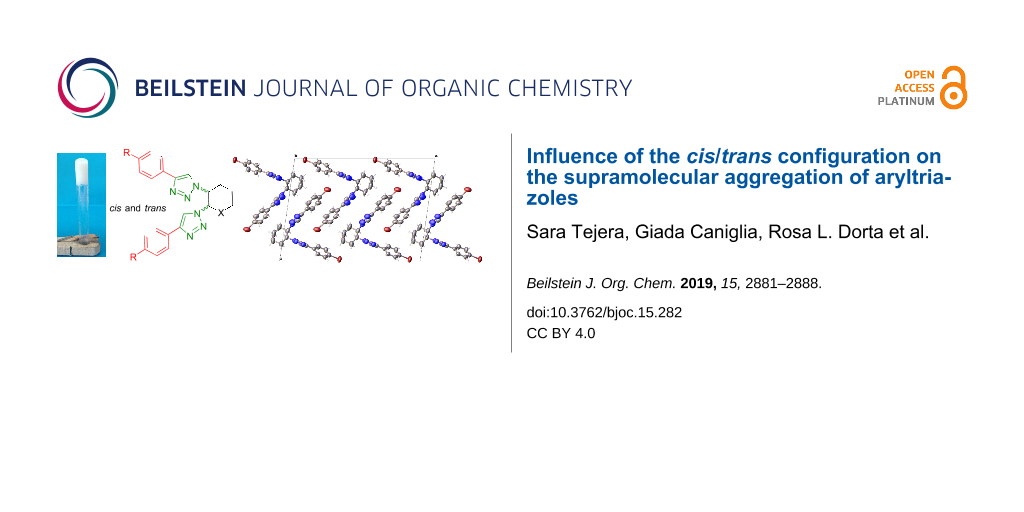
Abstract
The ability of trans- and cis-1,2-glucopyranosyl and cyclohexyl ditriazoles, synthesized by CuAAC "click" chemistry, to form gels was studied, their physical properties determined, and the self-aggregation behavior investigated by SEM, X-ray, and EDC studies. The results revealed that self-assembly was driven mainly by π–π stacking interactions, in addition to hydrogen bonding, with the aromatic rings adopting a high degree of parallelism, as seen in crystal packings and ECD data. Furthermore, π–bromine interactions between the bromine atom of the aryl substituents and the triazole units might also contribute to an overall stabilization of the supramolecular aggregation of bis(4-bromophenyl)triazoles. The trans or cis spatial disposition of the triazole rings is highly important for gelation, with the cis configuration having higher propensity.

Graphical Abstract
Introduction
Structures self-assembled by noncovalent interactions give rise to supramolecular architectures with specific physical and/or chemical properties. Gels are colloid systems in which the dispersed phase has combined with the dispersion medium to yield a semisolid material. Gels from low-molecular-weight gelators have potential applications in high-tech materials [1-3] and biomedical sciences [3-6]. Triazole derivatives have shown excellent gelation properties [7-10], in addition to a broad range of biological activities [11-14].
During the synthesis and characterization process of glucosyl mono- and ditriazole derivatives, which we carried out in order to analyze the use of aryltriazoles for the determination of absolute configuration [15], we serendipitously discovered that these glucosyl ditriazoles led to gels. Therefore, we carried out the corresponding supramolecular studies and report herein the ability of ditriazoles to form gels, their physical properties, as well as the dependence of these properties on the cis/trans relative configuration.
Results and Discussion
A large set of mono- and ditriazoles was synthesized using cycloaddition reactions based on "click" chemistry [16-18] of azides and alkynes catalyzed by Cu(I) salts, the CuAAC reaction. Self-assembling properties were not observed for any of the prepared monotriazoles, namely the 4-substituted 1-glucopyranosyltriazoles 1a–g and 2a–g (Scheme 1) [15]. However, most ditriazoles 7a–g and 8a–g (Scheme 2) showed supramolecular features, i.e., their DMSO solutions prepared for NMR analyses spontaneously transformed into gels inside the NMR tubes.
![[1860-5397-15-282-i1]](/bjoc/content/inline/1860-5397-15-282-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structures of 4-substituted 1-glucopyranosyltriazoles 1a–g and 2a–g [15].
Scheme 1: Structures of 4-substituted 1-glucopyranosyltriazoles 1a–g and 2a–g [15].
![[1860-5397-15-282-i2]](/bjoc/content/inline/1860-5397-15-282-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1,2-cis-/trans-bistriazoles 7a–7g and 8a–8g [15].
Scheme 2: Synthesis of 1,2-cis-/trans-bistriazoles 7a–7g and 8a–8g [15].
Three cis-/trans-configured pairs of compounds were chosen for a gelation ability study. Thus, analogue pairs 8f/7f (Scheme 2), 10/9 (Scheme 3), and 14/12 (Scheme 4) were selected. The pair 8f/7f was chosen for having different anomeric configurations in the glucosyl system, and therefore different cis/trans relationships with the equatorial substituent in position 2. Compounds 9 and 10 were selected to monitor the effects of the unprotected hydroxy groups, and the last pair, 14/12, for being a non-glycosyl system, yet with the same cis/trans relationship as the former compounds. In addition, the available compounds 8a, 8b, and 8e (cis) [15] were also tested for gelation to support the results.
![[1860-5397-15-282-i3]](/bjoc/content/inline/1860-5397-15-282-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Compounds 9 (trans) and 10 (cis) [15].
Scheme 3: Compounds 9 (trans) and 10 (cis) [15].
![[1860-5397-15-282-i4]](/bjoc/content/inline/1860-5397-15-282-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of (1R,2R)- and (1R,2S)-1,2-bis-(4-(4-bromophenyl)-1H-triazol-1-yl)cyclohexane (12 and 14).
Scheme 4: Synthesis of (1R,2R)- and (1R,2S)-1,2-bis-(4-(4-bromophenyl)-1H-triazol-1-yl)cyclohexane (12 and 14...
Compounds 7 and 8 (Scheme 2) were successfully prepared from the corresponding alkynes and diazides 5 and 6, respectively. In this context, it was required to isolate 5 and 6 before the coupling reaction could be performed.
Compounds 9 and 10 (Scheme 3) were obtained in good yields by treatment of 7f and 8f, respectively, with sodium methoxide in CH2Cl2/MeOH.
The trans- and cis-1,2-di(triazol-1-yl)cyclohexanes 12 [14] and 14 (Scheme 4), respectively, were prepared from 1-bromo-4-ethynylbenzene and their corresponding diazides, 11 and 13, through CuAAC reactions [16-18].
As can be seen in Figure 1 and Table 1, all these compounds except 9 (having a trans configuration) formed gels in DMSO or DMSO/H2O mixtures. While the ditriazole species with an α-anomeric configuration, 8f (cis), was able to form gels at different concentrations and DMSO/H2O ratios, its β-anomer 7f (trans) only formed a gel when the sample was exposed to a temperature of 8 °C in a refrigerator. This temperature gave rise to an opaque gel that melted above 18 °C (Table 2). The same approach, along with reduction of the minimum gelation concentration to 1.1% w/w (in DMSO) was used to obtain a gel based on 8f. In addition, this compound proved to be an effective translucent/transparent low-molecular-weight gelator in aqueous solutions of DMSO, forming gels in DMSO/H2O, 2:1, v/v at a minimum concentration of 0.8% w/w. The presence of water favored gel formation, decreased the gelation concentration, and the gel's appearance became translucent/transparent. In addition, these gels were stable for several months in sealed vials, although they could be disrupted by agitation or heating. However, the gel state could be restored through cooling.
![[1860-5397-15-282-1]](/bjoc/content/figures/1860-5397-15-282-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tube inversion test: gels formed by compounds 7f, 8f, 10, 12, and 14.
Figure 1: Tube inversion test: gels formed by compounds 7f, 8f, 10, 12, and 14.
Table 1: Gelation test results for compounds 7f, 8a, 8b, 8e, 8f, 9, 10, 12, and 14 (in % w/w).
| # | Config | DMSO |
DMSO/H2O
(9:1, v/v) |
DMSO/H2O
(3:1, v/v) |
DMSO/H2O
(2:1, v/v) |
DMSO/H2O
(1:1, v/v) |
DMSO/H2O
(1:2, v/v) |
|---|---|---|---|---|---|---|---|
| 7f | β (trans) | G 1.1a | – | p | p | p | p |
| 8f | α (cis) |
G 5.0
G 1.1a |
– | G 0.8 | G 0.8 | p | p |
| 9 | β (trans) | s | s | p | p | p | p |
| 10 | α (cis) | s | – | s | s | G 0.7 | G 0.5 |
| 12 | trans | G 1.1a | p | p | p | p | p |
| 14 | cis | s | G 0.8 | p | p | p | p |
| 8a | cis | – | – | – | G 1.0 | susp | susp |
| 8b | cis | – | – | – | weak G | susp | p |
| 8e | cis | – | – | – | G 1.0 | G 1.0 | susp |
aGelling at T = 8 °C (refrigerator); G = gel; p = precipitate; s = soluble; susp = suspension.
Table 2: Critical gelation concentration (CGC, in % w/w) and gel–sol transition temperature (Tgs, via dropping-ball method) for compounds 7f, 8a, 8b, 8e, 8f, 10, 12, and 14.
| # | Config | Conditions | CGC | Tgs (°C) | Appearance |
|---|---|---|---|---|---|
| 7f | β (trans) | DMSOa | 1.1 | 20 ± 2 | opaque |
| 8f | α (cis) | DMSO | 5.0 | 63 ± 1 | translucent |
| 8f | α (cis) | DMSOa | 1.1 | 20 ± 2 | opaque |
| 8f | α (cis) | DMSO/H2O (3:1, v/v) | 0.8 | 93 ± 3 | translucent |
| 8f | α (cis) | DMSO/H2O (2:1, v/v) | 0.8 | 95 ± 3 | transparent |
| 10 | α (cis) | DMSO/H2O (1:1, v/v) | 0.7 | 68 ± 1 | translucent |
| 10 | α (cis) | DMSO/H2O (1:2, v/v) | 0.5 | 94 ± 3 | transparent |
| 12 | trans | DMSOa | 1.1 | 20 ± 2 | opaque |
| 14 | cis | DMSO/H2O (9:1, v/v) | 0.8 | 50 ± 2b | opaque |
| 8a | cis | DMSO/H2O (2:1, v/v) | 1.0 | 87 ± 3 | opaque |
| 8b | cis | DMSO/H2O (2:1, v/v) | – | –c | – |
| 8e | cis | DMSO/H2O (1:1, v/v) | 1.0 | 96 ± 2 | opaque |
| 8e | cis | DMSO/H2O (2:1, v/v) | 1.0 | 92 ± 2 | translucent |
aGelling at T = 8 °C (refrigerator). bTube inversion test. cSoft gel: dropping-ball method failed.
The triol 9 (trans) did not lead to any gel, while its α-anomer 10 (cis) produced translucent and transparent gels, respectively, in 1:1 and 1:2 ratios of DMSO/H2O, v/v (Table 1 and Table 2). In addition, compound 10 (cis) exhibited the lowest minimum gelation concentration, 0.5% w/w in DMSO/H2O (1:2, v/v).
Compounds having an α-anomeric configuration (cis), independent of the hydroxy groups, were either acetylated (8f) or not (10), forming gels much easier than their corresponding β-anomers (trans). This showed that their ability to form gels was critically dependent on the cis/trans configuration present in the molecule.
To confirm this result, the supramolecular properties of trans- and cis-1,2-bis(4-(4-bromophenyl)-1H-triazol-1-yl)cyclohexanes 12 and 14, having the same configuration as compounds 7f and 8f, respectively, were analyzed. Both compounds formed gels, but the trans stereoisomer 12 did so only at a low temperature (8 °C) in DMSO (G = 1.1, w/w), similar to the β-anomer 7f, which possessed the same trans configuration. On the other hand, the cis stereoisomer 14 formed an opaque gel at room temperature in DMSO/H2O, 9:1, v/v (G = 0.8, w/w), showing a higher potency for gelation, similar to 8f. As such, this relationship mirrors the behavior observed for the 7f/8f pair. Table 2 also contains the different Tgs values obtained by the dropping ball method.
Supportive of these conclusions is the facility of cis-configured ditriazoles to form gels. Thus, compound 8a, with alkyl groups in position 4 on the triazolyl groups, and compound 8e, with tolyl groups in that position, gelled easily (Table 1 and Table 2).
In addition, 4-methoxycarbonyl-substituted ditriazole 8b also formed a gel, which was, however, too soft for the dropping ball method to be performed correctly. Therein, the ball dropped down immediately, and changing the DMSO/water ratio did not improve this result. In any case, the results obtained using 8a and 8e point at the fact that the presence of either the phenyl groups or the bromine atoms are not strictly necessary for gelation, although both might contribute to an overall stabilization of the supramolecular aggregation.
Optical micrographs of the xerogels formed from compounds 7f, 8f, 10, 12, and 14 were obtained by SEM (Figure 2). Analyses of these images revealed typically fibrous networks for all except 10. Compound 7f (trans) showed a dense, thin, short, and interlaced/tangled fiber network. The corresponding α-anomer 8f (cis) exhibited an irregular, porous, and dense structure having thin and short fibers. The xerogel of compound 12 showed long, thin, and relatively straight fibers, with lack of torsion, as well as regularity of the network. Its cis stereoisomer 14 showed a fibrous network, but of much shorter length. However, the gel of compound 10 showed a different morphology, with short and planar films or scales. This could be due to the high proportion of water in the gel and/or the presence of the three free hydroxy groups in its structure, changing its intermolecular self-assembly behavior.
![[1860-5397-15-282-2]](/bjoc/content/figures/1860-5397-15-282-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: SEM images of the xerogels of compounds 7f (DMSO, top left), 8f (DMSO/H2O, 3:1, v/v, top right), 10 (DMSO/H2O, 1:1, v/v, middle left), 10 (DMSO/H2O, 1:2, v/v, middle right), 12 (DMSO, bottom left), and 14 (DMSO/H2O, 9:1, v/v, bottom right).
Figure 2: SEM images of the xerogels of compounds 7f (DMSO, top left), 8f (DMSO/H2O, 3:1, v/v, top right), 10...
Various efforts were made to crystallize the obtained compounds; however, only 12 led to single crystals suitable for X-ray crystallography. Its analysis (Figure 3) not only confirmed the structure, but in addition showed a well-organized crystal packing (Figure 4) where the alternate disposition of the molecules displayed a parallelism between the p-bromophenyl groups as well as between the triazole rings, and therefore the presence of multiple intermolecular π–π stacking and π–bromine [19] interactions. This compound precipitated in the presence of any amount of water and gelled only in DMSO at a low temperature.
![[1860-5397-15-282-3]](/bjoc/content/figures/1860-5397-15-282-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: ORTEP representation of the molecular structure of compound 12 (trans configuration) obtained from X-ray diffraction data.
Figure 3: ORTEP representation of the molecular structure of compound 12 (trans configuration) obtained from ...
![[1860-5397-15-282-4]](/bjoc/content/figures/1860-5397-15-282-4.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 4: Crystal packing of compound 12 (trans configuration) in DMSO.
Figure 4: Crystal packing of compound 12 (trans configuration) in DMSO.
Compound 10 produced pseudo-crystals in DMSO/H2O (1:1, v/v) and its X-ray analysis was not accurate enough to resolve its structure. However, its crystal packing (Figure 5) deserves discussion. As with compound 12, this packing revealed a number of π–bromine interactions [19], together with a high degree of π–π stacking interactions between the aromatic rings, and also that they occured between specific phenyltriazoles. Specifically, these interactions formed between the phenyltriazole moieties in position 1 of a monosaccharide (A) and the phenyltriazole functions in position 1 of its vicinal monosaccharide (B, Figure 5, red lines). Similarly, the phenyltriazole substituents in position 2 of this monosaccharide (B) interacted with a corresponding phenyltriazole in position 2 of the vicinal monosaccharide (C, Figure 5, green lines). In addition, it is worth mentioning the presence of a number of DMSO solvate molecules in the crystal packing next to the hydroxymethyl group in position 6 in the gg rotamer, likely linked through hydrogen bonds.
![[1860-5397-15-282-5]](/bjoc/content/figures/1860-5397-15-282-5.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 5: Crystal packing of 10 (cis configuration) in DMSO/H2O (1:1, v/v). Colored lines: π–π stacking interactions between phenyltriazoles in positions 1 (red lines) and 2 (green lines).
Figure 5: Crystal packing of 10 (cis configuration) in DMSO/H2O (1:1, v/v). Colored lines: π–π stacking inter...
Electronic circular dichroism (ECD) is a powerful technique to study supramolecular systems [20,21], since many interactions responsible for the presence of CD Cotton effects occur through space, such as the well-known ECD exciton chirality method [22-24].
ECD of compound 10 in a DMSO/H2O (1:2, v/v) solution and in gel form were successfully measured (Figure 6). ECD of the solution exhibited negative first/positive second exciton Cotton effects at 262 (Δε = −8.6) and 242 nm (Δε = +3.3), respectively (Figure 6, in black), identical to those obtained for this compound in CH3CN [15], although of lower intensity. On the other hand, the ECD spectrum of the gel instead exhibited a normal Cotton effect at 253 nm (Δε = −3.8), which, at the same time, was the wavelength of its absorption maximum (Figure 6, in blue). The striking absence of exciton Cotton effects is in complete agreement with the high degree of parallelism between the chromophores, as observed in the crystal packing (Figure 5). Two chromophores can only be coupled under an exciton interaction if the angle between the 1La transitions is different from 0° or 180°, since this interaction is governed by an equation with a vectorial product. The lack of exciton pairwise interactions between the two chromophores in positions 1 and 2 of each molecule could be due to the intercalation of a third chromophore between the two former in a parallel disposition to one of them (see Figure 5).
![[1860-5397-15-282-6]](/bjoc/content/figures/1860-5397-15-282-6.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 6: CD spectra of compound 10 (cis) in DMSO/H2O (1:2, v/v) in solution (in black) and as gel (in blue).
Figure 6: CD spectra of compound 10 (cis) in DMSO/H2O (1:2, v/v) in solution (in black) and as gel (in blue).
Conclusion
In summary, the capacity of di-1,4-disubstituted 1,2,3-triazoles to form gels was studied taking the dependence on the trans/cis configuration of the molecules into account. The results clearly show that compounds having the cis configuration are more prone to forming thermoreversible gels than those with the trans configuration. In fact, compound 10, with an α-anomeric configuration (cis), showed the lowest gelation concentration at room temperature (G 0.5% w/w) in DMSO/H2O, 1:2, v/v.
The crystal structures obtained for either the cis or trans configuration through X-ray diffraction studies can be considered good approximations to the supramolecular structure of aryltriazoles in gels. Thus, these compounds showed a high degree of parallelism between the phenyltriazolyl rings, therefore revealing the presence of π–π stacking and π–bromine interactions. In addition, compound 10 (cis), with free hydroxy groups, formed hydrogen bonds between the hydroxymethyl groups in position 6 and DMSO molecules while in a gg rotamer conformation. The different types of EDC spectra obtained for compound 10 in solution (exciton) and in gel (non-exciton), using DMSO/H2O, 1:2, v/v, is in total agreement with the supramolecular structure of the crystal packing of this compound (Figure 5).
Experimental
General information
1H NMR spectra were recorded at 500 and 600 MHz, and 13C NMR spectra at 100, 125, and 150 MHz (VTU 300.0 K). Chemical shifts are reported in ppm. The residual solvent peak was used as an internal reference. HRMS was performed by HRTOFMS in positive mode (ES+). For analytical and preparative thin-layer chromatography, silica gel ready-foils and glass-backed plates (1 mm) were used, respectively. These were visualized using UV light at λ = 254 nm and/or developed by spraying with AcOH/H2O/H2SO4 (20:4:1, v/v/v) and heating to 150 °C. Column chromatography was performed using silica gel (0.015–0.04 mm) and n-hexane/EtOAc solvent systems. All reagents were obtained from commercial sources and used without further purification. Solvents were dried and distilled before use. ECD was recorded in the range of 200–400 nm at room temperature by using 10 mm quartz cells. SEM images were taken by a TEM, JEOL JEM 1010 belonging to the Electronic Microscopy Service (SEGAI) of the University of La Laguna, Spain.
Compounds 7f, 8f, 9, 10, 11, and 12 were prepared and characterized as described in reference [15].
Gelation test
Melting points were measured using the dropping ball method. For that, a stainless steel ball of ca. 3 mm diameter (m = 130.2 mg) was placed on the gel in a tube with a diameter of 1 cm. This was placed inside an oil bath together with a thermometer, and the temperature of the gel melting was recorded.
Synthesis and characterization
1,2-Bis[4-(4-bromophenyl)-1H-1,2,3-triazol-1-yl]-1,2-dideoxy-α-ᴅ-glucopyranoside (10): This compound was prepared as described before [15]. UV (CH3CN) λmax (ε) 253 nm (48000); CD (CH3CN) λext (Δε) 242 (+8.9), 261 (−26.5), 299 nm (+1.1); CD (DMSO:H2O, 1:2, v/v) λext (Δε) 242 (+3.3), 262 (−8.6), 295 nm (+3.0); CD of gel (DMSO:H2O, 1:2, v/v) λext (Δε) 253 nm (−3.8).
(1R,2S)-1,2-Diazidocyclohexane (13): For the synthesis of this compound, (1R,2S)-1,2-diaminocyclohexane and TfN3 were required. (a) The triflyl azide was prepared as follows: Sodium azide (922 mg, 14.2 mmol), dissolved in pyridine (15 mL), was cooled to 0 °C under vigorous stirring. Then, triflic anhydride (1.7 mL, 10.1 mmol) was added dropwise, and the reaction mixture was left for 2 h at 0 °C under vigorous stirring. During that time, a small amount of precipitate appeared, which was removed by filtration. The yellow solution was directly used in the next step. (b) To a solution of (1R,2S)-1,2-diaminocyclohexane (0.42 mL, 3.5 mmol) in 5 mL of pyridine, CuSO4·5H2O (15 mg, 0.06 mmol) was added while stirring. The mixture was cooled in an ice bath and the above-prepared solution of triflyl azide added dropwise. The resulting green reaction mixture was allowed to warm to room temperature and left for 20 h. The reaction mixture was diluted with CH2Cl2 (100 mL) and extracted using diluted HCl until the pH value was acidic (4 × 20 mL). The organic phase was washed with NaHCO3 (2 × 10 mL) and dried with sodium sulfate. Then, the organic solvent was removed at reduced pressure, keeping the temperature below 30 °C. The residue (437 mg, 75%) showed spectroscopic data according to the literature [25]. 1H NMR (600 MHz, CDCl3) δ 3.62 (d, J = 8.4 Hz, 2H), 1.85 (m, 2H), 1.64 (m, 4H), 1.38 ppm (m, 2H); 13C NMR (150 MHz, CDCl3) δ 61.4, 27.3, 21.6 ppm.
(1R,2S)-1,2-Bis(4-(4-bromophenyl)-1H-1,2,3-triazol-1-yl)cyclohexane (14): To a solution of (1R,2S)-1,2-diazidocyclohexane (13, 363 mg, 2.19 mmol) in H2O (22 mL, 22 mmol), 1-bromo-4-ethynylbenzene (913 mg, 5.04 mmol), CuSO4·5H2O (40 mg, 0.16 mmol) as a 1 M aq. solution, and sodium ascorbate (152 mg 0.77 mmol) as a 1 M aq. solution, were added. The reaction mixture was heated to 70 °C for 48 h. Then, the organic layer was extracted with EtOAc (3 × 100 mL) and the solvent removed at reduced pressure. The residue was chromatographed on silica gel using n-hexane/EtOAc to give compound 14 (782 mg, 68% yield). Rf 0.35 (n-hexane/EtOAc, 1:1, v/v); mp 286–288 °C; 1H NMR (500 MHz, CDCl3) δ 7.46–7.42 (m, 8H), 7.18 (s, 2H), 5.18 (m, 2H), 2.57 (m, 2H), 2.34 (m, 2H), 2.19 (m, 2H), 2.77 ppm (m, 2H); 13C NMR (150 MHz, CDCl3) δ 146.2, 131.9, 128.9, 127.1, 122.2, 119.9, 60.8, 28.0, 22.3 ppm; HRESIMS (m/z): [M + Na]+ calcd for C22H2079Br2N6Na, 549.0014; found, 549.0023; [M + Na]+ calcd for C22H2079Br81BrN6Na, 550.9993; found, 551.0006; [M + Na]+ calcd for C22H2081Br2N6Na, 552.9973; found, 552.9977; anal. calcd for C22H20Br2N6, C, 50.02; H, 3.82; N, 15.91%; found, C, 49.67; H, 3.90; N, 16.22%.
Supporting Information
| Supporting Information File 1: An SEM image collection of the xerogels and X-ray data for compound 12. | ||
| Format: PDF | Size: 3.6 MB | Download |
References
-
Babu, S. S.; Praveen, V. K.; Ajayaghosh, A. Chem. Rev. 2014, 114, 1973–2129. doi:10.1021/cr400195e
Return to citation in text: [1] -
Cornwell, D. J.; Smith, D. K. Mater. Horiz. 2015, 2, 279–293. doi:10.1039/c4mh00245h
Return to citation in text: [1] -
Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022
Return to citation in text: [1] [2] -
Lallana, E.; Fernández-Trillo, F.; Sousa-Herves, A.; Riguera, R.; Fernández-Megía, E. Pharm. Res. 2012, 29, 902–921. doi:10.1007/s11095-012-0683-y
Return to citation in text: [1] -
Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f
Return to citation in text: [1] -
Chai, Q.; Jiao, Y.; Yu, X. Gels 2017, 3, No. 6. doi:10.3390/gels3010006
Return to citation in text: [1] -
Mangunuru, H. P. R.; Yerabolu, J. R.; Liu, D.; Wang, G. Tetrahedron Lett. 2015, 56, 82–85. doi:10.1016/j.tetlet.2014.11.013
Return to citation in text: [1] -
Wang, G.; Chen, A.; Mangunuru, H. P. R.; Yerabolu, J. R. RSC Adv. 2017, 7, 40887–40895. doi:10.1039/c7ra06228a
Return to citation in text: [1] -
Okafor, I. S.; Wang, G. Carbohydr. Res. 2017, 451, 81–94. doi:10.1016/j.carres.2017.09.008
Return to citation in text: [1] -
Tautz, M.; Torras, J.; Grijalvo, S.; Eritja, R.; Saldías, C.; Alemán, C.; Díaz, D. D. RSC Adv. 2019, 9, 20841–20851. doi:10.1039/c9ra03316e
Return to citation in text: [1] -
Zhang, H.-Z.; Wei, J.-J.; Kumar, K. V.; Rasheed, S.; Zhou, C.-H. Med. Chem. Res. 2015, 24, 182–196. doi:10.1007/s00044-014-1123-9
Return to citation in text: [1] -
Dedola, S.; Hughes, D. L.; Nepogodiev, S. A.; Rejzek, M.; Field, R. A. Carbohydr. Res. 2010, 345, 1123–1134. doi:10.1016/j.carres.2010.03.041
Return to citation in text: [1] -
Bokor, É.; Docsa, T.; Gergely, P.; Somsák, L. Bioorg. Med. Chem. 2010, 18, 1171–1180. doi:10.1016/j.bmc.2009.12.043
Return to citation in text: [1] -
Goyard, D.; Docsa, T.; Gergely, P.; Praly, J.-P.; Vidal, S. Carbohydr. Res. 2015, 402, 245–251. doi:10.1016/j.carres.2014.10.009
Return to citation in text: [1] [2] -
Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5
Return to citation in text: [1] [2] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4
Return to citation in text: [1] [2] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] [2] -
Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484
Return to citation in text: [1] [2] -
Gottarelli, G.; Lena, S.; Masiero, S.; Pieraccini, S.; Spada, G. P. Chirality 2008, 20, 471–485. doi:10.1002/chir.20459
Return to citation in text: [1] -
Pescitelli, G.; Di Bari, L.; Berova, N. Chem. Soc. Rev. 2014, 43, 5211–5233. doi:10.1039/c4cs00104d
Return to citation in text: [1] -
Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N.; Nakanishi, K.; Woody, R. W., Eds.; Wiley-VCH: Weinheim, 2000.
Return to citation in text: [1] -
Lightner, D. A.; Gurst, J. E. Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy; Wiley-VCH: Weinheim, 2000.
Return to citation in text: [1] -
Pescitelli, G.; Di Bari, L.; Berova, N. Chem. Soc. Rev. 2011, 40, 4603–4625. doi:10.1039/c1cs15036g
Return to citation in text: [1] -
Magnus, P.; Lacour, J.; Evans, P. A.; Roe, M. B.; Hulme, C. J. Am. Chem. Soc. 1996, 118, 3406–3418. doi:10.1021/ja953906r
Return to citation in text: [1]
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 22. | Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N.; Nakanishi, K.; Woody, R. W., Eds.; Wiley-VCH: Weinheim, 2000. |
| 23. | Lightner, D. A.; Gurst, J. E. Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy; Wiley-VCH: Weinheim, 2000. |
| 24. | Pescitelli, G.; Di Bari, L.; Berova, N. Chem. Soc. Rev. 2011, 40, 4603–4625. doi:10.1039/c1cs15036g |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 1. | Babu, S. S.; Praveen, V. K.; Ajayaghosh, A. Chem. Rev. 2014, 114, 1973–2129. doi:10.1021/cr400195e |
| 2. | Cornwell, D. J.; Smith, D. K. Mater. Horiz. 2015, 2, 279–293. doi:10.1039/c4mh00245h |
| 3. | Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 19. | Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484 |
| 11. | Zhang, H.-Z.; Wei, J.-J.; Kumar, K. V.; Rasheed, S.; Zhou, C.-H. Med. Chem. Res. 2015, 24, 182–196. doi:10.1007/s00044-014-1123-9 |
| 12. | Dedola, S.; Hughes, D. L.; Nepogodiev, S. A.; Rejzek, M.; Field, R. A. Carbohydr. Res. 2010, 345, 1123–1134. doi:10.1016/j.carres.2010.03.041 |
| 13. | Bokor, É.; Docsa, T.; Gergely, P.; Somsák, L. Bioorg. Med. Chem. 2010, 18, 1171–1180. doi:10.1016/j.bmc.2009.12.043 |
| 14. | Goyard, D.; Docsa, T.; Gergely, P.; Praly, J.-P.; Vidal, S. Carbohydr. Res. 2015, 402, 245–251. doi:10.1016/j.carres.2014.10.009 |
| 20. | Gottarelli, G.; Lena, S.; Masiero, S.; Pieraccini, S.; Spada, G. P. Chirality 2008, 20, 471–485. doi:10.1002/chir.20459 |
| 21. | Pescitelli, G.; Di Bari, L.; Berova, N. Chem. Soc. Rev. 2014, 43, 5211–5233. doi:10.1039/c4cs00104d |
| 7. | Mangunuru, H. P. R.; Yerabolu, J. R.; Liu, D.; Wang, G. Tetrahedron Lett. 2015, 56, 82–85. doi:10.1016/j.tetlet.2014.11.013 |
| 8. | Wang, G.; Chen, A.; Mangunuru, H. P. R.; Yerabolu, J. R. RSC Adv. 2017, 7, 40887–40895. doi:10.1039/c7ra06228a |
| 9. | Okafor, I. S.; Wang, G. Carbohydr. Res. 2017, 451, 81–94. doi:10.1016/j.carres.2017.09.008 |
| 10. | Tautz, M.; Torras, J.; Grijalvo, S.; Eritja, R.; Saldías, C.; Alemán, C.; Díaz, D. D. RSC Adv. 2019, 9, 20841–20851. doi:10.1039/c9ra03316e |
| 16. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 17. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 18. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 3. | Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022 |
| 4. | Lallana, E.; Fernández-Trillo, F.; Sousa-Herves, A.; Riguera, R.; Fernández-Megía, E. Pharm. Res. 2012, 29, 902–921. doi:10.1007/s11095-012-0683-y |
| 5. | Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905–4979. doi:10.1021/cr200409f |
| 6. | Chai, Q.; Jiao, Y.; Yu, X. Gels 2017, 3, No. 6. doi:10.3390/gels3010006 |
| 19. | Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 14. | Goyard, D.; Docsa, T.; Gergely, P.; Praly, J.-P.; Vidal, S. Carbohydr. Res. 2015, 402, 245–251. doi:10.1016/j.carres.2014.10.009 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 16. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 17. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 18. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 15. | Tejera, S.; Dorta, R. L.; Vázquez, J. T. Tetrahedron: Asymmetry 2016, 27, 896–909. doi:10.1016/j.tetasy.2016.07.008 |
| 25. | Magnus, P.; Lacour, J.; Evans, P. A.; Roe, M. B.; Hulme, C. J. Am. Chem. Soc. 1996, 118, 3406–3418. doi:10.1021/ja953906r |
© 2019 Tejera et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)