaryl moiety of 1e was speculated to be associated with its agonist activity (vide supra) [24]. The alignment of the cis-isomer of 2a with agonist 1e is very different. The outer aromatic ring of cis-2a goes out of plane and is now occupying the space that is also occupied by the iodine atom of 1e. These calculations indicate that CXCR3 agonism is more likely to be associated with the cis-isomer than with the trans-isomer in our designed azobenzenes.
Synthesis of azobenzene analogues and exploration of substitution pattern on the outer aromatic ring
In addition to unsubstituted azobenzene analogue 2a, we explored the substitution pattern of the outer aromatic ring with chlorine atoms in the ortho, meta and para-position (compounds 2b–d, respectively) to also assess the possibility of agonism provided by a halogen bond. Compound 2e, which contains a bromine atom in the ortho-position, was also tested since this atom type provides full agonist activity of parent 1d. The synthesis of the compounds 2a–e was performed according to the strategies depicted in Scheme 1. The intermediate 7 was prepared as described previously by us [28] and was used in a reductive amination with 4-nitrobenzaldehyde (8a) to give the corresponding tertiary amine 9a in high yield. The nitro group of 9a was subsequently reduced by SnCl2 in high yield. The resulting aniline 10a was used to obtain the azo compounds 12a–e in varying yields through a Mills reaction with the corresponding nitroso compounds 11a–e, which were commercially available or prepared as described in our previous communication [7]. A final methylation of the tertiary amine 12a–e with MeI in DCM and subsequent precipitation with MTBE (methyl tert-butyl ether) gave 2a–e as orange powders with ≥99% trans-isomer in moderate to high yield.
![[1860-5397-15-244-i1]](/bjoc/content/inline/1860-5397-15-244-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic strategies for compounds 2a–e, 3a–e, 4a–d, 4f–i and 5b,c (Y = H, Cl). Reagents and conditions: (a) i) Et3N (1.1–1.6 equiv), DCM, rt, 10–30 min; ii) NaBH(OAc)3 (1.6 equiv), rt, 6–16 h, 91–97%; (b) SnCl2 (5.0 equiv), EtOH, 75 °C, 2 h, 87%–quant; (c) AcOH/DCM, rt, 1–5 d, 23–73%, (d) MeI (20 equiv), DCM, rt, 6–72 h, 41–97%.
Scheme 1: Synthetic strategies for compounds 2a–e, 3a–e, 4a–d, 4f–i and 5b,c (Y = H, Cl). Reagents and condit...
Next, we characterized the photochemical properties of 2a–e, including absorption maxima, wavelengths of illumination and percentage of conversion from trans to cis in the photostationary state (PSS cis). First, UV–vis absorption spectra were measured in the dark and after illumination with a wavelength of 360 nm. In all cases the spectrum measured in the dark shows a large band between 320 and 330 nm that corresponds to the π–π* transition of the trans-isomer (Figure S1A, Supporting Information File 1). After irradiating with different wavelengths, the proportion of trans and cis-isomer varies to reach the PPS. For example, after irradiating with 360 nm the π–π* transition band of the trans-isomer can barely be observed, but a less intense wide band around 420 nm appears, which corresponds to the n–π* transition of the cis-isomer (Figure S1A, Supporting Information File 1). This indicates that, using 360 nm light, a PPS of high percentage of cis-isomer (PPS360) can be reached. Literature evidence [7,29,30] suggests that this scaffold would have sufficiently long half-lives of the PSS state, i.e., the cis-isomer only slowly reverts to trans in the dark, which was confirmed by early-stage analyses on key compounds (data not shown). In fact, due to this bistable nature, the percentage of each photoisomer can be quantified by analytical chromatography (LC–MS). The integration wavelength (265 nm) was selected closely to the observed isosbestic point for most compounds to get an indication of mole ratios from the UV area ratios. For 2a–e after illumination at 360 nm, PPS360 values of 79–90% of cis-isomer were obtained. We also tested 434, 460 nm and/or 494 nm to revert the isomerization process to PSS trans (Figure S1A, Supporting Information File 1). After re-illuminating the samples at 434 nm or 460 nm, the π–π* transition band of the trans-isomer re-appears, indicating a high percentage of trans-isomer at that PSS. The use of 494 nm affords less trans compound and is less efficient in achieving PSS trans.
Next, the CXCR3 binding properties of 2a–e were measured (Table 1) in a competition binding assay versus displacement of a radiolabeled small-molecule CXCR3 antagonist ([3H]-VUF11211 [31]). Values reported are mean ± SEM (Standard Error of the Mean). These experiments were performed with samples under dark conditions to ensure ≥99% trans-isomer and with samples previously illuminated with 360 nm light to obtain a high percentage of cis compound in the PSS360. This assay setup is enabled by the bistable nature of the photoswitch (vide supra), maintaining integrity of both the trans and cis-isomer for the duration of the assay. Compounds 2a–e under dark conditions (trans-isomers) bind CXCR3 with Ki values in the high nanomolar range. In contrast, compounds 2a–e after 360 nm illumination bind CXCR3 with Ki values in the low micromolar range, although the observed photoinduced affinity shifts (PAS) are not large (<4.0-fold). To assess if the compounds have agonist or antagonist activity on CXCR3-mediated signaling, a single-dose functional [35S]-GTPγS accumulation assay was performed with the compounds 2a–e at 10 µM (Table 1). In this assay, we observed that most of the compounds are not activating the CXCR3 receptor in either cis or trans configuration, which indicates that the compounds bind to the receptor as antagonists. However, cis-2b and, more notably, cis-2e show a small partial agonist activity (11% and 23%, respectively) that gives a hint of a slight photoswitching of their efficacy. Both compounds have a halogen atom on the ortho-position of the outer ring, which seems to reaffirm the importance of the ortho-halogen atoms effect observed in the biaryl series [24]. However, the difference in efficacy between cis and trans-isomers (defined as PDE - photoinduced difference of efficacy) needed to be improved.
Table 1: Structures and results of photochemical, binding and functional characterization of compounds 2a–e and 3a–h.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-244-i5.svg?max-width=637&scale=1.0)
|
|||||||||||||||
| Cmpd | Photochemistry | CXCR3 binding affinity | Functional CXCR3 activity | ||||||||||||
| X |
λmax trans
π–π* a |
λmax cis
n–π* a |
PSS360 (area % cis)b | SEM |
pKi
transc |
SEM | pKi PSS360c | SEM | PASd | E (%) transe | SEM | E (%) PSS360f | SEM | PDE (%)g | |
| 2a | H | 321 | 422 | 88.3 | 0.8 | 6.3 | 0.0 | 5.7 | 0.1 | 4.0 | −11.0 | 2.3 | −8.3 | 0.7 | 2.7 |
| 2b | 2-Cl | 323 | 416 | 84.9 | 0.5 | 6.3 | 0.0 | 5.8 | 0.1 | 3.2 | 2.4 | 4.3 | 11.1 | 2.0 | 8.7 |
| 2c | 3-Cl | 320 | 421 | 79.2 | 0.9 | 6.5 | 0.0 | 6.3 | 0.0 | 1.6 | −10.3 | 3.5 | −9.6 | 2.7 | 0.7 |
| 2d | 4-Cl | 330 | 423 | 89.7 | 0.5 | 6.6 | 0.0 | 6.0 | 0.0 | 4.0 | −9.8 | 3.5 | −2.7 | 3.6 | 7.1 |
| 2e | 2-Br | 324 | 421 | 87.2 | 0.5 | 6.4 | 0.0 | 5.9 | 0.1 | 3.2 | 7.0 | 1.6 | 22.7 | 1.5 | 15.7 |
| 3a | H | 320 | 423 | 82.0 | 1.5 | 6.0 | 0.1 | 5.4 | 0.1 | 4.0 | −11.6 | 2.2 | −4.7 | 1.7 | 6.9 |
| 3b | 2-Cl | 323 | 419 | 85.0 | 0.5 | 6.3 | 0.0 | 5.6 | 0.0 | 5.0 | −4.7 | 1.7 | 17.4 | 3.7 | 22.1 |
| 3c | 3-Cl | 317 | 421 | 83.4 | 0.4 | 6.3 | 0.0 | 5.8 | 0.0 | 3.2 | −8.4 | 2.2 | −0.4 | 1.6 | 8.0 |
| 3d | 4-Cl | 325 | 424 | 92.0 | 0.3 | 6.4 | 0.0 | 5.7 | 0.0 | 5.0 | −8.1 | 2.6 | 1.7 | 2.3 | 9.8 |
| 3eh | 2-Br | 323 | 421 | 88.9 | 0.4 | 6.3 | 0.0 | 5.7 | 0.1 | 4.0 | −5.8 | 1.7 | 25.1 | 2.0 | 30.9 |
| 3f | 2-I | 323 | 422 | 80.9 | 0.2 | 6.2 | 0.0 | 5.8 | 0.1 | 2.5 | −4.0 | 1.2 | 15.8 | 1.3 | 19.8 |
| 3g | 3-I | 318 | 422 | 82.5 | 1.5 | 6.1 | 0.1 | 6.1 | 0.0 | 1.0 | −11.1 | 0.6 | −7.6 | 2.3 | 3.5 |
| 3h | 4-I | 337 | 422 | 91.1 | 1.1 | 6.0 | 0.0 | 5.7 | 0.0 | 2.0 | −15.3 | 1.1 | 5.4 | 1.5 | 20.7 |
aThe absorbance maxima were extracted from UV–vis spectra at 25 µM in PBS buffer with 1% DMSO. b% of cis-isomer at the photostationary state (PSS360) measured in 68% TRIS buffer and 32% DMSO (1 mM) after being pre-irradiated at 360 nm as obtained by LC–MS integration of the cis and trans-isomer signals at 265 nm. The mean and SEM of at least two experiments are shown. cBinding affinity of trans-isomer or PSS360 as measured using [3H]-VUF11211 displacement. The mean and SEM of at least three experiments are shown. dThe photoinduced affinity shift (PAS) is calculated as the ratio of the Ki PSS360 and Ki trans. eNormalized CXCR3 functional activity of trans-isomer (10 µM) in the dark (efficacy of 1d set at 100% activity). The mean and SEM of at least three experiments are shown. fNormalized CXCR3 functional activity of a sample (10 µM) pre-irradiated at 360 nm to reach the photostationary state (efficacy of 1d set at 100% activity). The mean and SEM of at least three experiments are shown. gThe photoinduced difference of efficacy (PDE) is obtained by subtracting E trans from E PSS360. hCompound was previously described by us [7].
Optimization of positioning of azobenzene unit
Aiming to improve the position and directionality of the halogen atom, we next designed a subseries with the azo group at the meta-position of the central ring (scaffold 3) instead of at the para-position as in 2a–e. The analogue without halogen substitution (3a) as well as Cl/Br analogues comparable to the first series (3b–e) were prepared. Moreover, since the importance of the presence of a halogen in the outer ring was suggested in 2, compounds 3f–h, which include an iodine atom on the ortho, meta and para-positions, respectively, were synthesized.
The synthesis of compounds 3a–e was performed following the strategies shown in Scheme 1 as disclosed for compounds 2a–e. Briefly, a reductive amination of 7 and 8b gave nitro compound 9b, which after reduction to 10b, coupling with nitroso compounds 11a–e to 13a–e and methylation gave iodide salts 3a–e with purities of trans-isomers ≥99% and overall yields similar to the ones obtained for 2a–e. However, 2-iodonitrosobenzene cannot be accessed through oxidation of the corresponding aniline owing to oxidation sensitivity of the iodine atom. Therefore, an alternative route had to be used to synthesize 3f–h (Scheme 2). The route began with the oxidation of methyl 3-aminobenzoate (17a) using Oxone® to obtain a crude nitroso product 18a, which was used in a Mills reaction with an iodoaniline (19a–c) at 100 °C to obtain azobenzenes 20g,h in high yields and ortho-analogue 20f in a decreased yield presumably due to steric hindrance. The methyl ester was selectively reduced with DIBAL-H to benzyl alcohols 23f–h, which were oxidized with Dess–Martin periodinane to the corresponding benzaldehyde 26f–h. Reductive amination of 26f–h with 7 gave the tertiary amines 13f–h. Methylation with iodomethane and subsequent precipitation gave 3f–h as orange powders with ≥99% trans-isomer in high yields.
![[1860-5397-15-244-i2]](/bjoc/content/inline/1860-5397-15-244-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic strategies for compounds 3f–h, 4e, 6b, and 6d (Y = H, F, Cl, Br). Reagents and conditions: (a) Oxone® (2.0 equiv), DCM/H2O 1:4, rt, 2 h, 91–98%; (b) AcOH, 100 °C, 16–20 h, 61–96%; (c) i) DIBAL-H (3.0–4.0 equiv), THF, 0–5 °C to rt, 2–4 h; ii) NH4Cl (aq), Rochelle salt (10% aq), EtOAc, 1–2 h, 76–99%; or for 23h i) DIBAL-H (1.2 equiv) , DCM, −78 °C, 1 h; ii) MeOH, −78 °C to rt, 0.5 h, iii) Rochelle salt (10% aq), 3 h, 45%; (d) Dess–Martin periodinane (1.0 equiv), DCM, rt, 1–2 h, 68–97%; (e) i) Et3N (1.1–1.6 equiv), DCM, rt, 10–30 min; ii) NaBH(OAc)3 (1.6 equiv), rt, 6–16 h, 69–96%; (f) MeI (20 equiv), DCM, rt, 6–72 h, 79–95%.
Scheme 2: Synthetic strategies for compounds 3f–h, 4e, 6b, and 6d (Y = H, F, Cl, Br). Reagents and conditions...
The photochemical properties of 3a–h are very similar to those of 2a–e. For the trans-isomers, the maximum of the π–π* band is located around 317–325 nm, with the exception of 3h which has an iodine atom on the para-position and arguably confers a larger electron delocalization of π-electrons that is translated to a bathochromic shifting of the band to lower energy wavelength (337 nm). PSS values of 81–92% are obtained when illuminating with 360 nm light. The binding properties of 3a–h also result in outcomes similar to those of 2a–e. That is, Ki values are in the high nanomolar range for the trans-isomers with no or low PAS values (1.0–5.0-fold) after isomerization. In single-dose functional assays, all trans-isomers do not substantially activate CXCR3, whereas three of the cis-isomers weakly to substantially activate CXCR3 (3b: 17%, 3e: 25% and 3f: 16%). Interestingly, these three compounds are the only ones to include a halogen atom on the ortho-position of the outer azobenzene ring, as is the case with para-compounds 2b (11%) and 2e (23%). However, compared to the latter, meta-compounds 3b,e,f display an agonist effect that is slightly higher. Increasing the size of the ortho-halogen does not guarantee a maximal agonist effect, as the absolute activity of 3f is lower than that of its Br analogue 3e. Nevertheless, evidence emerged that the ortho-position of the outer aromatic ring in scaffold 3 is important to achieve agonist activity of the cis-isomer, but it should be complemented with other strategies to further increase intrinsic activity.
Substituent effects on the outer ring
One of the postulated contributors to the CXCR3 agonism effect of parent biaryls such as 1d and 1e is the increased π-electron density of the aromatic rings [24]. A way to translate this effect to the azobenzene system is by including a π-donating substituent on the aromatic system. For synthetic access and thus rapid exploration, we chose a Cl atom as mildly π-EDG (electron-donating group) even though it is σ-EWG (electron-withdrawing group) on the para-position of the inner ring with respect to the azo bond (i.e., the ortho-position with respect to the benzylic position) to afford subseries 4. This π-electron delocalization would increase the electron density of the azobenzene unit, and was also expected to have an effect on the trans–cis azobenzene isomerization and PSS value. In terms of the outer ring, and given that ortho halogen atoms in subseries 2 and 3 play an important role in conferring the cis-isomer with partial agonism, in subseries 4 we explored halogen atoms and groups differing in, e.g., steric and electronic properties (Me, CF3, OMe, OCF3). The electron-donating groups in this series (Me and OMe) also increase the electron density of the azobenzene system.
The synthesis of 4a–d and 4f–i was performed following the route shown in Scheme 1. Briefly, 7 was used in a reductive amination with 2-chloro-3-nitrobenzaldehyde (8c) to give nitro compound 9c which was reduced to aniline 10c and used to obtain the azo compounds 14a–d,f–i in variable yields through a Mills reaction with the corresponding nitroso compounds 11a-b,e–j. Methylation of 14a–d,f–i with MeI yielded compounds 4a–d,f–g,i as orange powders with ≥99% trans-isomer in moderate to high yields. Salt 4h did not precipitate after treatment with MTBE and was isolated as an oil, which retained substantial amounts of MTBE solvate even after extensive drying. For 4e, we used the strategy as explained for iodo compounds 3f–h (vide supra). Briefly, the route (Scheme 2) consists of the oxidation of methyl 5-amino-2-chlorobenzoate (17c) with Oxone® to 18c, which was used in a Mills reaction with 2-iodoaniline (19a) to yield azobenzene 21. The methyl ester was selectively reduced with DIBAL-H and the resulting alcohol 24 oxidized with Dess–Martin periodinane to benzaldehyde 27. Reductive amination of 27 with 7 to 14e and subsequent methylation gave 4e as an orange powder with ≥99% trans-isomer.
Photochemical characterization of 4a–i (Table 2) gives similar results as observed for subseries 2 and 3 (Figure S2 (Supporting Information File 1) shows an exemplary time-resolved NMR and LC–MS analysis). That is, trans π–π* bands have a maximum between 326 and 332 nm and PSS values generally amount to over 90% of cis-isomer after illuminating with 360 nm light, as predicted from the electron localization provided by the chlorine atom in the central aromatic ring. Compounds 4e, 4i and most notably 4g deviate from this trend, since the percentage of cis-isomer in the PSS is significantly lower (58–80%). Trans-4a–i show a slight decrease in binding affinity compared to the subseries 2 and 3, amounting to low micromolar values (Table 2). Moreover, PSS affinity values are also modest, with only four compounds that have a PAS > 2.5 (4a–c,f). However, functional data from [35S]-GTPγS assays provide encouraging results. While all trans-isomers of 4a–i do not or only weakly activate the receptor, some of the cis-isomers clearly behave as partial agonists. The highest efficacy is exerted by cis-4c–f with E values between 30–50% at 10 µM. Interestingly, one of these compounds (4f) includes a methyl group as ortho-substituent on the outer ring and its agonist effect at PSS amounts to 32%, possibly questioning one of our hypotheses that a halogen bond is involved in inducing CXCR3 agonism.
Table 2: Structure and results of photochemical, binding and functional characterization of compounds 4a–i and 5b,c.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-244-i6.svg?max-width=637&scale=1.0)
|
|||||||||||||||
| Cmpd | Photochemistry | CXCR3 binding affinity | Functional CXCR3 activity | ||||||||||||
| X |
λmax trans
π–π* a |
λmax cis
n–π* a |
PSS360 (area % cis)b | SEM |
pKi
transc |
SEM | pKi PSS360c | SEM | PASd | E (%) transe | SEM | E (%) PSS360f | SEM | PDE (%)g | |
| 4a | H | 326 | 424 | 91.8 | 1.2 | 5.8 | 0.0 | 5.2 | 0.0 | 4.0 | −2.9 | 1.0 | 12.1 | 0.7 | 15.0 |
| 4b | 2-F | 330 | 420 | 93.0 | 0.2 | 5.6 | 0.0 | 5.0 | 0.0 | 4.0 | −4.9 | 0.5 | 14.6 | 0.8 | 19.5 |
| 4c | 2-Cl | 330 | 419 | 92.8 | 0.1 | 5.8 | 0.0 | 5.3 | 0.1 | 3.2 | 2.3 | 1.6 | 36.9 | 2.0 | 34.6 |
| 4dh | 2-Br | 328 | 422 | 92.6 | 0.2 | 5.9 | 0.0 | 5.6 | 0.0 | 2.0 | 13.6 | 2.4 | 49.6 | 2.6 | 36.0 |
| 4e | 2-I | 330 | 424 | 79.9 | 1.2 | 5.7 | 0.0 | 5.7 | 0.0 | 1.0 | 16.0 | 1.4 | 37.1 | 0.5 | 21.1 |
| 4f | 2-Me | 332 | 428 | 94.9 | 0.1 | 5.9 | 0.0 | 5.3 | 0.0 | 4.0 | 6.9 | 3.1 | 32.2 | 1.1 | 25.3 |
| 4g | 2-CF3 | 326 | 424 | 58.0 | 1.1 | 5.9 | 0.0 | 5.7 | 0.0 | 1.6 | 10.5 | 1.6 | 8.3 | 1.5 | −2.2 |
| 4h | 2-OMe | 328 | 427 | 92.6 | 0.2 | 5.4 | 0.0 | 5.0 | 0.0 | 2.5 | 4.9 | 1.2 | 18.7 | 1.1 | 13.8 |
| 4i | 2-OCF3 | 326 | 422 | 78.8 | 1.2 | 5.9 | 0.0 | 5.6 | 0.0 | 2.0 | −3.0 | 2.0 | -2.4 | 3.1 | 0.6 |
| 4dh | 2-Br | 328 | 422 | 92.6 | 0.2 | 5.9 | 0.0 | 5.6 | 0.0 | 2.0 | 13.6 | 2.4 | 49.6 | 2.6 | 36.0 |
| 5b | 3-Br | 324 | 420 | 87.4 | 1.2 | 5.7 | 0.1 | 5.6 | 0.0 | 1.3 | −1.7 | 1.9 | 10.5 | 0.5 | 12.2 |
| 5c | 4-Br | 335 | 427 | 93.8 | 0.5 | 6.2 | 0.0 | 5.6 | 0.0 | 4.0 | −7.2 | 1.8 | 1.8 | 2.5 | 9.0 |
aThe absorbance maxima were extracted from UV–vis spectra at 25 µM in PBS buffer with 1% DMSO. b% of cis-isomer at the photostationary state (PSS360) measured in 68% TRIS buffer and 32% DMSO (1 mM) after being pre-irradiated at 360 nm as obtained by LC–MS integration of the cis and trans-isomer signals at 265 nm. The mean and SEM of at least two experiments are shown. cBinding affinity of trans-isomer or PSS360 in PBS (25 µM) as measured using [3H]-VUF11211 displacement. The mean and SEM of at least three experiments are shown. dThe photoinduced affinity shift (PAS) is calculated as the ratio of the Ki PSS360 and Ki trans. eNormalized CXCR3 functional activity of trans-isomer 10 µM in the dark (efficacy of 1d set at 100% activity). The mean and SEM of at least three experiments are shown. fNormalized CXCR3 functional activity of a sample (10 µM) pre-irradiated at 360 nm to reach the photostationary state (efficacy of 1d set at 100% activity). The mean and SEM of at least three experiments are shown. gThe photoinduced difference of efficacy (PDE) is obtained by subtracting E trans from E PSS360. hCompound was previously described by us [7].
Similar to subseries 3, the substituent on the ortho-position of the outer aromatic ring appears to be a driver for the agonist activity of the corresponding cis-isomer. Starting from weak partial agonist 4a (X = H), the agonist response of the cis increases with increasing halogen atom (4b–d) to a maximal response with 4d (X = Br) having an agonist effect of 50%. However, when X = I (4e) the agonist activity is reduced, as also observed in the 3 subseries (compare 3d to 3e). In general, the agonist effects and PDE values exerted by subseries 4 are larger than those of 3 (Figure 3). This could be explained by the effect of the Cl atom present in the 4 subseries, that may give a rise in electron density to the azobenzene necessary to increase the efficacy of the cis-isomer.
![[1860-5397-15-244-3]](/bjoc/content/figures/1860-5397-15-244-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Comparison of compounds belonging to the subseries 3 or 4 with a halogen substitution on the ortho-position of the outer ring (X). (A) Activities of cis-isomers at 10 µM on CXCR3-mediated G protein activation. (B) PDE values at 10 µM.
Figure 3: Comparison of compounds belonging to the subseries 3 or 4 with a halogen substitution on the ortho-...
In both subseries 3 and 4, a bromine atom on the ortho-position of the outer ring gives optimal results in providing CXCR3 efficacy photoswitching (Figure 3). To confirm this for subseries 4, the analogues of 4d with the bromine on the meta and para position (5b and 5c, respectively) were also synthesized. The synthetic route (Scheme 1) utilized 10c and 3- and 4-bromonitrosobenzene (11k,l) to form azobenzenes 15b,c, which were methylated to obtain 5b,c. The binding affinities (Table 2) obtained for the trans and cis-isomers are all in the micromolar range. More importantly, when comparing functional results of 4d, 5b and 5d, the preference for a Br atom on the ortho-position can also be reaffirmed for subseries 4, because cis-5c is an antagonist, while cis-5b shows only a weak activity (Table 2).
Substituent effects on the central ring
As shown, substitution of the outer ring with an ortho-bromine in conjunction with a Cl substituent on the central ring appeared to pave the way for efficacy photoswitching but there was still room for improvement. Our strategy in the final optimization round was to replace the mildly π-EDG Cl atom with other groups. Thus, to aim for a full-agonist cis compound, the sub-series 6 was synthesized. In this subseries, different groups at the para-position of the central ring (Y) in combination with the ortho-Br atom on the outer aromatic ring are used to to explore optimal electron densities in the azobenzene system. Besides the H and Cl already explored (3e and 4d, respectively), other groups including an EWG halogen atom (F, 6b), EDG halogen atoms (Br, 6d and I, 6e) and stronger EDG moieties such as OMe (6f), OiPr (6g), SMe (6h) and NMe2 (6i) were introduced.
Compounds 6b and 6d were synthesized by the route shown in Scheme 2, using in this case halogenated methyl aminobenzoates 17b,c and 2-bromoaniline 19d. The synthetic route for compound 6e proved more challenging. The route of Scheme 1 could not be readily used because the corresponding iodinated nitro precursor 8 is not commercially available, nor could the second route (Scheme 2) since the iodine would likely be sensitive to the first oxidation step. Therefore, we designed a new route (Scheme 3). The starting point was the advanced intermediate 28b (Scheme 3), which was subjected to an aromatic nucleophilic substitution with potassium phthalimide prepared in situ from 29 and K2CO3. Presumably due to the alkaline medium, the phthalimide ring was partially opened as detected by HPLC–MS. Upon attempted re-closing under reflux in AcOH, completely deprotection took place and, after purification, aniline 30 was obtained with high purity. Compound 30 was used to introduce the iodine atom through a Sandmeyer reaction to give 28e albeit in low yield. Reductive amination with 7 afforded amine 16e followed by methylation to give trans-6e as an orange solid with 97% purity.
![[1860-5397-15-244-i3]](/bjoc/content/inline/1860-5397-15-244-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthetic strategy for compound 6e. Reagents and conditions: (a) i) K2CO3 (2.0 equiv), DMF, µW, 65 °C, 3 h; ii) AcOH, reflux, 1 h, 67%; (b) i) pTsOH·H2O (3.0 equiv), MeCN, 10–15 °C; ii) NaNO2 (2.0 equiv), KI (2.5 equiv), H2O, 10–15 °C to rt, 2 h, 13%; (c) i) Et3N (1.2 equiv), DCM, rt, 30 min; ii) NaBH(OAc)3 (1.6 equiv), rt, 16 h, 54%; (f) MeI (20 equiv), DCM, rt, 20 h, 57%.
Scheme 3: Synthetic strategy for compound 6e. Reagents and conditions: (a) i) K2CO3 (2.0 equiv), DMF, µW, 65 ...
The synthesis of compounds 6f–h was performed following a nucleophilic aromatic substitution route on 28b (Scheme 4) since precursors 8 or 17 with the required substituent Y were not available. We performed nucleophilic aromatic substitutions with the corresponding sodium salts of MeOH, 2-PrOH and MeSH under µW irradiation. Both 28f (Y = OMe) and 28h (Y = SMe) were formed in high yields, but the conversion of the reaction with NaOiPr was very low and partially gave reduction of the benzaldehyde. An alternative route utilized a method from Engle et al. proceeding through a tert-butylimine intermediate (31) formed under Dean–Stark conditions [32]. This imine was reacted with NaOiPr to form the ether 32, which was subsequently hydrolyzed to obtain the desired aldehyde 28g in high yield. Reductive amination of the aldehydes 28f–h with 7 furnished amines 16f–h which was followed by methylation to afford 6f–h as orange solids with ≥98% trans-isomer. The synthetic strategies for the synthesis of compound 6i were reported in our previous communication [7].
![[1860-5397-15-244-i4]](/bjoc/content/inline/1860-5397-15-244-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthetic strategies for compounds 6f–h (Y = OMe, OiPr, SMe). Reagents and conditions: (a) NaOMe or NaSMe (1.0–1.2 equiv), MeOH or DMF, 65 °C, 30–60 min, 88–90%. (b) PhMe, 110 °C, Dean–Stark, 20 h, 99%; (c) NaH (1.0 equiv), DMSO, 100 °C, 1 h; (d) THF/H2O/AcOH 50:15:1, rt, 16 h, 79% (two steps); (e) i) Et3N (1.3–1.4 equiv), DCM, rt, 10–30 min; ii) NaBH(OAc)3 (1.6 equiv), rt, 6–16 h, 84–95%; (f) MeI (20 equiv), DCM, rt, 20 h, 86–90%.
Scheme 4: Synthetic strategies for compounds 6f–h (Y = OMe, OiPr, SMe). Reagents and conditions: (a) NaOMe or...
In the subseries depicted in Table 3, several notable differences in both the UV–vis spectra (Figure S1, Supporting Information File 1) and the photoisomerization are observed. When the substituent Y is a halogen atom, we observe a slight bathochromic shift of the π–π* band to higher wavelengths with increasing size of the heteroatom, from 323 nm for Y = H (3e) to 339 nm for Y = I (6e) (Table 3, Figure 4A). When the group Y is OMe (6f), this shift is larger due to the higher EDG properties of MeO (λmax = 352 nm) and this is slightly increased with Y = OiPr (6g, λmax = 355 nm). The shift is highest for Y = NMe2 (6i, λmax = 387 nm). When the oxygen atom of 6f is replaced by a sulfur atom, the bathochromic shift of the π–π* band is also increased (6h, λmax = 373 nm, Table 3, Figure 4A). This high capacity of sulfur substituents to induce a bathochromic effect has already been reported in the azobenzene field [33]. The trans–cis photoisomerization for 3e, 4d, 6b and 6d–h in general gives a high percentage of cis-isomer (89–93%) with two exceptions. Compound 6h shows a PSS of only 65% cis-isomer due to a poor separation of the π–π* and n–π* bands as a result of the red-shifting by the SMe group (Figure S1A,B, Supporting Information File 1), whereas 6i decomposes upon irradiation with 360 nm light (as previously reported by us [7]) leading to its exclusion from further characterization. The PSS360 forms of exemplary compounds 4d, 6e and 6f have thermal half-lives of 55 [7], 28 and 29 [7] days, respectively, at 10 μM in HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer with 1% DMSO at 25 °C and we consider this to be in line with the expectations (vide supra).
Table 3: Structure and results of photochemical and binding characterization of compounds 3e, 4d, 6b and 6d–i.
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-244-i7.svg?max-width=637&scale=1.0)
|
||||||||||
| Compound | Photochemistry | CXCR3 binding affinity | ||||||||
| Y |
λmax trans
π–π*a |
λmax cis
n–π*a |
PSS360
(% area cis)b |
SEM |
pKi
transc |
SEM |
pKi
PSS360c |
SEM | PASd | |
| 3ee | H | 323 | 421 | 88.9 | 0.4 | 6.3 | 0.0 | 5.7 | 0.1 | 4.0 |
| 6be | F | 325 | 417 | 92.7 | 0.1 | 6.0 | 0.0 | 5.4 | 0.0 | 4.0 |
| 4de | Cl | 328 | 422 | 92.6 | 0.2 | 5.9 | 0.0 | 5.6 | 0.0 | 2.0 |
| 6de | Br | 333 | 420 | 92.6 | 0.2 | 5.8 | 0.0 | 5.4 | 0.0 | 2.5 |
| 6e | I | 339 | 424 | 90.5 | 1.0 | 5.5 | 0.1 | 5.5 | 0.0 | 1.0 |
| 6fe | OMe | 352 | 424 | 92.1 | 0.1 | 5.4 | 0.0 | 5.0 | 0.0 | 2.5 |
| 6g | OiPr | 355 | 424 | 89.0 | 0.4 | 5.1 | 0.2 | 5.0 | 0.0 | 1.3 |
| 6h | SMe | 373 | ≈425f | 64.5 | 1.7 | 5.3 | 0.0 | 5.5 | 0.1 | 0.6 |
| 6ie | NMe2 | 387 | dec.g | dec.g | ||||||
aThe absorbance maxima were extracted from UV–vis spectra at 25 µM in PBS buffer with 1% DMSO. b% of cis-isomer at the photostationary state (PSS360) in 68% TRIS buffer and 32% DMSO (1 mM) measured after being pre-irradiated at 360 nm as obtained by LC–MS integration of the cis and trans-isomer signals at 265 nm. The mean and SEM of at least two experiments are shown. cBinding affinity of trans-isomer or PSS360 as measured using [3H]-VUF11211 displacement. The mean and SEM of at least three experiments are shown. dThe photoinduced affinity shift (PAS) is calculated as the ratio of the Ki PSS360 and Ki trans. eCompound was previously described by us [7]. fCould not be determined accurately due to partial overlapping of the π–π* and n–π* bands. gCompound decomposes under illumination.
![[1860-5397-15-244-4]](/bjoc/content/figures/1860-5397-15-244-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Properties of subseries 3e, 4d, 6b and 6d-h. (A) UV–vis absorption spectra of (top) trans-isomers of 6b, 4d, 6d and 6e (having substituent Y = F, Cl, Br and I, respectively) and (bottom) 6f, 6g and 6h (having Y = OMe, OiPr and SMe, respectively). (B) Functional dose-response curves using [35S]-GTPγS assay exemplified for 4d, 6f and 6e, respectively. (C) Summary of the efficacies of compounds 3e, 4d, 6b and 6d–h at PSS360. (D) Correlation between the bathochromic shifting of the π–π* band and the intrinsic activity of PSS360 for the compounds in Table 4.
Figure 4: Properties of subseries 3e, 4d, 6b and 6d-h. (A) UV–vis absorption spectra of (top) trans-isomers o...
The binding affinity of trans-3e, 4d, 6b and 6d–h (Table 3) is in the low micromolar range with a low PAS value upon illumination. Initial pilot studies on [35S]-GTPγS binding after CXCR3 stimulation with single concentrations of subseries 6 (data not shown) showed substantial levels of CXCR3 agonism in this group of compounds. For subseries 6 (and associated 3e and 4d) we therefore generated dose-response curves for the trans and PSS360 forms (Figure S4, Supporting Information File 1) using the same [35S]-GTPγS functional assay and calculated the intrinsic activity (α) and potency (EC50). As reported in our previous communication for some of these compounds [7], the PSS360 forms give agonism with high nanomolar potencies while most trans compounds are antagonists or partial agonists with very low efficacies (Table 4). However, when the size and/or EDG properties of Y increase, remarkably partial agonism with substantial efficacies appears even for the trans-isomers, such as for 6e, 6g and 6h. The compounds illuminated to PSS360 follow a similar qualitative trend: when the Y substituent is H or F (3e and 6b), the cis-isomer behaves as a partial agonist with medium efficacy (α = 0.30–0.41), but when the Y substituent increases in size and/or EDG properties, the efficacy increases to full efficacy with compounds 6e–h (Y = I, OMe, OiPr and SMe). The trans-form of 6g shows anomalous behavior at 10−5 M and higher (Figure S4, Supporting Information File 1), preventing the extraction of accurate functional values. The PDE value, which reflects efficacies of both the trans and PSS form, appears to have an optimum for 6f (Figure 4B). Figure 4B also shows more subtle behavior for compounds 4d and 6e, which harbor a PDE value of 0.43 and 0.51, respectively, but in different parts of the efficacy window.
Table 4: Structure and results of functional characterization of compounds 3e, 4d, 6b and 6d–h.
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-244-i8.svg?max-width=637&scale=1.0)
|
||||||||||
| Compound | Y |
pEC50
transa |
SEM |
pEC50
PSS360b |
SEM | α transc | SEM | α PSS360d | SEM | PDEe |
| 1d | – | 6.9 | 0.0 | 6.9 | 0.0 | 1.04 | 0.08 | 1.03 | 0.07 | −0.01 |
| 3ef | H | n.m.g | n.m.g | 6.5 | 0.1 | 0.05 | 0.03 | 0.30 | 0.01 | 0.25 |
| 6bf | F | n.m.g | n.m.g | 6.2 | 0.1 | 0.12 | 0.00 | 0.41 | 0.03 | 0.29 |
| 4df | Cl | n.m.g | n.m.g | 6.3 | 0.1 | 0.11 | 0.01 | 0.54 | 0.01 | 0.43 |
| 6df | Br | n.m.g | n.m.g | 6.2 | 0.1 | 0.14 | 0.02 | 0.58 | 0.05 | 0.44 |
| 6e | I | 5.5 | 0.3 | 6.3 | 0.2 | 0.49 | 0.04 | 1.00 | 0.04 | 0.51 |
| 6ff | OMe | n.m.g | n.m.g | 6.4 | 0.1 | 0.16 | 0.01 | 0.99 | 0.04 | 0.83 |
| 6g | OiPr | n.m.h | n.m.h | 6.0 | 0.0 | n.m.h | n.m.h | 1.19 | 0.07 | n.m.h |
| 6h | SMe | 5.7 | 0.3 | 6.1 | 0.1 | 0.85 | 0.08 | 1.29 | 0.03 | 0.44 |
aPotency of trans-isomer in the dark. n.m. = not measurable. The mean and SEM of at least three experiments are shown. bPotency of a sample pre-irradiated at 360 nm to reach the photostationary state. The mean and SEM of at least three experiments are shown. cIntrinsic activity of trans-isomer in the dark (CXCL11 efficacy set at α = 1). The mean and SEM of at least three experiments are shown. dIntrinsic activity of a sample pre-irradiated at 360 nm to reach the photostationary state (CXCL11 efficacy set at α = 1). The mean and SEM of at least three experiments are shown. eThe photoinduced difference of efficacy (PDE) is obtained by subtracting α trans from α PSS360. fCompound was previously described by us [7]. gToo low window. hThe curve for trans-6g shows anomalous behavior at 10−5 M and higher.
The results reveal that the electron density of the aryl rings, especially of the inner one, plays a key role in inducing agonism in CXCR3. In general, the cis-isomers of the series 2–6 are better CXCR3 agonists. Indeed, the cis-isomers are generally assumed to have an intrinsically higher electron density due to the disruption of the conjugation between the two rings of the azobenzene through the N=N bond. Moreover, the electronic properties of the inner substituent Y have been proven to be of strategic use in increasing the intrinsic activity (α) of the cis-isomers (Figure 4C). This capacity of the Y substituent to alter the electron density is conceivably also related to its capacity to induce a bathochromic shift of the π–π* band of the trans-isomer. Potential evidence for this can be extracted from the significant correlation between the bathochromic shift and intrinsic activity of PSS360 (Figure 4D) for the subseries 3e, 4d, 6b and 6d–h, which only differ in the nature of Y group (Figure 4C, Table 4).
Conclusion
We report a toolbox of 31 photochromic small-molecule CXCR3 receptor ligands based on the modeling-assisted azologization of a biaryl series reported previously by us [24]. All compounds show affinity for CXCR3 from the high nanomolar to the low micromolar range. Our efforts, however, were focused on exploring the landscape in functional efficacy. To this end, the scaffold was subjected to positional and substitution changes in structure, necessitating extensive synthetic efforts through multiple routes. The presence of halogen substituents on the ortho-position of the outer ring (substituent X) provides partial agonism for the cis-isomer with a Br atom being the major exponent, while trans-isomers preserve antagonist behavior. The presence of a substituent on the para-position of the central ring (substituent Y) capable of delocalizing π-electrons increases the efficacy of the cis-isomer. The cis-isomers of compounds with Y = I, OMe, OiPr or SMe are all full agonists of CXCR3, however, the corresponding trans-isomers also activate the receptor to varying degrees. In all, our efforts deliver a spectrum of (subtle) efficacy differences. Notable tool compounds are VUF15888 (4d) switching from antagonism to partial agonism (PDE = 0.43), VUF16620 (6e) switching from partial agonism to full agonism (PDE = 0.51), and VUF16216 (6f), which represents the optimum balance and provides a CXCR3 photoswitch with a PDE value of 0.83, i.e., from antagonism to full agonism. Based on the pharmacological properties of these three compounds and the long half-lives of their PSS states, they will be valuable tools for future photopharmacological studies on the dynamic signaling of the chemokine receptor CXCR3.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.7 MB | Download |
Acknowledgements
All authors acknowledge the Netherlands Organization for Scientific Research for financial support (TOPPUNT, “7 ways to 7TMR modulation (7-to-7)”, 718.014.002). We thank Danny Scholten, Chris de Graaf, and Luc Roumen for helpful discussions, Hans Custers for recording HRMS spectra and Mounir Andaloussi for providing key building block 7.
References
-
Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. 2018, 118, 10710–10747. doi:10.1021/acs.chemrev.8b00037
Return to citation in text: [1] [2] -
Hoorens, M. W. H.; Szymanski, W. Trends Biochem. Sci. 2018, 43, 567–575. doi:10.1016/j.tibs.2018.05.004
Return to citation in text: [1] -
Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Kooistra, A. J.; Wijnen, L. M.; Vreeker, G. C. M.; Verweij, E. W. E.; De Boer, A. H.; Smit, M. J.; De Graaf, C.; Vischer, H. F.; de Esch, I. J. P.; Wijtmans, M.; Leurs, R. J. Am. Chem. Soc. 2018, 140, 4232–4243. doi:10.1021/jacs.7b11422
Return to citation in text: [1] [2] [3] [4] -
Donthamsetti, P. C.; Winter, N.; Schönberger, M.; Levitz, J.; Stanley, C.; Javitch, J. A.; Isacoff, E. Y.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 18522–18535. doi:10.1021/jacs.7b07659
Return to citation in text: [1] [2] [3] [4] -
Lachmann, D.; Konieczny, A.; Keller, M.; König, B. Org. Biomol. Chem. 2019, 17, 2467–2478. doi:10.1039/c8ob03221a
Return to citation in text: [1] [2] [3] -
Westphal, M. V.; Schafroth, M. A.; Sarott, R. C.; Imhof, M. A.; Bold, C. P.; Leippe, P.; Dhopeshwarkar, A.; Grandner, J. M.; Katritch, V.; Mackie, K.; Trauner, D.; Carreira, E. M.; Frank, J. A. J. Am. Chem. Soc. 2017, 139, 18206–18212. doi:10.1021/jacs.7b06456
Return to citation in text: [1] -
Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] -
Morstein, J.; Awale, M.; Reymond, J.-L.; Trauner, D. ACS Cent. Sci. 2019, 5, 607–618. doi:10.1021/acscentsci.8b00881
Return to citation in text: [1] [2] -
Hartley, G. S. Nature 1937, 140, 281. doi:10.1038/140281a0
Return to citation in text: [1] -
Ricart-Ortega, M.; Font, J.; Llebaria, A. Mol. Cell. Endocrinol. 2019, 488, 36–51. doi:10.1016/j.mce.2019.03.003
Return to citation in text: [1] [2] -
Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Lion, K.; Huppelschoten, Y.; Vischer, H. F.; De Esch, I. J. P.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. doi:10.1002/anie.201813110
Return to citation in text: [1] [2] [3] [4] -
Hauser, A. S.; Attwood, M. M.; Rask-Andersen, M.; Schiöth, H. B.; Gloriam, D. E. Nat. Rev. Drug Discovery 2017, 16, 829–842. doi:10.1038/nrd.2017.178
Return to citation in text: [1] [2] -
Schönberger, M.; Trauner, D. Angew. Chem., Int. Ed. 2014, 53, 3264–3267. doi:10.1002/anie.201309633
Return to citation in text: [1] [2] -
Pittolo, S.; Gómez-Santacana, X.; Eckelt, K.; Rovira, X.; Dalton, J.; Goudet, C.; Pin, J.-P.; Llobet, A.; Giraldo, J.; Llebaria, A.; Gorostiza, P. Nat. Chem. Biol. 2014, 10, 813–815. doi:10.1038/nchembio.1612
Return to citation in text: [1] -
Bahamonde, M. I.; Taura, J.; Paoletta, S.; Gakh, A. A.; Chakraborty, S.; Hernando, J.; Fernández-Dueñas, V.; Jacobson, K. A.; Gorostiza, P.; Ciruela, F. Bioconjugate Chem. 2014, 25, 1847–1854. doi:10.1021/bc5003373
Return to citation in text: [1] -
Levitz, J.; Pantoja, C.; Gaub, B.; Janovjak, H.; Reiner, A.; Hoagland, A.; Schoppik, D.; Kane, B.; Stawski, P.; Schier, A. F.; Trauner, D.; Isacoff, E. Y. Nat. Neurosci. 2013, 16, 507–516. doi:10.1038/nn.3346
Return to citation in text: [1] -
Rovira, X.; Trapero, A.; Pittolo, S.; Zussy, C.; Faucherre, A.; Jopling, C.; Giraldo, J.; Pin, J.-P.; Gorostiza, P.; Goudet, C.; Llebaria, A. Cell Chem. Biol. 2016, 23, 929–934. doi:10.1016/j.chembiol.2016.06.013
Return to citation in text: [1] -
Zussy, C.; Gómez-Santacana, X.; Rovira, X.; De Bundel, D.; Ferrazzo, S.; Bosch, D.; Asede, D.; Malhaire, F.; Acher, F.; Giraldo, J.; Valjent, E.; Ehrlich, I.; Ferraguti, F.; Pin, J.-P.; Llebaria, A.; Goudet, C. Mol. Psychiatry 2018, 23, 509–520. doi:10.1038/mp.2016.223
Return to citation in text: [1] -
Agnetta, L.; Bermudez, M.; Riefolo, F.; Matera, C.; Claro, E.; Messerer, R.; Littmann, T.; Wolber, G.; Holzgrabe, U.; Decker, M. J. Med. Chem. 2019, 62, 3009–3020. doi:10.1021/acs.jmedchem.8b01822
Return to citation in text: [1] -
Rustler, K.; Maleeva, G.; Bregestovski, P.; König, B. Beilstein J. Org. Chem. 2019, 15, 780–788. doi:10.3762/bjoc.15.74
Return to citation in text: [1] -
Broichhagen, J.; Podewin, T.; Meyer-Berg, H.; von Ohlen, Y.; Johnston, N. R.; Jones, B. J.; Bloom, S. R.; Rutter, G. A.; Hoffmann-Röder, A.; Hodson, D. J.; Trauner, D. Angew. Chem., Int. Ed. 2015, 54, 15565–15569. doi:10.1002/anie.201506384
Return to citation in text: [1] -
Broichhagen, J.; Johnston, N. R.; von Ohlen, Y.; Meyer-Berg, H.; Jones, B. J.; Bloom, S. R.; Rutter, G. A.; Trauner, D.; Hodson, D. J. Angew. Chem., Int. Ed. 2016, 55, 5865–5868. doi:10.1002/anie.201600957
Return to citation in text: [1] -
Gómez-Santacana, X.; Pittolo, S.; Rovira, X.; Lopez, M.; Zussy, C.; Dalton, J. A. R.; Faucherre, A.; Jopling, C.; Pin, J.-P.; Ciruela, F.; Goudet, C.; Giraldo, J.; Gorostiza, P.; Llebaria, A. ACS Cent. Sci. 2017, 3, 81–91. doi:10.1021/acscentsci.6b00353
Return to citation in text: [1] -
Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Wijtmans, M.; Scholten, D.; Mooij, W.; Smit, M. J.; de Esch, I. J. P.; de Graaf, C.; Leurs, R. Exploring the CXCR3 Chemokine Receptor with Small-Molecule Antagonists and Agonists. In Chemokines: Chemokines and Their Receptors in Drug Discovery; Tschammer, N., Ed.; Topics in Medicinal Chemistry; Springer International Publishing: Cham, Switzerland, 2015; pp 119–185. doi:10.1007/7355_2014_75
Return to citation in text: [1] [2] -
MOE, Version 2016.0802; Chemical Computing Group, Inc.: Chemical Computing Group, Inc, 2016.
Return to citation in text: [1] -
Fliegl, H.; Köhn, A.; Hättig, C.; Ahlrichs, R. J. Am. Chem. Soc. 2003, 125, 9821–9827. doi:10.1021/ja034433o
Return to citation in text: [1] -
Wijtmans, M.; Verzijl, D.; Bergmans, S.; Lai, M.; Bosch, L.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. Bioorg. Med. Chem. 2011, 19, 3384–3393. doi:10.1016/j.bmc.2011.04.035
Return to citation in text: [1] -
García-Amorós, J.; Velasco, D. Beilstein J. Org. Chem. 2012, 8, 1003–1017. doi:10.3762/bjoc.8.113
Return to citation in text: [1] -
Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g
Return to citation in text: [1] -
Scholten, D. J.; Wijtmans, M.; van Senten, J. R.; Custers, H.; Stunnenberg, A.; de Esch, I. J. P.; Smit, M. J.; Leurs, R. Mol. Pharmacol. 2015, 87, 639–648. doi:10.1124/mol.114.095265
Return to citation in text: [1] -
Engle, K. M.; Luo, S.-X.; Grubbs, R. H. J. Org. Chem. 2015, 80, 4213–4220. doi:10.1021/acs.joc.5b00563
Return to citation in text: [1] -
Nishioka, H.; Liang, X.; Kato, T.; Asanuma, H. Angew. Chem., Int. Ed. 2012, 51, 1165–1168. doi:10.1002/anie.201106093
Return to citation in text: [1]
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 28. | Wijtmans, M.; Verzijl, D.; Bergmans, S.; Lai, M.; Bosch, L.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. Bioorg. Med. Chem. 2011, 19, 3384–3393. doi:10.1016/j.bmc.2011.04.035 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 1. | Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. 2018, 118, 10710–10747. doi:10.1021/acs.chemrev.8b00037 |
| 2. | Hoorens, M. W. H.; Szymanski, W. Trends Biochem. Sci. 2018, 43, 567–575. doi:10.1016/j.tibs.2018.05.004 |
| 10. | Ricart-Ortega, M.; Font, J.; Llebaria, A. Mol. Cell. Endocrinol. 2019, 488, 36–51. doi:10.1016/j.mce.2019.03.003 |
| 32. | Engle, K. M.; Luo, S.-X.; Grubbs, R. H. J. Org. Chem. 2015, 80, 4213–4220. doi:10.1021/acs.joc.5b00563 |
| 8. | Morstein, J.; Awale, M.; Reymond, J.-L.; Trauner, D. ACS Cent. Sci. 2019, 5, 607–618. doi:10.1021/acscentsci.8b00881 |
| 4. | Donthamsetti, P. C.; Winter, N.; Schönberger, M.; Levitz, J.; Stanley, C.; Javitch, J. A.; Isacoff, E. Y.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 18522–18535. doi:10.1021/jacs.7b07659 |
| 11. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Lion, K.; Huppelschoten, Y.; Vischer, H. F.; De Esch, I. J. P.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. doi:10.1002/anie.201813110 |
| 23. | Gómez-Santacana, X.; Pittolo, S.; Rovira, X.; Lopez, M.; Zussy, C.; Dalton, J. A. R.; Faucherre, A.; Jopling, C.; Pin, J.-P.; Ciruela, F.; Goudet, C.; Giraldo, J.; Gorostiza, P.; Llebaria, A. ACS Cent. Sci. 2017, 3, 81–91. doi:10.1021/acscentsci.6b00353 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 6. | Westphal, M. V.; Schafroth, M. A.; Sarott, R. C.; Imhof, M. A.; Bold, C. P.; Leippe, P.; Dhopeshwarkar, A.; Grandner, J. M.; Katritch, V.; Mackie, K.; Trauner, D.; Carreira, E. M.; Frank, J. A. J. Am. Chem. Soc. 2017, 139, 18206–18212. doi:10.1021/jacs.7b06456 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 5. | Lachmann, D.; Konieczny, A.; Keller, M.; König, B. Org. Biomol. Chem. 2019, 17, 2467–2478. doi:10.1039/c8ob03221a |
| 13. | Schönberger, M.; Trauner, D. Angew. Chem., Int. Ed. 2014, 53, 3264–3267. doi:10.1002/anie.201309633 |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 3. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Kooistra, A. J.; Wijnen, L. M.; Vreeker, G. C. M.; Verweij, E. W. E.; De Boer, A. H.; Smit, M. J.; De Graaf, C.; Vischer, H. F.; de Esch, I. J. P.; Wijtmans, M.; Leurs, R. J. Am. Chem. Soc. 2018, 140, 4232–4243. doi:10.1021/jacs.7b11422 |
| 4. | Donthamsetti, P. C.; Winter, N.; Schönberger, M.; Levitz, J.; Stanley, C.; Javitch, J. A.; Isacoff, E. Y.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 18522–18535. doi:10.1021/jacs.7b07659 |
| 5. | Lachmann, D.; Konieczny, A.; Keller, M.; König, B. Org. Biomol. Chem. 2019, 17, 2467–2478. doi:10.1039/c8ob03221a |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 21. | Broichhagen, J.; Podewin, T.; Meyer-Berg, H.; von Ohlen, Y.; Johnston, N. R.; Jones, B. J.; Bloom, S. R.; Rutter, G. A.; Hoffmann-Röder, A.; Hodson, D. J.; Trauner, D. Angew. Chem., Int. Ed. 2015, 54, 15565–15569. doi:10.1002/anie.201506384 |
| 22. | Broichhagen, J.; Johnston, N. R.; von Ohlen, Y.; Meyer-Berg, H.; Jones, B. J.; Bloom, S. R.; Rutter, G. A.; Trauner, D.; Hodson, D. J. Angew. Chem., Int. Ed. 2016, 55, 5865–5868. doi:10.1002/anie.201600957 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 12. | Hauser, A. S.; Attwood, M. M.; Rask-Andersen, M.; Schiöth, H. B.; Gloriam, D. E. Nat. Rev. Drug Discovery 2017, 16, 829–842. doi:10.1038/nrd.2017.178 |
| 13. | Schönberger, M.; Trauner, D. Angew. Chem., Int. Ed. 2014, 53, 3264–3267. doi:10.1002/anie.201309633 |
| 14. | Pittolo, S.; Gómez-Santacana, X.; Eckelt, K.; Rovira, X.; Dalton, J.; Goudet, C.; Pin, J.-P.; Llobet, A.; Giraldo, J.; Llebaria, A.; Gorostiza, P. Nat. Chem. Biol. 2014, 10, 813–815. doi:10.1038/nchembio.1612 |
| 15. | Bahamonde, M. I.; Taura, J.; Paoletta, S.; Gakh, A. A.; Chakraborty, S.; Hernando, J.; Fernández-Dueñas, V.; Jacobson, K. A.; Gorostiza, P.; Ciruela, F. Bioconjugate Chem. 2014, 25, 1847–1854. doi:10.1021/bc5003373 |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 3. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Kooistra, A. J.; Wijnen, L. M.; Vreeker, G. C. M.; Verweij, E. W. E.; De Boer, A. H.; Smit, M. J.; De Graaf, C.; Vischer, H. F.; de Esch, I. J. P.; Wijtmans, M.; Leurs, R. J. Am. Chem. Soc. 2018, 140, 4232–4243. doi:10.1021/jacs.7b11422 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 11. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Lion, K.; Huppelschoten, Y.; Vischer, H. F.; De Esch, I. J. P.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. doi:10.1002/anie.201813110 |
| 3. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Kooistra, A. J.; Wijnen, L. M.; Vreeker, G. C. M.; Verweij, E. W. E.; De Boer, A. H.; Smit, M. J.; De Graaf, C.; Vischer, H. F.; de Esch, I. J. P.; Wijtmans, M.; Leurs, R. J. Am. Chem. Soc. 2018, 140, 4232–4243. doi:10.1021/jacs.7b11422 |
| 4. | Donthamsetti, P. C.; Winter, N.; Schönberger, M.; Levitz, J.; Stanley, C.; Javitch, J. A.; Isacoff, E. Y.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 18522–18535. doi:10.1021/jacs.7b07659 |
| 11. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Lion, K.; Huppelschoten, Y.; Vischer, H. F.; De Esch, I. J. P.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. doi:10.1002/anie.201813110 |
| 16. | Levitz, J.; Pantoja, C.; Gaub, B.; Janovjak, H.; Reiner, A.; Hoagland, A.; Schoppik, D.; Kane, B.; Stawski, P.; Schier, A. F.; Trauner, D.; Isacoff, E. Y. Nat. Neurosci. 2013, 16, 507–516. doi:10.1038/nn.3346 |
| 17. | Rovira, X.; Trapero, A.; Pittolo, S.; Zussy, C.; Faucherre, A.; Jopling, C.; Giraldo, J.; Pin, J.-P.; Gorostiza, P.; Goudet, C.; Llebaria, A. Cell Chem. Biol. 2016, 23, 929–934. doi:10.1016/j.chembiol.2016.06.013 |
| 18. | Zussy, C.; Gómez-Santacana, X.; Rovira, X.; De Bundel, D.; Ferrazzo, S.; Bosch, D.; Asede, D.; Malhaire, F.; Acher, F.; Giraldo, J.; Valjent, E.; Ehrlich, I.; Ferraguti, F.; Pin, J.-P.; Llebaria, A.; Goudet, C. Mol. Psychiatry 2018, 23, 509–520. doi:10.1038/mp.2016.223 |
| 19. | Agnetta, L.; Bermudez, M.; Riefolo, F.; Matera, C.; Claro, E.; Messerer, R.; Littmann, T.; Wolber, G.; Holzgrabe, U.; Decker, M. J. Med. Chem. 2019, 62, 3009–3020. doi:10.1021/acs.jmedchem.8b01822 |
| 20. | Rustler, K.; Maleeva, G.; Bregestovski, P.; König, B. Beilstein J. Org. Chem. 2019, 15, 780–788. doi:10.3762/bjoc.15.74 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 1. | Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. 2018, 118, 10710–10747. doi:10.1021/acs.chemrev.8b00037 |
| 10. | Ricart-Ortega, M.; Font, J.; Llebaria, A. Mol. Cell. Endocrinol. 2019, 488, 36–51. doi:10.1016/j.mce.2019.03.003 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 29. | García-Amorós, J.; Velasco, D. Beilstein J. Org. Chem. 2012, 8, 1003–1017. doi:10.3762/bjoc.8.113 |
| 30. | Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g |
| 8. | Morstein, J.; Awale, M.; Reymond, J.-L.; Trauner, D. ACS Cent. Sci. 2019, 5, 607–618. doi:10.1021/acscentsci.8b00881 |
| 12. | Hauser, A. S.; Attwood, M. M.; Rask-Andersen, M.; Schiöth, H. B.; Gloriam, D. E. Nat. Rev. Drug Discovery 2017, 16, 829–842. doi:10.1038/nrd.2017.178 |
| 31. | Scholten, D. J.; Wijtmans, M.; van Senten, J. R.; Custers, H.; Stunnenberg, A.; de Esch, I. J. P.; Smit, M. J.; Leurs, R. Mol. Pharmacol. 2015, 87, 639–648. doi:10.1124/mol.114.095265 |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 3. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Kooistra, A. J.; Wijnen, L. M.; Vreeker, G. C. M.; Verweij, E. W. E.; De Boer, A. H.; Smit, M. J.; De Graaf, C.; Vischer, H. F.; de Esch, I. J. P.; Wijtmans, M.; Leurs, R. J. Am. Chem. Soc. 2018, 140, 4232–4243. doi:10.1021/jacs.7b11422 |
| 4. | Donthamsetti, P. C.; Winter, N.; Schönberger, M.; Levitz, J.; Stanley, C.; Javitch, J. A.; Isacoff, E. Y.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 18522–18535. doi:10.1021/jacs.7b07659 |
| 5. | Lachmann, D.; Konieczny, A.; Keller, M.; König, B. Org. Biomol. Chem. 2019, 17, 2467–2478. doi:10.1039/c8ob03221a |
| 11. | Hauwert, N. J.; Mocking, T. A. M.; Da Costa Pereira, D.; Lion, K.; Huppelschoten, Y.; Vischer, H. F.; De Esch, I. J. P.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. doi:10.1002/anie.201813110 |
| 33. | Nishioka, H.; Liang, X.; Kato, T.; Asanuma, H. Angew. Chem., Int. Ed. 2012, 51, 1165–1168. doi:10.1002/anie.201106093 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 26. | MOE, Version 2016.0802; Chemical Computing Group, Inc.: Chemical Computing Group, Inc, 2016. |
| 27. | Fliegl, H.; Köhn, A.; Hättig, C.; Ahlrichs, R. J. Am. Chem. Soc. 2003, 125, 9821–9827. doi:10.1021/ja034433o |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 25. | Wijtmans, M.; Scholten, D.; Mooij, W.; Smit, M. J.; de Esch, I. J. P.; de Graaf, C.; Leurs, R. Exploring the CXCR3 Chemokine Receptor with Small-Molecule Antagonists and Agonists. In Chemokines: Chemokines and Their Receptors in Drug Discovery; Tschammer, N., Ed.; Topics in Medicinal Chemistry; Springer International Publishing: Cham, Switzerland, 2015; pp 119–185. doi:10.1007/7355_2014_75 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 24. | Wijtmans, M.; Scholten, D. J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M. C. A.; de Kanter, F. J. J.; de Graaf, C.; Smit, M. J.; de Esch, I. J. P.; Leurs, R. J. Med. Chem. 2012, 55, 10572–10583. doi:10.1021/jm301240t |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
| 25. | Wijtmans, M.; Scholten, D.; Mooij, W.; Smit, M. J.; de Esch, I. J. P.; de Graaf, C.; Leurs, R. Exploring the CXCR3 Chemokine Receptor with Small-Molecule Antagonists and Agonists. In Chemokines: Chemokines and Their Receptors in Drug Discovery; Tschammer, N., Ed.; Topics in Medicinal Chemistry; Springer International Publishing: Cham, Switzerland, 2015; pp 119–185. doi:10.1007/7355_2014_75 |
| 7. | Gómez-Santacana, X.; de Munnik, S. M.; Vijayachandran, P.; Da Costa Pereira, D.; Bebelman, J. P. M.; de Esch, I. J. P.; Vischer, H. F.; Wijtmans, M.; Leurs, R. Angew. Chem., Int. Ed. 2018, 57, 11608–11612. doi:10.1002/anie.201804875 |
© 2019 Gómez-Santacana et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)