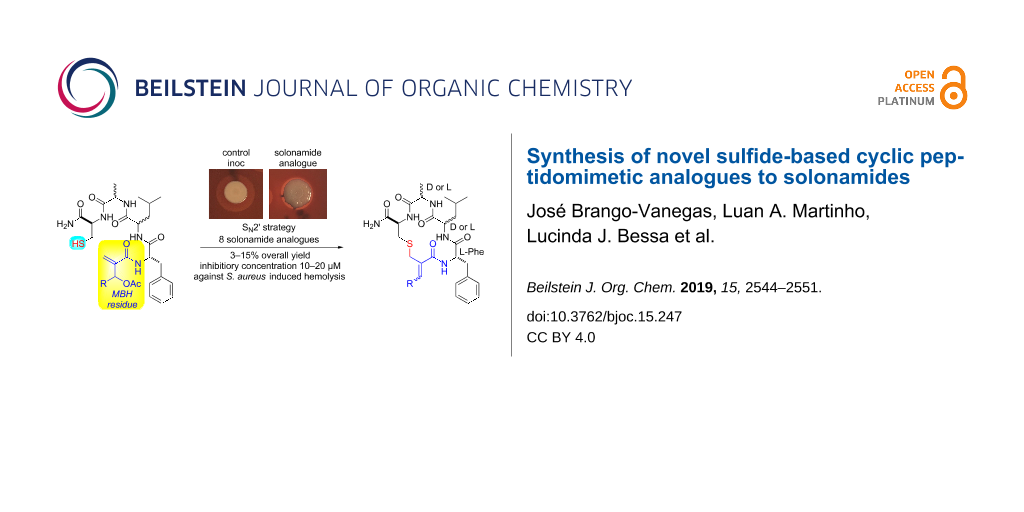
Abstract
Eight new sulfide-based cyclic peptidomimetic analogues of solonamides A and B have been synthesized via solid-phase peptide synthesis and SN2’ reaction on a Morita–Baylis–Hillman (MBH) residue introduced at the N-terminal of a tetrapeptide. This last step takes advantage of the electrophilic feature of the MBH residue and represents a new cyclization strategy occurring. The analogues were prepared in moderate overall yields and did not show toxic effects on Staphylococcus aureus growth and were not toxic to human fibroblasts. Two of them inhibited the hemolytic activity of S. aureus, suggesting an interfering action in the bacterial quorum sensing similar to the one already reported for solonamides.

Graphical Abstract
Introduction
The cyclodepsipeptides called solonamides A and B are natural molecules extracted from the marine bacterium Photobacterium halotolerans [1,2] (Figure 1). They are able to prevent the expression of Staphylococcus aureus virulence factors such as α-hemolysin and phenol-soluble modulins without affecting the bacterial growth [3]. Particularly, solonamide B and its analogues revealed no detectable toxicity against erythrocytes or human neutrophils [3,4].
![[1860-5397-15-247-1]](/bjoc/content/figures/1860-5397-15-247-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of solonamides and autoinducing peptides (AIP).
Figure 1: Chemical structure of solonamides and autoinducing peptides (AIP).
Staphylococcus aureus is an opportunistic Gram-positive bacterium found in human commensal microbiota. Once epithelial barriers or immune systems become compromised, S. aureus can cause skin and soft tissue infections besides severe invasive diseases such as endocarditis, pneumonia, and septicemia [5-7]. In particular, methicillin-resistant S. aureus (MRSA) is considered an endemic cause of nosocomial infections and has spread into the community and livestock animals as well [8]. Expression of many S. aureus virulence factors is controlled by a sophisticated intercellular chemical signalling pathway named quorum-sensing (QS) circuit Agr (accessory gene regulator) [8-11]. Four native thiolactonic cyclopeptides, named autoinducing peptides (AIPs, Figure 1), were found to be the chemical signals for the QS circuit Agr. Their chemical structures are remarkably alike to solonamides, and the synthesis of new molecules structurally related to these natural peptidomimetics has been used as a promising strategy for the attenuation of bacterial virulence in strains of S. aureus [12-15].
Herein, we report the synthesis of new sulfide-based cyclic peptidomimetics through the allylic nucleophilic substitution (SN2’) of cysteine sulfhydryl side chain to electrophilic Cβ of an O-acetylated Morita–Baylis–Hillman (MBH) adduct residue (Scheme 1). Despite reports describing the use of amino acid residues with nucleophilic side chains to prompt the macrocyclization of peptides and their mimetics, to the best of our knowledge, this is the first report on the participation of MBH residues as electrophilic sites to allow an SN2’-based macrocyclization of peptidomimetics [16-30]. Two of these new compounds were able to interfere with the hemolytic activity of S. aureus, and since the hemolysin expression is activated by S. aureus QS, we can suppose that the reported activity may be related to the inhibition of this bacterial communication system.
![[1860-5397-15-247-i1]](/bjoc/content/inline/1860-5397-15-247-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Macrocyclization strategy based on SN2’.
Scheme 1: Macrocyclization strategy based on SN2’.
Results and Discussion
Rational design and synthesis of the solonamide analogues
The rational design of our solonamide analogues was based on the conservation of the 16-membered macrocyclic scaffold and the apolar tripeptidyl moiety found in the solonamides. Both features are important to guarantee the interference with S. aureus QS [12-15]. The ester linkage of the lactone core was substituted by the sulfide group. Cyclic thioether peptides have been found in the chemical skeletons of natural products and synthetic ones that display a wide variety of activities, including antibiotics [31], vascular cell adhesion molecule-1 antagonists [32] and anticardiolipin antibodies [33,34].
Two MBH adducts (2) (R = Me, heptyl) and their respective carboxylic acids 3 were obtained in good yields based on previously reported procedures (Scheme 2) [35,36].
![[1860-5397-15-247-i2]](/bjoc/content/inline/1860-5397-15-247-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chemical synthesis of the MBH adducts 2 and their carboxylic acids 3.
Scheme 2: Chemical synthesis of the MBH adducts 2 and their carboxylic acids 3.
Starting from Rink Amide AM resin-bound orthogonally protected Fmoc-Cys-(Trt) 4, solonamide analogues were synthesized following stepwise Fmoc deprotection and standard repetitive Fmoc-amino acid couplings yielding the linear resin-bound tetrapeptides 5 (Scheme 3) [37,38]. The MBH acids 3 were coupled to the free amine at the N-terminal of 5 to afford the resin-bound linear peptidomimetics 6, which subsequently had the hydroxy group of the MBH residue acylated with acetic anhydride to yield 7. Despite the good results for the acetylation of peptides 6 with R = Me at room temperature, the reaction of the ones with R = n-hexyl needed to be conducted under heat conditions (50 °C). Acidic treatment of the acetates 7 with trifluoroacetic acid: triisopropylsilane decoupling cocktail afforded the resin-free linear peptidomimetics 8.
![[1860-5397-15-247-i3]](/bjoc/content/inline/1860-5397-15-247-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Chemical synthesis of the linear peptidomimetics 8.
Scheme 3: Chemical synthesis of the linear peptidomimetics 8.
The SN2’ macrocyclization step was performed immediately after the cleavage procedure to avoid the oxidative disulfide dimerization observed when preparative HPLC purification of the resin-free linear peptidomimetics 8 was tried. Thus, a 1 mM solution of peptides 8 in dry THF/CH2Cl2 (1:1) containing Et3N was vigorously stirred for 48 h at room temperature (Scheme 4) as previously described in a similar procedure for the intermolecular SN2’ reaction between thiophenol and O-acetylated MBH adducts [39]. The solonamide analogues 9 were obtained after preparative HPLC purification and lyophilization. The overall yield for this 11-step synthesis ranged from 7% to 15% for almost all solonamide analogues 9, based on the initial resin’s molarity. The exception relays on those containing ᴅ-Ala and ʟ-Leu amino acid residues sequentially attached to an ʟ-Cys residue, which were obtained in yields lower than 5%. We ascribed this result to the steric strains imposed by the spatial accommodation of these amino acid residue side chains on the resin-free linear peptidomimetics 8, which seems to disfavour the approximation of the nucleophilic sulfhydryl group to the electrophilic MBH residue.
![[1860-5397-15-247-i4]](/bjoc/content/inline/1860-5397-15-247-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Macrocyclization strategy based on SN2’ reaction to affords the solonamide analogues 9 and their overall yields based on the initial resin’s molarity.
Scheme 4: Macrocyclization strategy based on SN2’ reaction to affords the solonamide analogues 9 and their ov...
Spectral characterization of the chemical structures of the solonamide analogues 9
The compounds were characterized by one- and two-dimensional NMR spectroscopy, infrared spectroscopy (IR) and mass spectrometry. The high-resolution MS/MS analysis allowed the observation of a fragmentation pattern that does not coincide with the one found in linear peptides, where we would observe the loss of the ʟ-Cys residue. In addition to the sequential loss of ammonia (17 amu) and CO (28 amu) to yield the iminium [M − NH3 − CO]+ (Schemes S4–S11, Supporting Information File 1), as expected for the C-terminal carboxamide, we observed three ions derived from the breaking of two amide bonds, starting from the opening of the macrocycle by the loss of one, two or three amino acid residues as neutral fragments. This fragmentation pattern agrees with the one expected for cyclic peptides [40]. Noteworthy, the detected ions A–C always showed the –SCH2CH=NH moiety, confirming the formation of the sulfide group (Schemes S4–S11, Supporting Information File 1).
The NMR experiments also confirmed the macrocyclic structure (Supporting Information File 1). The 1H NMR spectra of compounds 9 had the characteristic signal in δ 3.2–3.6 ppm, resonating as a dd, assigned to C1 methylene protons (Scheme 4); this signal only changes in the multiplicity for compounds 9g and 9h that is presented as a broad singlet. Additionally, we observed another signal around δ 5.8–6.5 ppm, that depending on the compound can be a q or t, integrating for one hydrogen, assigned to C3 methine hydrogen. The 1H,13C-HMBC spectra allows us to observe an important strong correlation between the signals assigned to the protons on C1 and the carbon Cβ of the cysteine, which strongly suggest the formation of the new C–S bond. The configuration of the double bond could be assigned as Z for all compounds due to the 1H,1H-NOESY correlations between the C3 hydrogen and the NH hydrogen of the amino acid residue attached to the adduct residue.
The IR spectra of analogues 9 were quite similar (Supporting Information File 1). Three main absorption bands could be readily observed around 3280, 1650 and 1520 cm−1. The first one was assigned to the stretch for N–H bonds of the peptide linkage. The stretch for the lactam and lactone C=O bonds gives rise to the broad absorption close to 1650 cm−1. The lowering on the wavenumber values for the lactone C=O stretch was also observed for bands assigned to the C=C bonds as consequence of their conjugation.
Evaluation of the growth inhibition and hemolytic activity of S. aureus for the solonamide analogues
Initially, the antibacterial activity of all analogues 9 was tested by determining the minimum inhibitory concentration against two antibiotic-susceptible reference strains of S. aureus, S. aureus ATCC 25923 and S. aureus ATCC 29213 (see Supporting Information File 1, assay 1) [41]. Two-fold serial dilutions were performed, allowing to test several concentrations within the range of 300–0.3 µM. None of the compounds presented antibacterial activity against S. aureus, since no MIC value could be obtained in the range of concentrations tested (MIC > 300 µM).
Subsequently, a screening assay was carried out to evaluate the effect of these compounds on the hemolysis of defibrinated sheep blood promoted by S. aureus ATCC 25923, a strain that produces hemolysins under the control of QS (see Supporting Information File 1, assay 2) [42]. Among all compounds, 9e and 9g showed the best results, inhibiting the hemolytic activity of S. aureus at lower concentrations (Table 1 and Figure 2). Analogues 9e and 9g were then tested at lower concentrations (300–5 µM), and 9e was able to hamper the hemolysis by the strain at 10 µM (see Supporting Information File 1, Table S1).
Table 1: Halos of hemolysis or inhibition of hemolysis of S. aureus ATCC 25923 on sheep blood agar plates (Assay 2).a
| Analogue | 20 mM | 1 mM | 200 μM |
|---|---|---|---|
| 9a | + | + | + |
| 9b | + | + | + |
| 9c | + | + | + |
| 9d | + | + | + |
| 9e | – | – | – |
| 9f | + | + | + |
| 9g | – | +* | +* |
| 9h | – | + | + |
a(+): Hemolysis halo; (–): no hemolysis halo; *visibly smaller hemolysis halo when compared to controls.
![[1860-5397-15-247-2]](/bjoc/content/figures/1860-5397-15-247-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Effect of compounds 9e and 9g at three concentrations on the hemolysis production by S. aureus ATCC 25923 (Assay 2, Inoc = inoculum). Regarding compound 9e, no hemolysis halo was observed around the bacterial spot.
Figure 2: Effect of compounds 9e and 9g at three concentrations on the hemolysis production by S. aureus ATCC...
The antihemolytic activity of 9e and 9g was also tested in a quantitative microdilution assay using human red blood cells (see Supporting Information File 1, assay 3) [43]. Analogue 9e showed better capacity to inhibit the hemolysis than 9g at the same concentration (Figure 3). At concentrations of 10 µM and 200 µM of 9e, the hemolysis production by S. aureus was inhibited by 63% and 84%, respectively.
![[1860-5397-15-247-3]](/bjoc/content/figures/1860-5397-15-247-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Inhibition of hemolysis (%) on human red blood cells caused by S. aureus ATCC 25923 after being in presence of several concentrations of compounds 9e and 9g (assay 3). The hemolysis is scored in % of hemolysis relative to the control (hemolysis caused by S. aureus ATCC 25923 in absence of any compound; only DMSO), which was set to 100%. Two independent experiments were performed in duplicate. Error bars represent standard deviations.
Figure 3: Inhibition of hemolysis (%) on human red blood cells caused by S. aureus ATCC 25923 after being in ...
Also, a cell viability assay on human fibroblasts (Detroit 551 cell line) was performed for compounds 9e and 9g in order to observe their eventual cytotoxicity to normal cells (see Supporting Information File 1, assay 4). The concentration range tested was 6.25 to 200 µM for 9e and 9.38 to 300 µM for 9g, chosen based on the optimal dose observed in the hemolysis inhibition assay. We observed that these two compounds did not affect the fibroblast viability in the concentrations tested (Figure 4). Furthermore, no statistically significant difference was observed between exposition times of 24 and 48 h. These data suggest that 9e and 9g are not toxic to normal cells within the tested experimental conditions.
![[1860-5397-15-247-4]](/bjoc/content/figures/1860-5397-15-247-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Cell viability of human fibroblast exposed to compounds 9e (A) and 9g (B) for 24 and 48 hours. The values are expressed as mean ± SEM. ****p < 0.0001, **p < 0.005 and ***p < 0.001 versus DMEM control group.
Figure 4: Cell viability of human fibroblast exposed to compounds 9e (A) and 9g (B) for 24 and 48 hours. The ...
Solonamide B has been shown to inhibit agr-controlled phenotypes in S. aureus (including hemolysis) at concentrations ranging between 8.5 µM and 17 µM [3]. Similar inhibitory concentrations (10 to 20 µM) were displayed by our analogues, 9e and 9g, with the advantage that sulfide linkage is less prompted to hydrolysis than the ester found at solonamides [44,45]. Comparing the chemical structure of analogues 9e–g and 9h to the previously reported analogues of solonamides and S. aureus AIPs, some observations can be made: (1) 9e is the only one among all the analogues derived from the acetaldehyde adduct that, regardless of the configuration of the stereogenic centres, has the same amino acid residue sequence found in solonamides; (2) despite the close similarity on the amino acid residue sequence displayed by 9e and 9h, the stereochemistry of the Phe residue is detrimental to the antihemolytic activity; (3) in a similar sense, for 9f and 9g, diastereosiomers on the Ile residue, the L-configuration on this residue is necessary to the observed hemolysis inhibition by S. aureus.
Conclusion
A new, effective and simple cyclization strategy using MBH adducts for the preparation of eight new sulfide-based cyclic peptidomimetics structurally similar to solonamides was established. Two of these new compounds had the capacity to inhibit the hemolytic activity of S. aureus ATCC 25923 without affecting its growth, at very low concentrations, namely 10 μM, and were not cytotoxic to human fibroblasts at the same concentration. Due to the well-known relationship of the α-hemolysins expression with the accessory gene regulator (agr) of the S. aureus, we can suggest that these two peptidomimetics may exert an effect on S. aureus quorum sensing. To evaluate this hypothesis, we are now rationally designing new solonamide analogues, based on the SN2’ strategy reported here, to be tested for AgrC-inhibitory activity.
Supporting Information
| Supporting Information File 1: Detailed synthetic procedures, biological assay procedures and copies of NMR and MS spectra of all compounds. | ||
| Format: PDF | Size: 6.0 MB | Download |
Acknowledgements
This work has been supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) - Finance Code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico – Brasil (CNPq) – J. B-V graduate fellow and L. A. M. undergraduate fellow, Fundação de Apoio à Pesquisa do Distrito Federal – Brasil 0193.001.560/2017, Decanato de Pesquisa e Pós-graduação (DPG-UnB). We also acknowledge Prof. Dr. Guilherme D. Brand (IQ-UnB), Prof. Dr. Carlos K. Z. Andrade (IQ-UnB), Prof. Dr. Peter Bakuzis (IQ-UnB), Departamento de Polícia Federal (SEPLAB-INC-DPF) and CAIQ-UnB.
References
-
Mansson, M.; Nielsen, A.; Kjærulff, L.; Gotfredsen, C. H.; Wietz, M.; Ingmer, H.; Gram, L.; Larsen, T. O. Mar. Drugs 2011, 9, 2537–2552. doi:10.3390/md9122537
Return to citation in text: [1] -
Kitir, B.; Baldry, M.; Ingmer, H.; Olsen, C. A. Tetrahedron 2014, 70, 7721–7732. doi:10.1016/j.tet.2014.05.107
Return to citation in text: [1] -
Nielsen, A.; Månsson, M.; Bojer, M. S.; Gram, L.; Larsen, T. O.; Novick, R. P.; Frees, D.; Frøkiær, H.; Ingmer, H. PLoS One 2014, 9, e84992. doi:10.1371/journal.pone.0084992
Return to citation in text: [1] [2] [3] -
Baldry, M.; Kitir, B.; Frøkiær, H.; Christensen, S. B.; Taverne, N.; Meijerink, M.; Franzyk, H.; Olsen, C. A.; Wells, J. M.; Ingmer, H. PLoS One 2016, 11, e0145618. doi:10.1371/journal.pone.0145618
Return to citation in text: [1] -
Broderick, A. H.; Stacy, D. M.; Tal-Gan, Y.; Kratochvil, M. J.; Blackwell, H. E.; Lynn, D. M. Adv. Healthcare Mater. 2014, 3, 97–105. doi:10.1002/adhm.201300119
Return to citation in text: [1] -
Grundmann, H.; Aires-de-Sousa, M.; Boyce, J.; Tiemersma, E. Lancet 2006, 368, 874–885. doi:10.1016/s0140-6736(06)68853-3
Return to citation in text: [1] -
European Centre for Disease Prevention and Control. Surveillance of antimicrobial resistance in Europe – Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017; ECDC: Stockholm, Sweden, 2018. doi:10.2900/230516
Return to citation in text: [1] -
Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R. P.; Vandenesch, F. Mol. Microbiol. 1998, 28, 655–662. doi:10.1046/j.1365-2958.1998.00830.x
Return to citation in text: [1] [2] -
Novick, R. P.; Geisinger, E. Annu. Rev. Genet. 2008, 42, 541–564. doi:10.1146/annurev.genet.42.110807.091640
Return to citation in text: [1] -
Greenberg, E. P. J. Clin. Invest. 2003, 112, 1288–1290. doi:10.1172/jci20099
Return to citation in text: [1] -
Camilli, A.; Bassler, B. L. Science 2006, 311, 1113–1116. doi:10.1126/science.1121357
Return to citation in text: [1] -
Tal-Gan, Y.; Stacy, D. M.; Foegen, M. K.; Koenig, D. W.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 7869–7882. doi:10.1021/ja3112115
Return to citation in text: [1] [2] -
Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Cornilescu, C. C.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 18436–18444. doi:10.1021/ja407533e
Return to citation in text: [1] [2] -
Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Blackwell, H. E. Org. Biomol. Chem. 2016, 14, 113–121. doi:10.1039/c5ob01735a
Return to citation in text: [1] [2] -
Hansen, A. M.; Peng, P.; Baldry, M.; Perez-Gassol, I.; Christensen, S. B.; Vinther, J. M. O.; Ingmer, H.; Franzyk, H. Eur. J. Med. Chem. 2018, 152, 370–376. doi:10.1016/j.ejmech.2018.04.053
Return to citation in text: [1] [2] -
Feng, Y.; Pattarawarapan, M.; Wang, Z.; Burgess, K. Org. Lett. 1999, 1, 121–124. doi:10.1021/ol990597r
Return to citation in text: [1] -
Feng, Y.; Burgess, K. Biotechnol. Bioeng. 2000, 71, 3–8. doi:10.1002/(sici)1097-0290(200024)71:1<3::aid-bit2>3.0.co;2-k
Return to citation in text: [1] -
Park, C.; Burgess, K. J. Comb. Chem. 2001, 3, 257–266. doi:10.1021/cc010003n
Return to citation in text: [1] -
Burgess, K. Acc. Chem. Res. 2001, 34, 826–835. doi:10.1021/ar9901523
Return to citation in text: [1] -
Lindman, S.; Lindeberg, G.; Gogoll, A.; Nyberg, F.; Karlén, A.; Hallberg, A. Bioorg. Med. Chem. 2001, 9, 763–772. doi:10.1016/s0968-0896(00)00294-7
Return to citation in text: [1] -
Johannesson, P.; Lindeberg, G.; Johansson, A.; Nikiforovich, G. V.; Gogoll, A.; Synnergren, B.; Le Grèves, M.; Nyberg, F.; Karlén, A.; Hallberg, A. J. Med. Chem. 2002, 45, 1767–1777. doi:10.1021/jm011063a
Return to citation in text: [1] -
Giulianotti, M.; Nefzi, A. Tetrahedron Lett. 2003, 44, 5307–5309. doi:10.1016/s0040-4039(03)01219-x
Return to citation in text: [1] -
Reyes, S. J.; Burgess, K. Tetrahedron: Asymmetry 2005, 16, 1061–1069. doi:10.1016/j.tetasy.2005.01.008
Return to citation in text: [1] -
Grieco, P.; Cai, M.; Liu, L.; Mayorov, A.; Chandler, K.; Trivedi, D.; Lin, G.; Campiglia, P.; Novellino, E.; Hruby, V. J. J. Med. Chem. 2008, 51, 2701–2707. doi:10.1021/jm701181n
Return to citation in text: [1] -
Derbel, S.; Ghedira, K.; Nefzi, A. Tetrahedron Lett. 2010, 51, 3607–3609. doi:10.1016/j.tetlet.2010.05.029
Return to citation in text: [1] -
Nefzi, A.; Arutyunyan, S.; Fenwick, J. E. J. Org. Chem. 2010, 75, 7939–7941. doi:10.1021/jo1016822
Return to citation in text: [1] -
Nefzi, A.; Fenwick, J. E. Tetrahedron Lett. 2011, 52, 817–819. doi:10.1016/j.tetlet.2010.12.043
Return to citation in text: [1] -
Wu, Z.-M.; Liu, S.-Z.; Cheng, X.-Z.; Ding, W.-Z.; Zhu, T.; Chen, B. Chin. Chem. Lett. 2016, 27, 1731–1739. doi:10.1016/j.cclet.2016.04.024
Return to citation in text: [1] -
Nishihara, T.; Kitada, H.; Fujiwara, D.; Fujii, I. Biopolymers 2016, 106, 415–421. doi:10.1002/bip.22826
Return to citation in text: [1] -
Sutherland, B. P.; El-Zaatari, B. M.; Halaszynski, N. I.; French, J. M.; Bai, S.; Kloxin, C. J. Bioconjugate Chem. 2018, 29, 3987–3992. doi:10.1021/acs.bioconjchem.8b00751
Return to citation in text: [1] -
Jung, G. Angew. Chem., Int. Ed. Engl. 1991, 30, 1051–1068. doi:10.1002/anie.199110513
Return to citation in text: [1] -
Fotouhi, N.; Joshi, P.; Tilley, J. W.; Rowan, K.; Schwinge, V.; Wolitzky, B. Bioorg. Med. Chem. Lett. 2000, 10, 1167–1169. doi:10.1016/s0960-894x(00)00175-x
Return to citation in text: [1] -
Jones, D. S.; Gamino, C. A.; Randow, M. E.; Victoria, E. J.; Yu, L.; Coutts, S. M. Tetrahedron Lett. 1998, 39, 6107–6110. doi:10.1016/s0040-4039(98)01292-1
Return to citation in text: [1] -
Roberts, K. D.; Lambert, J. N.; Ede, N. J.; Bray, A. M. Tetrahedron Lett. 1998, 39, 8357–8360. doi:10.1016/s0040-4039(98)01843-7
Return to citation in text: [1] -
Silva, V. S.; Tolentino, T. A.; Rodrigues, T. C. A. F.; Santos, F. F. M.; Machado, D. F. S.; Silva, W. A.; de Oliveira, H. C. B.; Machado, A. H. L. Org. Biomol. Chem. 2019, 17, 4498–4511. doi:10.1039/c9ob00533a
Return to citation in text: [1] -
Mateus, C. R.; Feltrin, M. P.; Costa, A. M.; Coelho, F.; Almeida, W. P. Tetrahedron 2001, 57, 6901–6908. doi:10.1016/s0040-4020(01)00647-0
Return to citation in text: [1] -
Kim, Y.-W.; Grossmann, T. N.; Verdine, G. L. Nat. Protoc. 2011, 6, 761–771. doi:10.1038/nprot.2011.324
Return to citation in text: [1] -
AAPPTEC. Practical Guide to Solid Phase Peptide Synthesis. https://www.peptide.com/custdocs/aapptec%20synthesis%20guide%202-0%20%282%29.pdf (accessed May 10, 2019).
Return to citation in text: [1] -
Kamimura, A.; Morita, R.; Matsuura, K.; Mitsudera, H.; Shirai, M. Tetrahedron 2003, 59, 9931–9938. doi:10.1016/j.tet.2003.10.035
Return to citation in text: [1] -
Liu, W.-T.; Ng, J.; Meluzzi, D.; Bandeira, N.; Gutierrez, M.; Simmons, T. L.; Schultz, A. W.; Linington, R. G.; Moore, B. S.; Gerwick, W. H.; Pevzner, P. A.; Dorrestein, P. C. Anal. Chem. (Washington, DC, U. S.) 2009, 81, 4200–4209. doi:10.1021/ac900114t
Return to citation in text: [1] -
Staphylococcus aureus subsp. Aureus Rosenbach (ATCC® 29213™). https://www.lgcstandards-atcc.org/products/all/29213.aspx?geo_country=pt#characteristics (accessed May 10, 2019).
Return to citation in text: [1] -
Nakayama, J.; Uemura, Y.; Nishiguchi, K.; Yoshimura, N.; Igarashi, Y.; Sonomoto, K. Antimicrob. Agents Chemother. 2009, 53, 580–586. doi:10.1128/aac.00995-08
Return to citation in text: [1] -
Sakoulas, G.; Eliopoulos, G. M.; Moellering, R. C., Jr.; Wennersten, C.; Venkataraman, L.; Novick, R. P.; Gold, H. S. Antimicrob. Agents Chemother. 2002, 46, 1492–1502. doi:10.1128/aac.46.5.1492-1502.2002
Return to citation in text: [1] -
Webster, A. M.; Cobb, S. L. Chem. – Eur. J. 2018, 24, 7560–7573. doi:10.1002/chem.201705340
Return to citation in text: [1] -
Sivanathan, S.; Scherkenbeck, J. Molecules 2014, 19, 12368–12420. doi:10.3390/molecules190812368
Return to citation in text: [1]
| 43. | Sakoulas, G.; Eliopoulos, G. M.; Moellering, R. C., Jr.; Wennersten, C.; Venkataraman, L.; Novick, R. P.; Gold, H. S. Antimicrob. Agents Chemother. 2002, 46, 1492–1502. doi:10.1128/aac.46.5.1492-1502.2002 |
| 41. | Staphylococcus aureus subsp. Aureus Rosenbach (ATCC® 29213™). https://www.lgcstandards-atcc.org/products/all/29213.aspx?geo_country=pt#characteristics (accessed May 10, 2019). |
| 42. | Nakayama, J.; Uemura, Y.; Nishiguchi, K.; Yoshimura, N.; Igarashi, Y.; Sonomoto, K. Antimicrob. Agents Chemother. 2009, 53, 580–586. doi:10.1128/aac.00995-08 |
| 1. | Mansson, M.; Nielsen, A.; Kjærulff, L.; Gotfredsen, C. H.; Wietz, M.; Ingmer, H.; Gram, L.; Larsen, T. O. Mar. Drugs 2011, 9, 2537–2552. doi:10.3390/md9122537 |
| 2. | Kitir, B.; Baldry, M.; Ingmer, H.; Olsen, C. A. Tetrahedron 2014, 70, 7721–7732. doi:10.1016/j.tet.2014.05.107 |
| 8. | Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R. P.; Vandenesch, F. Mol. Microbiol. 1998, 28, 655–662. doi:10.1046/j.1365-2958.1998.00830.x |
| 39. | Kamimura, A.; Morita, R.; Matsuura, K.; Mitsudera, H.; Shirai, M. Tetrahedron 2003, 59, 9931–9938. doi:10.1016/j.tet.2003.10.035 |
| 5. | Broderick, A. H.; Stacy, D. M.; Tal-Gan, Y.; Kratochvil, M. J.; Blackwell, H. E.; Lynn, D. M. Adv. Healthcare Mater. 2014, 3, 97–105. doi:10.1002/adhm.201300119 |
| 6. | Grundmann, H.; Aires-de-Sousa, M.; Boyce, J.; Tiemersma, E. Lancet 2006, 368, 874–885. doi:10.1016/s0140-6736(06)68853-3 |
| 7. | European Centre for Disease Prevention and Control. Surveillance of antimicrobial resistance in Europe – Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017; ECDC: Stockholm, Sweden, 2018. doi:10.2900/230516 |
| 40. | Liu, W.-T.; Ng, J.; Meluzzi, D.; Bandeira, N.; Gutierrez, M.; Simmons, T. L.; Schultz, A. W.; Linington, R. G.; Moore, B. S.; Gerwick, W. H.; Pevzner, P. A.; Dorrestein, P. C. Anal. Chem. (Washington, DC, U. S.) 2009, 81, 4200–4209. doi:10.1021/ac900114t |
| 3. | Nielsen, A.; Månsson, M.; Bojer, M. S.; Gram, L.; Larsen, T. O.; Novick, R. P.; Frees, D.; Frøkiær, H.; Ingmer, H. PLoS One 2014, 9, e84992. doi:10.1371/journal.pone.0084992 |
| 4. | Baldry, M.; Kitir, B.; Frøkiær, H.; Christensen, S. B.; Taverne, N.; Meijerink, M.; Franzyk, H.; Olsen, C. A.; Wells, J. M.; Ingmer, H. PLoS One 2016, 11, e0145618. doi:10.1371/journal.pone.0145618 |
| 35. | Silva, V. S.; Tolentino, T. A.; Rodrigues, T. C. A. F.; Santos, F. F. M.; Machado, D. F. S.; Silva, W. A.; de Oliveira, H. C. B.; Machado, A. H. L. Org. Biomol. Chem. 2019, 17, 4498–4511. doi:10.1039/c9ob00533a |
| 36. | Mateus, C. R.; Feltrin, M. P.; Costa, A. M.; Coelho, F.; Almeida, W. P. Tetrahedron 2001, 57, 6901–6908. doi:10.1016/s0040-4020(01)00647-0 |
| 3. | Nielsen, A.; Månsson, M.; Bojer, M. S.; Gram, L.; Larsen, T. O.; Novick, R. P.; Frees, D.; Frøkiær, H.; Ingmer, H. PLoS One 2014, 9, e84992. doi:10.1371/journal.pone.0084992 |
| 37. | Kim, Y.-W.; Grossmann, T. N.; Verdine, G. L. Nat. Protoc. 2011, 6, 761–771. doi:10.1038/nprot.2011.324 |
| 38. | AAPPTEC. Practical Guide to Solid Phase Peptide Synthesis. https://www.peptide.com/custdocs/aapptec%20synthesis%20guide%202-0%20%282%29.pdf (accessed May 10, 2019). |
| 12. | Tal-Gan, Y.; Stacy, D. M.; Foegen, M. K.; Koenig, D. W.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 7869–7882. doi:10.1021/ja3112115 |
| 13. | Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Cornilescu, C. C.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 18436–18444. doi:10.1021/ja407533e |
| 14. | Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Blackwell, H. E. Org. Biomol. Chem. 2016, 14, 113–121. doi:10.1039/c5ob01735a |
| 15. | Hansen, A. M.; Peng, P.; Baldry, M.; Perez-Gassol, I.; Christensen, S. B.; Vinther, J. M. O.; Ingmer, H.; Franzyk, H. Eur. J. Med. Chem. 2018, 152, 370–376. doi:10.1016/j.ejmech.2018.04.053 |
| 32. | Fotouhi, N.; Joshi, P.; Tilley, J. W.; Rowan, K.; Schwinge, V.; Wolitzky, B. Bioorg. Med. Chem. Lett. 2000, 10, 1167–1169. doi:10.1016/s0960-894x(00)00175-x |
| 16. | Feng, Y.; Pattarawarapan, M.; Wang, Z.; Burgess, K. Org. Lett. 1999, 1, 121–124. doi:10.1021/ol990597r |
| 17. | Feng, Y.; Burgess, K. Biotechnol. Bioeng. 2000, 71, 3–8. doi:10.1002/(sici)1097-0290(200024)71:1<3::aid-bit2>3.0.co;2-k |
| 18. | Park, C.; Burgess, K. J. Comb. Chem. 2001, 3, 257–266. doi:10.1021/cc010003n |
| 19. | Burgess, K. Acc. Chem. Res. 2001, 34, 826–835. doi:10.1021/ar9901523 |
| 20. | Lindman, S.; Lindeberg, G.; Gogoll, A.; Nyberg, F.; Karlén, A.; Hallberg, A. Bioorg. Med. Chem. 2001, 9, 763–772. doi:10.1016/s0968-0896(00)00294-7 |
| 21. | Johannesson, P.; Lindeberg, G.; Johansson, A.; Nikiforovich, G. V.; Gogoll, A.; Synnergren, B.; Le Grèves, M.; Nyberg, F.; Karlén, A.; Hallberg, A. J. Med. Chem. 2002, 45, 1767–1777. doi:10.1021/jm011063a |
| 22. | Giulianotti, M.; Nefzi, A. Tetrahedron Lett. 2003, 44, 5307–5309. doi:10.1016/s0040-4039(03)01219-x |
| 23. | Reyes, S. J.; Burgess, K. Tetrahedron: Asymmetry 2005, 16, 1061–1069. doi:10.1016/j.tetasy.2005.01.008 |
| 24. | Grieco, P.; Cai, M.; Liu, L.; Mayorov, A.; Chandler, K.; Trivedi, D.; Lin, G.; Campiglia, P.; Novellino, E.; Hruby, V. J. J. Med. Chem. 2008, 51, 2701–2707. doi:10.1021/jm701181n |
| 25. | Derbel, S.; Ghedira, K.; Nefzi, A. Tetrahedron Lett. 2010, 51, 3607–3609. doi:10.1016/j.tetlet.2010.05.029 |
| 26. | Nefzi, A.; Arutyunyan, S.; Fenwick, J. E. J. Org. Chem. 2010, 75, 7939–7941. doi:10.1021/jo1016822 |
| 27. | Nefzi, A.; Fenwick, J. E. Tetrahedron Lett. 2011, 52, 817–819. doi:10.1016/j.tetlet.2010.12.043 |
| 28. | Wu, Z.-M.; Liu, S.-Z.; Cheng, X.-Z.; Ding, W.-Z.; Zhu, T.; Chen, B. Chin. Chem. Lett. 2016, 27, 1731–1739. doi:10.1016/j.cclet.2016.04.024 |
| 29. | Nishihara, T.; Kitada, H.; Fujiwara, D.; Fujii, I. Biopolymers 2016, 106, 415–421. doi:10.1002/bip.22826 |
| 30. | Sutherland, B. P.; El-Zaatari, B. M.; Halaszynski, N. I.; French, J. M.; Bai, S.; Kloxin, C. J. Bioconjugate Chem. 2018, 29, 3987–3992. doi:10.1021/acs.bioconjchem.8b00751 |
| 33. | Jones, D. S.; Gamino, C. A.; Randow, M. E.; Victoria, E. J.; Yu, L.; Coutts, S. M. Tetrahedron Lett. 1998, 39, 6107–6110. doi:10.1016/s0040-4039(98)01292-1 |
| 34. | Roberts, K. D.; Lambert, J. N.; Ede, N. J.; Bray, A. M. Tetrahedron Lett. 1998, 39, 8357–8360. doi:10.1016/s0040-4039(98)01843-7 |
| 12. | Tal-Gan, Y.; Stacy, D. M.; Foegen, M. K.; Koenig, D. W.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 7869–7882. doi:10.1021/ja3112115 |
| 13. | Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Cornilescu, C. C.; Blackwell, H. E. J. Am. Chem. Soc. 2013, 135, 18436–18444. doi:10.1021/ja407533e |
| 14. | Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Blackwell, H. E. Org. Biomol. Chem. 2016, 14, 113–121. doi:10.1039/c5ob01735a |
| 15. | Hansen, A. M.; Peng, P.; Baldry, M.; Perez-Gassol, I.; Christensen, S. B.; Vinther, J. M. O.; Ingmer, H.; Franzyk, H. Eur. J. Med. Chem. 2018, 152, 370–376. doi:10.1016/j.ejmech.2018.04.053 |
| 3. | Nielsen, A.; Månsson, M.; Bojer, M. S.; Gram, L.; Larsen, T. O.; Novick, R. P.; Frees, D.; Frøkiær, H.; Ingmer, H. PLoS One 2014, 9, e84992. doi:10.1371/journal.pone.0084992 |
| 8. | Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R. P.; Vandenesch, F. Mol. Microbiol. 1998, 28, 655–662. doi:10.1046/j.1365-2958.1998.00830.x |
| 9. | Novick, R. P.; Geisinger, E. Annu. Rev. Genet. 2008, 42, 541–564. doi:10.1146/annurev.genet.42.110807.091640 |
| 10. | Greenberg, E. P. J. Clin. Invest. 2003, 112, 1288–1290. doi:10.1172/jci20099 |
| 11. | Camilli, A.; Bassler, B. L. Science 2006, 311, 1113–1116. doi:10.1126/science.1121357 |
| 31. | Jung, G. Angew. Chem., Int. Ed. Engl. 1991, 30, 1051–1068. doi:10.1002/anie.199110513 |
| 44. | Webster, A. M.; Cobb, S. L. Chem. – Eur. J. 2018, 24, 7560–7573. doi:10.1002/chem.201705340 |
| 45. | Sivanathan, S.; Scherkenbeck, J. Molecules 2014, 19, 12368–12420. doi:10.3390/molecules190812368 |
© 2019 Brango-Vanegas et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)