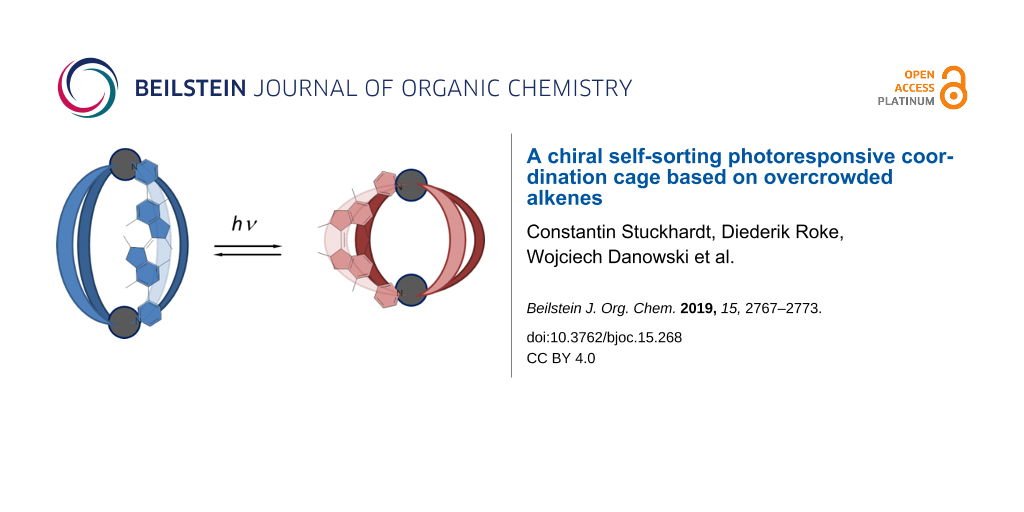
Abstract
In recent years, increasing efforts have been devoted to designing new functional stimuli-responsive supramolecular assemblies. Here, we present three isomeric supramolecular coordination complexes consisting of a Pd2L4 stoichiometry. As shown by NMR, CD and X-ray studies, as well as DFT calculations, these complexes form cage-like structures by chiral self-sorting. Photochromic ligands derived from first generation molecular motors enable light-driven interconversion between the three isomers. Two of the isomers were able to form host–guest complexes opening up new prospects toward stimuli-controlled substrate binding and release.

Graphical Abstract
Introduction
Supramolecular coordination complexes (SCCs) represent a promising class of compounds which have been used in a variety of molecular systems [1-6]. Taking advantage of the vacant cavity inside these complexes, SCCs have been applied in drug delivery [6-8], supramolecular catalysis [9-12], X-ray structure determination [13,14] and stabilization of reactive species [15-17]. The use of reversible metal–ligand coordination bonds gives rise to systems that allow for adaption to external stimuli such as pH, anions, electric potential, concentration and light [4,8,18-21]. Using light to dynamically control the shape and function of SCCs is a very promising strategy as it is a noninvasive stimulus that can be easily applied with high spatiotemporal control, without producing any waste. Systems have been reported where photoisomerization of azobenzene-derived anions encapsulated in supramolecular palladium complexes caused immediate crystallization [22]. Moreover, azobenzenes have been used to functionalize both the interior [23] and exterior [24] of SCCs to photochemically control guest binding and release. Furthermore, incorporation of dithienylethene into the ligands connecting the metal centers has been used to control host–guest interactions [25-28], structural composition [29], and sol–gel transition [30]. However, dithienylethene undergoes a limited structural change upon photoisomerization and, up to now, photoswitchable ligands based on other types of photochromic switches have not been reported in the literature.
In chiral self-sorting SCCs, either homo- or heterochiral complexes are formed exclusively through high fidelity recognition of the components within the complex [31-33]. The formation of such complexes with high selectivity and well-defined chirality is essential for the application of SCCs towards chiral recognition and sensing [34-37]. To the best of our knowledge no self-sorted responsive SCCs have been reported so far.
Molecular motors based on overcrowded alkenes are unique photoswitches that are able to undergo unidirectional rotation upon irradiation with light [38-40]. Moreover, they can be used as chiroptical multistate switches to control various functions in areas such as catalysis [41-43], soft materials [43-45] and supramolecular chemistry [46,47]. Employing molecular motors as ligands in SCCs provides an interesting strategy to form responsive coordination complexes, as they feature a large geometric change upon switching [47].
Herein, we report a photoresponsive coordination cage with ligands based on a first generation molecular motor (Figure 1). Cages with a Pd2L4 composition are formed from bidentate bispyridyl ligands and Pd(II) ions with a square planar geometry, which have been widely studied [6,48-50]. The photochromic ligands can be switched between three states, forming separate discrete cage complexes, allowing cage-to-cage transformations (Scheme 1). Interestingly, only homochiral cages are formed revealing that a chiral self-sorting process takes place. In addition, two of the cage isomers can bind a tosylate anion in solution by formation of a host–guest complex.
![[1860-5397-15-268-1]](/bjoc/content/figures/1860-5397-15-268-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic representation of a photoresponsive cage with ligands based on overcrowded alkenes.
Figure 1: Schematic representation of a photoresponsive cage with ligands based on overcrowded alkenes.
![[1860-5397-15-268-i1]](/bjoc/content/inline/1860-5397-15-268-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cage formation of overcrowded alkene switches E/Z-1 and their isomerization behavior. Note that the intermediate unstable E-1 is not shown; due to the low barrier of thermal helix inversion and therefore fast isomerization to the corresponding stable isomer.
Scheme 1: Cage formation of overcrowded alkene switches E/Z-1 and their isomerization behavior. Note that the...
Results and Discussion
Ligands Z-1 and E-1 (Scheme 1) were synthesized by a Suzuki cross-coupling reaction of 3-pyridinylboronic acid with an E/Z mixture of reported overcrowded alkene precursors [51] (see Supporting Information File 1 for full experimental details). Heating a 2:1 mixture of ligands (S,S)-Z-1 or (S,S)-E-1 with Pd(NO3)2 in acetonitrile at reflux led to the quantitative formation of cage Pd2(stable Z-1)4 or Pd2(stable E-1)4, respectively, as evidenced by 1H NMR, DOSY and HRMS. The 1H NMR signals of the pyridine moieties of the ligands (Ha–d) in the assembled cages are shifted downfield compared to those of the free ligands, as expected due to metal coordination (Figure 2) [28]. As the ligand exchange in Pd2L4 complexes is slow on the NMR timescale, the discrete signals do not represent an average of quickly interconverting isomers [52,53]. The formation of cage complexes using a racemic mixture of ligands stable Z-1 or stable E-1, resulted in the exact same 1H NMR spectrum as was obtained with the enantiopure ligands. Using a racemic mixture of ligands, four different diastereomeric cages can be formed ((S,S)4, (S,S)3(R,R), (S,S)2(R,R)2 and (S,S)(R,R)(S,S)(R,R) and their enantiomeric pairs). However, in both cases, only one set of signals is observed, which is a strong indication that only one species with high symmetry is formed by chiral self-sorting without any sign of the formation of diastereomeric mixtures. To further confirm the self-sorting behavior of these cage structures, CD spectroscopy was performed. A linear dependence of CD amplitude on the ee of ligand stable Z-1 was found, which can be expected when the homochiral enantiomers of cage structure Pd2(stable Z-1)4 are the only optically active species in solution (Figure S1, Supporting Information File 1).
![[1860-5397-15-268-2]](/bjoc/content/figures/1860-5397-15-268-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Aromatic region of stacked 1H NMR spectra (in CD3CN) of stable Z-1 and cage complex Pd2(stable Z-1)4 (top) and E-1 and cage complex Pd2(stable E-1)4 (bottom).
Figure 2: Aromatic region of stacked 1H NMR spectra (in CD3CN) of stable Z-1 and cage complex Pd2(stable Z-1)4...
Additionally, DOSY NMR spectroscopy revealed that the signals correspond to a single type of assembly in each case (see Supporting Information File 1, section 2). The measured diffusion coefficients (D = 8.7 × 10−10 m2 s−1 for Pd2(stable Z-1)4 and D = 7.9 × 10−10 m2 s−1 for cage Pd2(stable E-1)4 in CD3CN at 23 °C) can be translated into hydrodynamic radii of rH = 7.2 Å for Pd2(stable Z-1)4 and rH = 8.0 Å for Pd2(stable E-1)4 by using the Stokes–Einstein equation [54]. By means of ESI high-resolution mass spectrometry, we were able to verify the Pd2L4 constitution of both cages. The HRMS spectrum of Pd2(stable Z-1)4 shows the signals for the cations [Pd2(stable Z-1)4(NO3)3]+, [Pd2(stable Z-1)4(NO3)2]2+, [Pd2(stable Z-1)4(NO3)]3+, [Pd2(stable Z-1)4]4+ (Figure 3). For Pd2(stable E-1)4, the peaks corresponding to the cations [Pd2(stable E-1)4(NO3)2]2+ and [Pd2(stable E-1)4(NO3)]3+ were observed (Figure 3). For both isomers, the experimental isotopic patterns and exact m/z values match the simulated patterns.
![[1860-5397-15-268-3]](/bjoc/content/figures/1860-5397-15-268-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: HRMS spectra of cage complex Pd2(stable Z-1)4 (top) and cage complex Pd2(stable E-1)4 (bottom); Insets: comparison of simulated and measured isotopic patterns of Pd2L4(NO3)3+ ions.
Figure 3: HRMS spectra of cage complex Pd2(stable Z-1)4 (top) and cage complex Pd2(stable E-1)4 (bottom); Ins...
A single crystal of Pd2(stable E-1)4 formed from a racemic mixture of ligand E-1 suitable for X-ray structure determination was grown by vapor diffusion of a 1:1 mixture of benzene and diethyl ether into a solution of the cage in a 1:1 mixture of acetonitrile and chloroform. The crystal structure shows cage structures with a Pd2L4 stoichiometry with one NO3− counterion and one molecule of acetonitrile located inside each cage. In addition, a chloride ion is located close to the metal centers outside of the cage. This counterion most likely originates from the solvent, as chloroform can contain considerable amounts of HCl. The additional anions required to balance the charge of the tetracationic Pd2L4 cage could not be unambiguously located in the difference Fourier map (see Supporting Information File 1 for details). The structure belongs to the P4/n space group and the unit cell is occupied by a pair of enantiomeric cages in which the Pd–Pd axis of each cage is located at the 4-fold rotation axis. This means that the cage structure is made up with exclusively (R,R) or (S,S) enantiomer of the ligand and that the elementary cell is built from the racemic pair of cages.
DFT calculations were performed to gain insight in the self-sorting behavior of cages Pd2(stable Z-1)4 and Pd2(stable E-1)4. The structures of all possible cage diastereomers were optimized using B3LYP/6-31G(d) for C,H,N and LANL2DZ with ECP for Pd in the gas phase without counter ions. The optimized structure of Pd2(stable E-1)4 is in good agreement with the solved X-ray structure (Figure 4). Moreover, the calculations revealed that the homochiral cage Pd2((S,S)-stable E-1)4 (and its enantiomer) are energetically favored by at least 61 kJ mol−1 compared to the other possible diastereomers (Table S1, Supporting Information File 1). Similar calculations on the diastereomers of Pd2(stable Z-1)4 revealed that the homochiral cage diastereomers Pd2((S,S)-stable Z-1)4 are energetically favored as well, by at least 19 kJ mol−1 (Table S2, Supporting Information File 1). These calculations support that these cages are formed by chiral narcissistic self-sorting.
![[1860-5397-15-268-4]](/bjoc/content/figures/1860-5397-15-268-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Crystal structure of cage complex Pd2(stable E-1)4 (top left) and DFT optimized structures of cage complexes Pd2(stable E-1)4, Pd2(stable Z-1)4 and Pd2(unstable Z-1)4. For clarity only one of the enantiomeric cages was depicted on the figure (carbon – black, nitrogen – blue, oxygen – red, palladium – cyan, chlorine – green, hydrogen – white).
Figure 4: Crystal structure of cage complex Pd2(stable E-1)4 (top left) and DFT optimized structures of cage ...
Next, we were interested in the guest binding abilities of cages Pd2(stable Z-1)4 and Pd2(stable E-1)4. The tosylate anion was chosen as it has the appropriate size to fit inside the cages. A Job plot analysis revealed a 1:1 binding stoichiometry between both cage isomers and OTs− (Figures S3–S5, Supporting Information File 1), which corresponds to the model in which OTs− serves as a guest molecule which is encapsulated inside the cages [55,56]. 1H NMR titrations with tetrabutylammonium tosylate revealed that both cages are able to bind OTs−, showing similar binding strengths (KB = 1604 ± 39 M−1 for Pd2(stable Z-1)4; KB = 1758 ± 39 M−1 for Pd2(stable E-1)4 at 293 K).
Finally, the photochemical and thermal isomerization behavior of the complexes was studied. Initially, the behavior of ligand 1 was studied by UV–vis and NMR spectroscopy, showing similar behavior as related first generation molecular motors (see Supporting Information File 1 for full details). Irradiation with 312 nm light at −70 °C isomerizes stable Z-1 to unstable E-1, which undergoes a thermal helix inversion (THI) when warmed to room temperature to form stable E-1. The second half of the rotation cycle is similar, as irradiation with 312 nm light isomerizes stable E-1 to unstable Z-1, which undergoes THI to form stable Z-1 when left for several days at room temperature.
Subsequently, the photochemical and thermal isomerizations of cages Pd2(stable Z-1)4 and Pd2(stable E-1)4 were followed by 1H NMR studies (Figure 5). Irradiation of Pd2(stable Z-1)4 in a CD3CN/CD2Cl2 1:1 mixture at 312 nm at −70 °C was performed to isomerize ligand stable Z-1 to unstable E-1 (vide infra), followed by allowing the sample to warm to room temperature to form stable E-1 (Figure 5ii). The 1H NMR spectrum of this newly formed complex is identical to the spectrum of Pd2(stable E-1)4 prepared directly from E-1 (Figure 5iii), showing that cage Pd2(stable Z-1)4 is effectively converted to Pd2(stable E-1)4. An intermediate complex containing unstable E-1 ligands was not observed, even at low temperatures, most likely due to the low barrier for THI of this isomer. Conversion of cage Pd2(stable E-1)4 to Pd2(unstable Z-1)4 by photochemical E/Z isomerization of ligand stable E-1 to unstable Z-1 was performed by irradiation of a sample of Pd2(stable E-1)4 at 312 nm at −20 °C (Figure 5iv). Signals of cage Pd2(stable Z-1)4 disappeared and the formation of a new set of signals was observed. DOSY NMR confirmed the formation of an assembly with a hydrodynamic radius which was similar to that of the cage Pd2(stable Z-1)4. Precipitation of the metal centers in this assembly using tetrabutylammonium glutarate liberates the ligands and they were identified as unstable Z-1. Combined, these results confirm that the photogenerated complex is indeed Pd2(unstable Z-1)4. Subsequent irradiation of this sample containing Pd2(unstable Z-1)4 at −20 °C at 365 nm converts the unstable Z-1 ligands back to stable E-1, reforming Pd2(stable E-1)4 (Figure 5v). These experiments highlight the reversible formation of Pd2(unstable Z-1)4 through photochemical E/Z isomerization of the ligands.
![[1860-5397-15-268-5]](/bjoc/content/figures/1860-5397-15-268-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Aromatic region of stacked 1H NMR spectra (CD3CN/CD2Cl2 1:1) of i) Pd2(stable Z-1)4; ii) Pd2(stable E-1)4 generated by irradiation of Pd2(stable Z-1)4 at 312 nm; iii) Pd2(stable E-1)4 prepared directly from ligand stable E-1; iv) Pd2(unstable Z-1)4 generated by irradiation of Pd2(stable E-1)4 at 312 nm and v) Pd2(stable E-1)4 generated by irradiation of Pd2(unstable Z-1)4 at 365 nm.
Figure 5: Aromatic region of stacked 1H NMR spectra (CD3CN/CD2Cl2 1:1) of i) Pd2(stable Z-1)4; ii) Pd2(stable ...
On the other hand, allowing the THI of ligands unstable Z-1 in cage Pd2(unstable Z-1)4 to take place by leaving the solution at room temperature for 5 d did not lead to the formation of Pd2(stable Z-1)4, but to disassembly of the cage and formation of ill-defined complexes. Precipitation of the metal centers in these complexes identified the ligands as a mixture of both stable Z-1 and stable E-1 (originating from the PSS mixture), indicating that the THI does take place. A possible explanation could be that the mixture of stable Z-1 and stable E-1 does not form separate well-defined cage structures, but forms mixed complexes.
Conclusion
In summary, a new photoresponsive supramolecular coordination complex based on overcrowded alkenes is presented, allowing switching between three different cage structures. Interestingly, the cage structures with Pd2L4 constitution were shown to be homochiral, forming single diastereomers as evident from NMR, CD and X-ray analysis, supported by DFT calculations. Additionally, the cage structures were able to bind OTs− inside their cavity. Although photoswitching affords a large geometric change of the ligands, only minor changes were observed in binding constants of the different cage structures. These results show that by incorporation of overcrowded alkenes into SCCs the geometry of cage structures can be controlled by light. Different designs might be considered to translate these geometrical changes to changes in properties such as guest binding affinity and selectivity.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization, CD spectroscopy, binding studies, NMR studies of the photochemical and thermal isomerizations, X-ray crystallography, computational details and Cartesian coordinates of DFT optimized structures. | ||
| Format: PDF | Size: 3.7 MB | Download |
Acknowledgements
Financial support from the Ministry of Education, Culture and Science (Gravitation program 024.001.035) and European Research Council (Advanced Investigator Grant No. 694345 to B.L.F.) are gratefully acknowledged. We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster.
References
-
Cook, T. R.; Zheng, Y.-R.; Stang, P. J. Chem. Rev. 2013, 113, 734–777. doi:10.1021/cr3002824
Return to citation in text: [1] -
Smulders, M. M. J.; Riddell, I. A.; Browne, C.; Nitschke, J. R. Chem. Soc. Rev. 2013, 42, 1728–1754. doi:10.1039/c2cs35254k
Return to citation in text: [1] -
Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666
Return to citation in text: [1] -
Harris, K.; Fujita, D.; Fujita, M. Chem. Commun. 2013, 49, 6703–6712. doi:10.1039/c3cc43191f
Return to citation in text: [1] [2] -
Han, M.; Engelhard, D. M.; Clever, G. H. Chem. Soc. Rev. 2014, 43, 1848–1860. doi:10.1039/c3cs60473j
Return to citation in text: [1] -
Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037
Return to citation in text: [1] [2] [3] -
Ma, Z.; Moulton, B. Coord. Chem. Rev. 2011, 255, 1623–1641. doi:10.1016/j.ccr.2011.01.031
Return to citation in text: [1] -
Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/c2sc00899h
Return to citation in text: [1] [2] -
Vriezema, D. M.; Comellas Aragonès, M.; Elemans, J. A. A. W.; Cornelissen, J. J. L. M.; Rowan, A. E.; Nolte, R. J. M. Chem. Rev. 2005, 105, 1445–1490. doi:10.1021/cr0300688
Return to citation in text: [1] -
Pluth, M. D.; Bergman, R. G.; Raymond, K. N. Acc. Chem. Res. 2009, 42, 1650–1659. doi:10.1021/ar900118t
Return to citation in text: [1] -
Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340
Return to citation in text: [1] -
Kaphan, D. M.; Levin, M. D.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Science 2015, 350, 1235–1238. doi:10.1126/science.aad3087
Return to citation in text: [1] -
Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. Nature 2013, 495, 461–466. doi:10.1038/nature11990
Return to citation in text: [1] -
Yan, K.; Dubey, R.; Arai, T.; Inokuma, Y.; Fujita, M. J. Am. Chem. Soc. 2017, 139, 11341–11344. doi:10.1021/jacs.7b06607
Return to citation in text: [1] -
Ziegler, M.; Brumaghim, J. L.; Raymond, K. N. Angew. Chem., Int. Ed. 2000, 39, 4119–4121. doi:10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1
Return to citation in text: [1] -
Kawano, M.; Kobayashi, Y.; Ozeki, T.; Fujita, M. J. Am. Chem. Soc. 2006, 128, 6558–6559. doi:10.1021/ja0609250
Return to citation in text: [1] -
Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J. R. Science 2009, 324, 1697–1699. doi:10.1126/science.1175313
Return to citation in text: [1] -
McConnell, A. J.; Wood, C. S.; Neelakandan, P. P.; Nitschke, J. R. Chem. Rev. 2015, 115, 7729–7793. doi:10.1021/cr500632f
Return to citation in text: [1] -
Mahata, K.; Frischmann, P. D.; Würthner, F. J. Am. Chem. Soc. 2013, 135, 15656–15661. doi:10.1021/ja4083039
Return to citation in text: [1] -
Scherer, M.; Caulder, D. L.; Johnson, D. W.; Raymond, K. N. Angew. Chem., Int. Ed. 1999, 38, 1587–1592. doi:10.1002/(sici)1521-3773(19990601)38:11<1587::aid-anie1587>3.0.co;2-r
Return to citation in text: [1] -
Mal, P.; Schultz, D.; Beyeh, K.; Rissanen, K.; Nitschke, J. R. Angew. Chem., Int. Ed. 2008, 47, 8297–8301. doi:10.1002/anie.200803066
Return to citation in text: [1] -
Clever, G. H.; Tashiro, S.; Shionoya, M. J. Am. Chem. Soc. 2010, 132, 9973–9975. doi:10.1021/ja103620z
Return to citation in text: [1] -
Murase, T.; Sato, S.; Fujita, M. Angew. Chem., Int. Ed. 2007, 46, 5133–5136. doi:10.1002/anie.200700793
Return to citation in text: [1] -
Park, J.; Sun, L.-B.; Chen, Y.-P.; Perry, Z.; Zhou, H.-C. Angew. Chem., Int. Ed. 2014, 53, 5842–5846. doi:10.1002/anie.201310211
Return to citation in text: [1] -
Feringa, B. L.; Browne, W. R. Molecular Switches, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Vol. 1.
Return to citation in text: [1] -
Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p
Return to citation in text: [1] -
Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/b302356g
Return to citation in text: [1] -
Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373
Return to citation in text: [1] [2] -
Han, M.; Luo, Y.; Damaschke, B.; Gómez, L.; Ribas, X.; Jose, A.; Peretzki, P.; Seibt, M.; Clever, G. H. Angew. Chem., Int. Ed. 2016, 55, 445–449. doi:10.1002/anie.201508307
Return to citation in text: [1] -
Wei, S.-C.; Pan, M.; Fan, Y.-Z.; Liu, H.; Zhang, J.; Su, C.-Y. Chem. – Eur. J. 2015, 21, 7418–7427. doi:10.1002/chem.201406517
Return to citation in text: [1] -
Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h
Return to citation in text: [1] -
He, Z.; Jiang, W.; Schalley, C. A. Chem. Soc. Rev. 2015, 44, 779–789. doi:10.1039/c4cs00305e
Return to citation in text: [1] -
Jędrzejewska, H.; Szumna, A. Chem. Rev. 2017, 117, 4863–4899. doi:10.1021/acs.chemrev.6b00745
Return to citation in text: [1] -
Huang, W.-H.; Zavalij, P. Y.; Isaacs, L. Angew. Chem., Int. Ed. 2007, 46, 7425–7427. doi:10.1002/anie.200702189
Return to citation in text: [1] -
Haino, T.; Shio, H.; Takano, R.; Fukazawa, Y. Chem. Commun. 2009, 2481–2483. doi:10.1039/b900599d
Return to citation in text: [1] -
Xuan, W.; Zhang, M.; Liu, Y.; Chen, Z.; Cui, Y. J. Am. Chem. Soc. 2012, 134, 6904–6907. doi:10.1021/ja212132r
Return to citation in text: [1] -
Gidron, O.; Ebert, M.-O.; Trapp, N.; Diederich, F. Angew. Chem., Int. Ed. 2014, 53, 13614–13618. doi:10.1002/anie.201406585
Return to citation in text: [1] -
Koumura, N.; Zijlstra, R. W. J.; van Delden, R. A.; Harada, N.; Feringa, B. L. Nature 1999, 401, 152–155. doi:10.1038/43646
Return to citation in text: [1] -
Kassem, S.; van Leeuwen, T.; Lubbe, A. S.; Wilson, M. R.; Feringa, B. L.; Leigh, D. A. Chem. Soc. Rev. 2017, 46, 2592–2621. doi:10.1039/c7cs00245a
Return to citation in text: [1] -
Roke, D.; Wezenberg, S. J.; Feringa, B. L. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9423–9431. doi:10.1073/pnas.1712784115
Return to citation in text: [1] -
Wang, J.; Feringa, B. L. Science 2011, 331, 1429–1432. doi:10.1126/science.1199844
Return to citation in text: [1] -
Zhao, D.; Neubauer, T. M.; Feringa, B. L. Nat. Commun. 2015, 6, 6652. doi:10.1038/ncomms7652
Return to citation in text: [1] -
Chen, J.; Leung, F. K.-C.; Stuart, M. C. A.; Kajitani, T.; Fukushima, T.; van der Giessen, E.; Feringa, B. L. Nat. Chem. 2018, 10, 132–138. doi:10.1038/nchem.2887
Return to citation in text: [1] [2] -
Li, Q.; Fuks, G.; Moulin, E.; Maaloum, M.; Rawiso, M.; Kulic, I.; Foy, J. T.; Giuseppone, N. Nat. Nanotechnol. 2015, 10, 161–165. doi:10.1038/nnano.2014.315
Return to citation in text: [1] -
Foy, J. T.; Li, Q.; Goujon, A.; Colard-Itté, J.-R.; Fuks, G.; Moulin, E.; Schiffmann, O.; Dattler, D.; Funeriu, D. P.; Giuseppone, N. Nat. Nanotechnol. 2017, 12, 540–545. doi:10.1038/nnano.2017.28
Return to citation in text: [1] -
van Leeuwen, T.; Heideman, G. H.; Zhao, D.; Wezenberg, S. J.; Feringa, B. L. Chem. Commun. 2017, 53, 6393–6396. doi:10.1039/c7cc03188b
Return to citation in text: [1] -
Zhao, D.; van Leeuwen, T.; Cheng, J.; Feringa, B. L. Nat. Chem. 2017, 9, 250–256. doi:10.1038/nchem.2668
Return to citation in text: [1] [2] -
McMorran, D. A.; Steel, P. J. Angew. Chem., Int. Ed. 1998, 37, 3295–3297. doi:10.1002/(sici)1521-3773(19981217)37:23<3295::aid-anie3295>3.0.co;2-5
Return to citation in text: [1] -
Chand, D. K.; Biradha, K.; Fujita, M. Chem. Commun. 2001, 1652–1653. doi:10.1039/b104853h
Return to citation in text: [1] -
Su, C.-Y.; Cai, Y.-P.; Chen, C.-L.; Smith, M. D.; Kaim, W.; zur Loye, H.-C. J. Am. Chem. Soc. 2003, 125, 8595–8613. doi:10.1021/ja034267k
Return to citation in text: [1] -
Neubauer, T. M.; van Leeuwen, T.; Zhao, D.; Lubbe, A. S.; Kistemaker, J. C. M.; Feringa, B. L. Org. Lett. 2014, 16, 4220–4223. doi:10.1021/ol501925f
Return to citation in text: [1] -
Clever, G. H.; Shionoya, M. Chem. – Eur. J. 2010, 16, 11792–11796. doi:10.1002/chem.201002013
Return to citation in text: [1] -
Sato, S.; Ishido, Y.; Fujita, M. J. Am. Chem. Soc. 2009, 131, 6064–6065. doi:10.1021/ja900676f
Return to citation in text: [1] -
Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Chem. Soc. Rev. 2008, 37, 479–489. doi:10.1039/b615067p
Return to citation in text: [1] -
Performed with the use of Bindfit software – http://supramolecular.com
Return to citation in text: [1] -
Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k
Return to citation in text: [1]
| 1. | Cook, T. R.; Zheng, Y.-R.; Stang, P. J. Chem. Rev. 2013, 113, 734–777. doi:10.1021/cr3002824 |
| 2. | Smulders, M. M. J.; Riddell, I. A.; Browne, C.; Nitschke, J. R. Chem. Soc. Rev. 2013, 42, 1728–1754. doi:10.1039/c2cs35254k |
| 3. | Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666 |
| 4. | Harris, K.; Fujita, D.; Fujita, M. Chem. Commun. 2013, 49, 6703–6712. doi:10.1039/c3cc43191f |
| 5. | Han, M.; Engelhard, D. M.; Clever, G. H. Chem. Soc. Rev. 2014, 43, 1848–1860. doi:10.1039/c3cs60473j |
| 6. | Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037 |
| 15. | Ziegler, M.; Brumaghim, J. L.; Raymond, K. N. Angew. Chem., Int. Ed. 2000, 39, 4119–4121. doi:10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1 |
| 16. | Kawano, M.; Kobayashi, Y.; Ozeki, T.; Fujita, M. J. Am. Chem. Soc. 2006, 128, 6558–6559. doi:10.1021/ja0609250 |
| 17. | Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J. R. Science 2009, 324, 1697–1699. doi:10.1126/science.1175313 |
| 38. | Koumura, N.; Zijlstra, R. W. J.; van Delden, R. A.; Harada, N.; Feringa, B. L. Nature 1999, 401, 152–155. doi:10.1038/43646 |
| 39. | Kassem, S.; van Leeuwen, T.; Lubbe, A. S.; Wilson, M. R.; Feringa, B. L.; Leigh, D. A. Chem. Soc. Rev. 2017, 46, 2592–2621. doi:10.1039/c7cs00245a |
| 40. | Roke, D.; Wezenberg, S. J.; Feringa, B. L. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9423–9431. doi:10.1073/pnas.1712784115 |
| 13. | Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. Nature 2013, 495, 461–466. doi:10.1038/nature11990 |
| 14. | Yan, K.; Dubey, R.; Arai, T.; Inokuma, Y.; Fujita, M. J. Am. Chem. Soc. 2017, 139, 11341–11344. doi:10.1021/jacs.7b06607 |
| 41. | Wang, J.; Feringa, B. L. Science 2011, 331, 1429–1432. doi:10.1126/science.1199844 |
| 42. | Zhao, D.; Neubauer, T. M.; Feringa, B. L. Nat. Commun. 2015, 6, 6652. doi:10.1038/ncomms7652 |
| 43. | Chen, J.; Leung, F. K.-C.; Stuart, M. C. A.; Kajitani, T.; Fukushima, T.; van der Giessen, E.; Feringa, B. L. Nat. Chem. 2018, 10, 132–138. doi:10.1038/nchem.2887 |
| 9. | Vriezema, D. M.; Comellas Aragonès, M.; Elemans, J. A. A. W.; Cornelissen, J. J. L. M.; Rowan, A. E.; Nolte, R. J. M. Chem. Rev. 2005, 105, 1445–1490. doi:10.1021/cr0300688 |
| 10. | Pluth, M. D.; Bergman, R. G.; Raymond, K. N. Acc. Chem. Res. 2009, 42, 1650–1659. doi:10.1021/ar900118t |
| 11. | Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340 |
| 12. | Kaphan, D. M.; Levin, M. D.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Science 2015, 350, 1235–1238. doi:10.1126/science.aad3087 |
| 31. | Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h |
| 32. | He, Z.; Jiang, W.; Schalley, C. A. Chem. Soc. Rev. 2015, 44, 779–789. doi:10.1039/c4cs00305e |
| 33. | Jędrzejewska, H.; Szumna, A. Chem. Rev. 2017, 117, 4863–4899. doi:10.1021/acs.chemrev.6b00745 |
| 6. | Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037 |
| 7. | Ma, Z.; Moulton, B. Coord. Chem. Rev. 2011, 255, 1623–1641. doi:10.1016/j.ccr.2011.01.031 |
| 8. | Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/c2sc00899h |
| 34. | Huang, W.-H.; Zavalij, P. Y.; Isaacs, L. Angew. Chem., Int. Ed. 2007, 46, 7425–7427. doi:10.1002/anie.200702189 |
| 35. | Haino, T.; Shio, H.; Takano, R.; Fukazawa, Y. Chem. Commun. 2009, 2481–2483. doi:10.1039/b900599d |
| 36. | Xuan, W.; Zhang, M.; Liu, Y.; Chen, Z.; Cui, Y. J. Am. Chem. Soc. 2012, 134, 6904–6907. doi:10.1021/ja212132r |
| 37. | Gidron, O.; Ebert, M.-O.; Trapp, N.; Diederich, F. Angew. Chem., Int. Ed. 2014, 53, 13614–13618. doi:10.1002/anie.201406585 |
| 24. | Park, J.; Sun, L.-B.; Chen, Y.-P.; Perry, Z.; Zhou, H.-C. Angew. Chem., Int. Ed. 2014, 53, 5842–5846. doi:10.1002/anie.201310211 |
| 29. | Han, M.; Luo, Y.; Damaschke, B.; Gómez, L.; Ribas, X.; Jose, A.; Peretzki, P.; Seibt, M.; Clever, G. H. Angew. Chem., Int. Ed. 2016, 55, 445–449. doi:10.1002/anie.201508307 |
| 23. | Murase, T.; Sato, S.; Fujita, M. Angew. Chem., Int. Ed. 2007, 46, 5133–5136. doi:10.1002/anie.200700793 |
| 30. | Wei, S.-C.; Pan, M.; Fan, Y.-Z.; Liu, H.; Zhang, J.; Su, C.-Y. Chem. – Eur. J. 2015, 21, 7418–7427. doi:10.1002/chem.201406517 |
| 22. | Clever, G. H.; Tashiro, S.; Shionoya, M. J. Am. Chem. Soc. 2010, 132, 9973–9975. doi:10.1021/ja103620z |
| 4. | Harris, K.; Fujita, D.; Fujita, M. Chem. Commun. 2013, 49, 6703–6712. doi:10.1039/c3cc43191f |
| 8. | Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/c2sc00899h |
| 18. | McConnell, A. J.; Wood, C. S.; Neelakandan, P. P.; Nitschke, J. R. Chem. Rev. 2015, 115, 7729–7793. doi:10.1021/cr500632f |
| 19. | Mahata, K.; Frischmann, P. D.; Würthner, F. J. Am. Chem. Soc. 2013, 135, 15656–15661. doi:10.1021/ja4083039 |
| 20. | Scherer, M.; Caulder, D. L.; Johnson, D. W.; Raymond, K. N. Angew. Chem., Int. Ed. 1999, 38, 1587–1592. doi:10.1002/(sici)1521-3773(19990601)38:11<1587::aid-anie1587>3.0.co;2-r |
| 21. | Mal, P.; Schultz, D.; Beyeh, K.; Rissanen, K.; Nitschke, J. R. Angew. Chem., Int. Ed. 2008, 47, 8297–8301. doi:10.1002/anie.200803066 |
| 25. | Feringa, B. L.; Browne, W. R. Molecular Switches, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Vol. 1. |
| 26. | Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p |
| 27. | Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/b302356g |
| 28. | Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373 |
| 47. | Zhao, D.; van Leeuwen, T.; Cheng, J.; Feringa, B. L. Nat. Chem. 2017, 9, 250–256. doi:10.1038/nchem.2668 |
| 43. | Chen, J.; Leung, F. K.-C.; Stuart, M. C. A.; Kajitani, T.; Fukushima, T.; van der Giessen, E.; Feringa, B. L. Nat. Chem. 2018, 10, 132–138. doi:10.1038/nchem.2887 |
| 44. | Li, Q.; Fuks, G.; Moulin, E.; Maaloum, M.; Rawiso, M.; Kulic, I.; Foy, J. T.; Giuseppone, N. Nat. Nanotechnol. 2015, 10, 161–165. doi:10.1038/nnano.2014.315 |
| 45. | Foy, J. T.; Li, Q.; Goujon, A.; Colard-Itté, J.-R.; Fuks, G.; Moulin, E.; Schiffmann, O.; Dattler, D.; Funeriu, D. P.; Giuseppone, N. Nat. Nanotechnol. 2017, 12, 540–545. doi:10.1038/nnano.2017.28 |
| 46. | van Leeuwen, T.; Heideman, G. H.; Zhao, D.; Wezenberg, S. J.; Feringa, B. L. Chem. Commun. 2017, 53, 6393–6396. doi:10.1039/c7cc03188b |
| 47. | Zhao, D.; van Leeuwen, T.; Cheng, J.; Feringa, B. L. Nat. Chem. 2017, 9, 250–256. doi:10.1038/nchem.2668 |
| 54. | Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Chem. Soc. Rev. 2008, 37, 479–489. doi:10.1039/b615067p |
| 55. | Performed with the use of Bindfit software – http://supramolecular.com |
| 56. | Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k |
| 28. | Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373 |
| 52. | Clever, G. H.; Shionoya, M. Chem. – Eur. J. 2010, 16, 11792–11796. doi:10.1002/chem.201002013 |
| 53. | Sato, S.; Ishido, Y.; Fujita, M. J. Am. Chem. Soc. 2009, 131, 6064–6065. doi:10.1021/ja900676f |
| 6. | Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037 |
| 48. | McMorran, D. A.; Steel, P. J. Angew. Chem., Int. Ed. 1998, 37, 3295–3297. doi:10.1002/(sici)1521-3773(19981217)37:23<3295::aid-anie3295>3.0.co;2-5 |
| 49. | Chand, D. K.; Biradha, K.; Fujita, M. Chem. Commun. 2001, 1652–1653. doi:10.1039/b104853h |
| 50. | Su, C.-Y.; Cai, Y.-P.; Chen, C.-L.; Smith, M. D.; Kaim, W.; zur Loye, H.-C. J. Am. Chem. Soc. 2003, 125, 8595–8613. doi:10.1021/ja034267k |
| 51. | Neubauer, T. M.; van Leeuwen, T.; Zhao, D.; Lubbe, A. S.; Kistemaker, J. C. M.; Feringa, B. L. Org. Lett. 2014, 16, 4220–4223. doi:10.1021/ol501925f |
© 2019 Stuckhardt et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)