Abstract
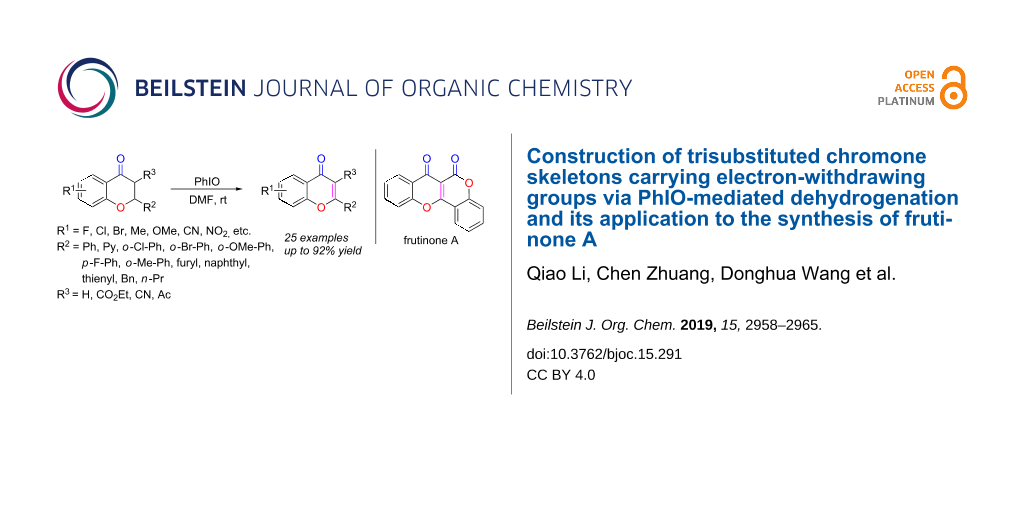
The construction of the biologically interesting chromone skeleton was realized by PhIO-mediated dehydrogenation of chromanones under mild conditions. Interestingly, this method also found application in the synthesis of the naturally occurring frutinone A.

Graphical Abstract
Introduction
The chromone system and its derivatives are an important class of heterocyclic compounds, the skeleton of which widely exists in a variety of natural products and medicinal agents [1-3]. It was found that chromone derivatives exhibit a wide range of pharmacological effects, including antibacterical [4], antifungal [5,6], anticancer [7], antioxidant [8], anti-HIV [9], antiulcer, immunostimulator [10], anti-inflammatory [11], as well as biocidal [12], wound-healing [13], and immune-stimulatory activities [14]. For instance, flavoxate [15,16] is a chromone derivative that was employed as an anticholinergic agent for its antimuscarinic effects [3,17]; apigenin can function as an antiviral drug for the treatment of HIV [18,19], cancer [20-22], and other viral infections [23]; pranlukast [24] can be used in the treatment of allergic rhinitis [25] and asthma [26]; and khellin [27] has been proved to possess antiviral and antispasmodic effects (Figure 1) [28]. All of these pharmaceutical agents bear a chromone framework in their respective chemical structure.
![[1860-5397-15-291-1]](/bjoc/content/figures/1860-5397-15-291-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active chromone derivatives.
Figure 1: Biologically active chromone derivatives.
Since the chromone derivatives have a variety of biological activities, developing synthetic methods for efficient construction of the chromone skeleton has been a research field of great interest and long history [2]. Among the numerous synthetic approaches [29-54], the dehydrogenative oxidation of the readily available chromanones is a highly efficient method for the synthesis of this privileged class of heterocycles. For example, Kim and co-workers reported that chromones could be prepared via a Pd(II)-catalyzed [55-57] dehydrogenation of chromanone derivatives at 100 °C in DMSO (Scheme 1a) [58]. The synthesis of chromones could also be realized by DDQ-mediated dehydrogenation of chromanones under heating in dioxane (Scheme 1b) [3,59,60]. In 2005, Yang and co-workers reported that chromones could be formed by microwave irradiation of the corresponding chromanone reactants and N-bromosuccinimide (NBS) in the presence of a catalytic amount of azobis(isobutyronitrile) (AIBN) in CCl4 (Scheme 1c) [61,62]. In 2002, Nicolaou and co-workers found that ortho-iodoxybenzoic acid (IBX) could also effectively dehydrogenate chromanones to chromones (Scheme 1d, method 1) [63]. Moreover, active MnO2 was also found useful in the oxidative dehydrogenation of chromanones at a relatively high temperature of 110 °C (Scheme 1d, method 2) [64,65]. Although all of the above methods have their respective merits in the preparation of the corresponding chromone derivatives, it is obvious that some of them suffer from drawbacks, such as the requirement of a high reaction temperature, extended reaction time, involvement of transition metal catalysts, and low yield. In these regards, the development of alternative approaches that can realize an efficient synthesis of chromones under mild conditions is desirable.
![[1860-5397-15-291-i1]](/bjoc/content/inline/1860-5397-15-291-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Methods for the synthesis of chromones via dehydrogenative oxidation of chromanones.
Scheme 1: Methods for the synthesis of chromones via dehydrogenative oxidation of chromanones.
In recent decades, hypervalent iodine reagents have emerged as a class of efficient and environmentally benign nonmetal “green” oxidants [66-73]. For instance, iodosobenzene (PhIO) [74] has been widely used in many synthetic transformations. It was found that PhIO is efficient in realizing epoxidation of olefins [75-77], converting alkynes and alkenes to ketones [78], oxidizing alcohols to aldehydes [79,80], as well as in the direct α-hydroxylation of ketones [81]. Furthermore, it could also be used to realize oxidative C–C [82], C–N [83], and C–O [84] bond formations. However, to the best of our knowledge, PhIO has never been utilized for the dehydrogenative oxidation reaction. In this letter, we report a facile PhIO-mediated dehydrogenation of chromanones, resulting in the efficient synthesis of biologically interesting chromone compounds under metal-free conditions.
Results and Discussion
We initially studied the feasibility of converting chromanone ethyl 4-oxo-2-phenylchromane-3-carboxylate (1a) to chromone 2a via PhIO-mediated dehydrogenation. To our delight, when 1a was treated with PhIO in DCE at room temperature, the desired product 2a could be obtained in 66% yield (Table 1, entry 1), with the generation of some unidentified byproducts. A solvent screening identified DMF to be the most appropriate solvent for this transformation (Table 1, entries 1–9). Other commonly employed oxidants, including phenyliodine(III) diacetate (PIDA), phenyliodine(III) bis(trifluoroacetate) (PIFA), and iodylbenzene (PhIO2), were found to be less efficient for this transformation, as the desired product 2a was generated in significantly lower yield in each case (Table 1, entries 10–12). Further studies indicated that the reaction gave the best result if performed at room temperature, while reduced or elevated temperature (0 or 50 °C) was not beneficial for the reaction (Table 1, entries 13 and 14). In addition, the amount of PhIO was proved to be critical for this transformation, as a decreased yield of the desired product, accompanied with more byproducts, was observed when increasing the PhIO amount from 2.0 to 3.0 mmol. When the amount of PhIO was reduced to 1.0 mmol, the yield also decreased due to the fact that the starting material could not be completely consumed (Table 1, entries 15 and 16). Based on the above screening results, the optimal reaction conditions were concluded to be 1.0 mmol of substrate, 2.0 mmol of PhIO, DMF as solvent, and 10 minutes reaction time at room temperature.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-291-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | oxidant | solvent | T (°C) | yield (%)b |
| 1 | PhIO | DCE | rt | 66 |
| 2 | PhIO | THF | rt | 57 |
| 3 | PhIO | toluene | rt | 42 |
| 4 | PhIO | 1,4-dioxane | rt | 33 |
| 5 | PhIO | DMSO | rt | 53 |
| 6 | PhIO | DMF | rt | 89 |
| 7 | PhIO | MeOH | rt | 33 |
| 8 | PhIO | MeCN | rt | 37 |
| 9 | PhIO | EtOAc | rt | 35 |
| 10 | PIDA | DMF | rt | 62 |
| 11 | PIFA | DMF | rt | 21 |
| 12 | PhIO2 | DMF | rt | 46 |
| 13 | PhIO | DMF | 0 | 38 |
| 14 | PhIO | DMF | 50 | 65 |
| 15c | PhIO | DMF | rt | 64 |
| 16d | PhIO | DMF | rt | 78 |
aReaction conditions: 1a (1.0 mmol), oxidant (2.0 mmol), solvent (6 mL). bIsolated yield. c1a (1.0 mmol), PhIO (1.0 mmol), DMF (6 mL). d1a (1.0 mmol), PhIO (3.0 mmol), DMF (6 mL), 10 min.
With the optimized conditions in hand, we explored the scope and generality of the newly established method. As shown in Scheme 2, a wide range of chromanones could be well tolerated under the standard conditions, affording the expected products 2a–x in satisfactory to good yields. Substrates bearing various R1 substituents (F, Cl, Br, CN, NO2, Me, OMe) were all efficiently converted to the corresponding products 2a–i, with the substrates carrying electron-donating groups affording the desired products 2h and 2i in noticeably higher yields than those with electron-withdrawing groups, 2b–g. The product 2g, with an electron-withdrawing group in ortho position, was formed in a much lower yield, most likely due to steric hindrance. When the phenyl group was replaced by a naphthyl group, less of the corresponding substrate was converted to the desired product 2j. The substrates bearing various R2 substituents, including electron-withdrawing, electron-donating, sterically hindered, and heterocyclic groups, all reacted smoothly and afforded the desired chromone derivatives 2k–v in acceptable to good yields. Furthermore, when the ethoxycarbonyl group R3 was replaced by an acetyl substituent, the resulting substrate was successfully converted to 2w with a high yield. However, when the substrate bore a strong electron-withdrawing cyano group, the reaction was less efficient. and the product 2x was produced in a much lower yield. When the electron-withdrawing group (R3) in substrate 1 was replaced by hydrogen, the corresponding product 2y was obtained in a relatively low yield. To our disappointment, the method was not applicable to the synthesis of 4-chromones, as the reaction of 4-chromanones did not occur under the standard conditions (2z). But when R3 was a carbonyl group and R2 was a hydrogen atom, the corresponding compound 1aa could also be converted to the desired product with 35% yield. On the basis of these results, we tentatively proposed that the failure of 1z formation might have been caused by the absence of a carbonyl group in its chemical structure [85].
![[1860-5397-15-291-i2]](/bjoc/content/inline/1860-5397-15-291-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope studies. Reaction conditions: 1 (1.0 mmol), PhIO (2.0 mmol), DMF (6 mL), rt. Isolated yields are given.
Scheme 2: Substrate scope studies. Reaction conditions: 1 (1.0 mmol), PhIO (2.0 mmol), DMF (6 mL), rt. Isolat...
Control experiments were designed and conducted to elucidate the reaction mechanism of this transformation. When 3 equiv of TEMPO, a radical-trapping reagent, were added to the reaction mixture, product 2a was obtained in 55% yield (Scheme 3, method a). When another radical-trapping reagent, BHT, was used, the reaction gave 50% yield of the desired product 2a (Scheme 3, method b). Since the reaction was not greatly suppressed in both cases, we tentatively propose that this dehydrogenative oxidation reaction may undergo both radical and nonradical pathways.
![[1860-5397-15-291-i3]](/bjoc/content/inline/1860-5397-15-291-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Control experiments for mechanistic studies.
Scheme 3: Control experiments for mechanistic studies.
Based on previous reports [63,86] and our own experimental results, two plausible mechanisms for this dehydrogenation reaction are proposed (Scheme 4): In pathway a, substrate 1a is first tautomerized to its enol form A, which would be stabilized by an internal H-bond. It is highly likely that the enolization is a necessary process for the reaction to occur, as compound 2z could not undergo the transformation under the standard conditions. Then, nucleophilic attack on the iodine center of PhIO [87] by the enol moiety of intermediate A affords the O–I enol form C (via intermediate B), which is subsequently converted to I–C intermediate D via radical migration [88]. The homogeneous cleavage of the C–I bond in D leads to the stable carbon radical E and an iodine radical. Finally, the reaction between E and the iodine radical produces product 2a, with the concomitant release of water and iodobenzene. In pathway b, the O–I intermediate C is converted to a C–I intermediate D via 1,3-migration [89]. Then, intermediate D carries through a five-membered ring transition state F to afford the title product 2a, accompanied by the release of iodobenzene and water.
![[1860-5397-15-291-i4]](/bjoc/content/inline/1860-5397-15-291-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
One practical application of the obtained chromone derivatives was their conversion to chromone-derived natural products. Frutinone A, isolated from the leaves and root bark of Polygala fruticosa, shows various biological activities, including antibacterial, antioxidant, and potent cytochrome P450 1A2 inhibition (CYP1A2, IC50 = 5.3 nM) properties [90-92]. Treating the obtained chromene-3-carboxylate 2l with LiOH [93] led to the formation of the chromene-3-carboxylic acid G. Heating compound G overnight in the presence of AgNO3 and K2S2O8 afforded frutinone A in an isolated yield of 45% (Scheme 5).
![[1860-5397-15-291-i5]](/bjoc/content/inline/1860-5397-15-291-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Application of the reported method to the synthesis of frutinone A.
Scheme 5: Application of the reported method to the synthesis of frutinone A.
Conclusion
In summary, we have developed an efficient metal-free approach for the synthesis of chromone derivatives via PhIO-mediated dehydrogenative oxidation of chromanones. Compared with the existing methods, the new reaction features mild conditions, high efficiency, and is metal-free. Moreover, the product 2l could be further applied to the synthesis of the naturally occurring frutinone A.
Supporting Information
| Supporting Information File 1: Synthetic details and compound characterization data. | ||
| Format: PDF | Size: 7.3 MB | Download |
References
-
Gaspar, A.; Matos, M. J.; Garrido, J.; Uriarte, E.; Borges, F. Chem. Rev. 2014, 114, 4960–4992. doi:10.1021/cr400265z
Return to citation in text: [1] -
Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s
Return to citation in text: [1] [2] -
Keri, R. S.; Budagumpi, S.; Pai, R. K.; Balakrishna, R. G. Eur. J. Med. Chem. 2014, 78, 340–374. doi:10.1016/j.ejmech.2014.03.047
Return to citation in text: [1] [2] [3] -
Li, X.; Xu, L.; Cui, Y. Q.; Pang, M.; Wang, F.; Qi, J. IOP Conf. Ser.: Mater. Sci. Eng. 2017, 275, No. 012013. doi:10.1088/1757-899x/275/1/012013
Return to citation in text: [1] -
Jovanovic, S. V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M. G. J. Am. Chem. Soc. 1994, 116, 4846–4851. doi:10.1021/ja00090a032
Return to citation in text: [1] -
Grindlay, D.; Reynolds, T. J. Ethnopharmacol. 1986, 16, 117–151. doi:10.1016/0378-8741(86)90085-1
Return to citation in text: [1] -
Martens, S.; Mithöfer, A. Phytochemistry 2005, 66, 2399–2407. doi:10.1016/j.phytochem.2005.07.013
Return to citation in text: [1] -
Kuroda, M.; Uchida, S.; Watanabe, K.; Mimaki, Y. Phytochemistry 2009, 70, 288–293. doi:10.1016/j.phytochem.2008.12.002
Return to citation in text: [1] -
Zhou, T.; Shi, Q.; Lee, K. H. Tetrahedron Lett. 2010, 51, 4382–4386. doi:10.1016/j.tetlet.2010.06.058
Return to citation in text: [1] -
Gamal-Eldeen, A. M.; Djemgou, P. C.; Tchuendem, M.; Ngadjui, B. T.; Tane, P.; Toshifumi, H. Z. Naturforsch., C: J. Biosci. 2007, 62, 331–338. doi:10.1515/znc-2007-5-622
Return to citation in text: [1] -
Crombie, A. L. Br. J. Ophthalmol. 1989, 73, 79. doi:10.1136/bjo.73.1.79-b
Return to citation in text: [1] -
Binbuga, N.; Ruhs, C.; Hasty, J. K.; Henry, W. P.; Schultz, T. P. Holzforschung 2008, 62, 264–269. doi:10.1515/hf.2008.038
Return to citation in text: [1] -
Sumiyoshi, M.; Kimura, Y. Phytomedicine 2010, 17, 820–829. doi:10.1016/j.phymed.2010.01.010
Return to citation in text: [1] -
Fu, Z.; Sun, W. C.; Luan, X. C.; Gao, J. X.; Zhang, Z. H. J. J. Med. Univ. 2018, 39, 88–92.
Return to citation in text: [1] -
Asimakopoulos, A. D.; Cerruto, M. A.; Del Popolo, G.; La Martina, M.; Artibani, W.; Carone, R.; Finazzi-Agrò, E. Urol. Int. 2012, 89, 259–269. doi:10.1159/000339600
Return to citation in text: [1] -
Yamaguchi, O.; Nishizawa, O.; Takeda, M.; Yokoyama, O.; Homma, Y.; Kakizaki, H.; Obara, K.; Gotoh, M.; Igawa, Y.; Seki, N.; Yoshida, M. Int. J. Urol. 2009, 16, 126–142. doi:10.1111/j.1442-2042.2008.02177.x
Return to citation in text: [1] -
Tang, H. C.; Lam, W. P.; Zhang, X.; Leung, P. C.; Yew, D. T.; Liang, W. LUTS: Lower Urinary Tract Symptoms 2015, 7, 149–154. doi:10.1111/luts.12063
Return to citation in text: [1] -
Chinsembu, K. C. Braz. J. Pharm. 2019, 572–596.
Return to citation in text: [1] -
Li, H.-M.; Zhou, C.; Chen, C.-H.; Li, R.-T.; Lee, K.-H. Helv. Chim. Acta 2015, 98, 1177–1187. doi:10.1002/hlca.201500123
Return to citation in text: [1] -
Shukla, S.; Gupta, S. Pharm. Res. 2010, 27, 962–978. doi:10.1007/s11095-010-0089-7
Return to citation in text: [1] -
Li, Y.; Yang, B.; Bai, J.-y.; Xia, S.; Mao, M.; Li, X.; Li, N.; Chen, L. Int. Immunopharmacol. 2019, 73, 362–369. doi:10.1016/j.intimp.2019.05.024
Return to citation in text: [1] -
Erdogan, S.; Turkekul, K.; Dibirdik, I.; Doganlar, Z. B.; Doganlar, O.; Bilir, A. Invest. New Drugs 2019. doi:10.1007/s10637-019-00774-8
Return to citation in text: [1] -
Zhou, Q.; Cheng, K.-W.; Gong, J.; Li, E. T. S.; Wang, M. Biochem. Pharmacol. 2019, 166, 231–241. doi:10.1016/j.bcp.2019.05.027
Return to citation in text: [1] -
Ha, E.-S.; Kim, J.-S.; Kuk, D.-H.; Ha, D.-H.; Baek, I.-h.; Kim, M.-S. J. Mol. Liq. 2017, 225, 231–234. doi:10.1016/j.molliq.2016.11.082
Return to citation in text: [1] -
Huang, F. C.; Galemmo, R. A.; Poli, G. B.; Learn, K. S.; Morrissette, M. M.; Johnson, W. H.; Dankulich, W. P.; Campbell, H. F.; Carnathan, G. W.; Van Inwegen, R. G. J. Med. Chem. 1991, 34, 1704–1707. doi:10.1021/jm00109a025
Return to citation in text: [1] -
Darmani, N. A.; Chebolu, S.; Zhong, W.; Kim, W. D.; Narlesky, M.; Adams, J.; Dong, F. Eur. J. Pharmacol. 2017, 809, 20–31. doi:10.1016/j.ejphar.2017.05.014
Return to citation in text: [1] -
Sharma, R.; Williams, I. S.; Gatchie, L.; Sonawane, V. R.; Chaudhuri, B.; Bharate, S. B. ACS Omega 2018, 3, 8553–8566. doi:10.1021/acsomega.8b01088
Return to citation in text: [1] -
Machado, N. F. L.; Marques, M. P. M. Curr. Bioact. Compd. 2010, 6, 76–89. doi:10.2174/157340710791184859
Return to citation in text: [1] -
Ellis, G. P. General Methods of Preparing Chromones. In Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; Ellis, G. P., Ed.; John Wiley & Sons: Hoboken, NJ, 1977; Vol. 31, pp 495–555. doi:10.1002/9780470187012.ch9
Return to citation in text: [1] -
Singh, O. V.; Kapoor, R. P. Tetrahedron Lett. 1990, 31, 1459–1462. doi:10.1016/s0040-4039(00)88832-2
Return to citation in text: [1] -
Ciattini, P. G.; Morera, E.; Ortar, G. J. Heterocycl. Chem. 1982, 19, 395–400. doi:10.1002/jhet.5570190236
Return to citation in text: [1] -
Löwe, W.; Matzanke, N. J. Heterocycl. Chem. 1996, 33, 943–948. doi:10.1002/jhet.5570330364
Return to citation in text: [1] -
Turcas, R.; Kripli, B.; Attia, A. A. A.; Lakk-Bogáth, D.; Speier, G.; Giorgi, M.; Silaghi-Dumitrescu, R.; Kaizer, J. Dalton Trans. 2018, 47, 14416–14420. doi:10.1039/c8dt03519a
Return to citation in text: [1] -
Yoshii, D.; Jin, X.; Yatabe, T.; Hasegawa, J.-y.; Yamaguchi, K.; Mizuno, N. Chem. Commun. 2016, 52, 14314–14317. doi:10.1039/c6cc07846j
Return to citation in text: [1] -
Patonay, T.; Vasas, A.; Kiss-Szikszai, A.; Silva, A. M. S.; Cavaleiro, J. A. S. Aust. J. Chem. 2010, 63, 1582–1593. doi:10.1071/ch10295
Return to citation in text: [1] -
Bovicelli, P.; D'Angelo, V.; Collalto, D.; Verzina, A.; D'Antona, N.; Lambusta, D. J. Pharm. Pharmacol. 2007, 59, 1697–1701. doi:10.1211/jpp.59.12.0012
Return to citation in text: [1] -
Zacuto, M. J.; Cai, D. Tetrahedron Lett. 2005, 46, 447–450. doi:10.1016/j.tetlet.2004.11.092
Return to citation in text: [1] -
Muthukrishnan, M.; Patil, P. S.; More, S. V.; Joshi, R. A. Mendeleev Commun. 2005, 15, 100–101. doi:10.1070/mc2005v015n03abeh001997
Return to citation in text: [1] -
Prakash, O.; Pahuja, S.; Moriarty, R. M. Synth. Commun. 1990, 20, 1417–1422. doi:10.1080/00397919008052857
Return to citation in text: [1] -
Harvey, R. G.; Hahn, J. T.; Bukowska, M.; Jackson, H. J. Org. Chem. 1990, 55, 6161–6166. doi:10.1021/jo00312a023
Return to citation in text: [1] -
Takeno, N.; Fukushima, T.; Takeda, S.-i.; Kishimoto, K. Bull. Chem. Soc. Jpn. 1985, 58, 1599–1600. doi:10.1246/bcsj.58.1599
Return to citation in text: [1] -
Wu, L.-L.; Tang, L.; Zhou, S.-G.; Peng, Y.-J.; He, X.-D.; Guan, Z.; He, Y.-H. Tetrahedron 2017, 73, 6471–6478. doi:10.1016/j.tet.2017.09.050
Return to citation in text: [1] -
Patonay, T.; Lévai, A.; Nemes, C.; Timár, T.; Tóth, G.; Adam, W. J. Org. Chem. 1996, 61, 5375–5383. doi:10.1021/jo960163z
Return to citation in text: [1] -
Mal, K.; Kaur, A.; Haque, F.; Das, I. J. Org. Chem. 2015, 80, 6400–6410. doi:10.1021/acs.joc.5b00846
Return to citation in text: [1] -
Thomsen, I.; Torssell, K. B. G. Acta Chem. Scand., Ser. B 1988, 42, 303–308. doi:10.3891/acta.chem.scand.42b-0303
Return to citation in text: [1] -
Litkei, G.; Patonay, T.; Szilágyi, L.; Dinya, Z. Org. Prep. Proced. Int. 1991, 23, 741–747. doi:10.1080/00304949109458249
Return to citation in text: [1] -
Zanarotti, A. Heterocycles 1982, 19, 1585–1586. doi:10.3987/r-1982-09-1585
Return to citation in text: [1] -
Prakash, O.; Mendiratta, S. Synth. Commun. 1992, 22, 327–334. doi:10.1080/00397919208021310
Return to citation in text: [1] -
Somogyi, L. Synth. Commun. 1999, 29, 1857–1872. doi:10.1080/00397919908086175
Return to citation in text: [1] -
Marié, J.-C.; Xiong, Y.; Min, G. K.; Yeager, A. R.; Taniguchi, T.; Berova, N.; Schaus, S. E.; Porco, J. A., Jr. J. Org. Chem. 2010, 75, 4584–4590. doi:10.1021/jo100889c
Return to citation in text: [1] -
Kumar, D.; Singh, O. V.; Prakash, O.; Singh, S. P. Synth. Commun. 1994, 24, 2637–2644. doi:10.1080/00397919408010576
Return to citation in text: [1] -
Prakash, O.; Tanwar, M. P. Bull. Chem. Soc. Jpn. 1995, 68, 1168–1171. doi:10.1246/bcsj.68.1168
Return to citation in text: [1] -
Lambaa, M.; Makrandi, J. K. J. Chem. Res. 2008, 225–226. doi:10.3184/030823408x313591
Return to citation in text: [1] -
Varma, R. S.; Varma, M. Synth. Commun. 1982, 12, 927–930. doi:10.1080/00397918208061928
Return to citation in text: [1] -
Pan, G.-F.; Zhu, X.-Q.; Guo, R.-L.; Gao, Y.-R.; Wang, Y.-Q. Adv. Synth. Catal. 2018, 360, 4774–4783. doi:10.1002/adsc.201801058
Return to citation in text: [1] -
Chen, M.; Rago, A. J.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 16205–16209. doi:10.1002/anie.201811197
Return to citation in text: [1] -
Diao, T.; Stahl, S. S. J. Am. Chem. Soc. 2011, 133, 14566–14569. doi:10.1021/ja206575j
Return to citation in text: [1] -
Lee, J.; Yu, J.; Son, S. H.; Heo, J.; Kim, T.; An, J.-Y.; Inn, K.-S.; Kim, N.-J. Org. Biomol. Chem. 2016, 14, 777–784. doi:10.1039/c5ob01911g
Return to citation in text: [1] -
Shanker, C. G.; Mallaiah, B. V.; Srimannarayana, G. Synthesis 1983, 310–311. doi:10.1055/s-1983-30317
Return to citation in text: [1] -
Hoshino, Y.; Takeno, N. Bull. Chem. Soc. Jpn. 1987, 60, 4468–4470. doi:10.1246/bcsj.60.4468
Return to citation in text: [1] -
Zhou, Z.; Zhao, P.; Huang, W.; Yang, G. Adv. Synth. Catal. 2006, 348, 63–67. doi:10.1002/adsc.200505223
Return to citation in text: [1] -
Vaz, P. A. A. M.; Pinto, D. C. G. A.; Rocha, D. H. A.; Silva, A. M. S.; Cavaleiro, J. A. S. Synlett 2012, 23, 2353–2356. doi:10.1055/s-0032-1317159
Return to citation in text: [1] -
Nicolaou, K. C.; Montagnon, T.; Baran, P. S.; Zhong, Y.-L. J. Am. Chem. Soc. 2002, 124, 2245–2258. doi:10.1021/ja012127+
Return to citation in text: [1] [2] -
Mashraqui, S.; Keehn, P. Synth. Commun. 2006, 12, 637–645. doi:10.1080/13504620601054090
Return to citation in text: [1] -
Singh, O. V.; Muthukrishnan, M.; Raj, G. Synth. Commun. 2005, 35, 2723–2728. doi:10.1080/00397910500214490
Return to citation in text: [1] -
Gayen, K. S.; Chatterjee, N.; Khamarui, S.; Tarafdar, P. K. Eur. J. Org. Chem. 2018, 425–439. doi:10.1002/ejoc.201701306
Return to citation in text: [1] -
Narayan, R.; Manna, S.; Antonchick, A. P. Synlett 2015, 26, 1785–1803. doi:10.1055/s-0034-1379912
Return to citation in text: [1] -
Minatti, A. Synlett 2003, 140–141. doi:10.1055/s-2003-36242
Return to citation in text: [1] -
Yoshimura, A.; Jarvi, M. E.; Shea, M. T.; Makitalo, C. L.; Rohde, G. T.; Yusubov, M. S.; Saito, A.; Zhdankin, V. V. Eur. J. Org. Chem. 2019, 6682–6689. doi:10.1002/ejoc.201901258
Return to citation in text: [1] -
Xing, Q.; Liang, H.; Bao, M.; Li, X.; Zhang, J.; Bi, T.; Zhang, Y.; Xu, J.; Du, Y.; Zhao, K. Adv. Synth. Catal. 2019, 361, 4669–4673. doi:10.1002/adsc.201900652
Return to citation in text: [1] -
Zhdankin, V. V.; Protasiewicz, J. D. Coord. Chem. Rev. 2014, 275, 54–62. doi:10.1016/j.ccr.2014.04.007
Return to citation in text: [1] -
Reddy Kandimalla, S.; Parvathaneni, S. P.; Sabitha, G.; Subba Reddy, B. V. Eur. J. Org. Chem. 2019, 1687–1714. doi:10.1002/ejoc.201801469
Return to citation in text: [1] -
Kita, Y.; Dohi, T. Chem. Rec. 2015, 15, 886–906. doi:10.1002/tcr.201500020
Return to citation in text: [1] -
Saltzman, H.; Sharefkin, J. G. Org. Synth. 1963, 43, 62–64. doi:10.15227/orgsyn.043.0062
Return to citation in text: [1] -
Yang, Y.; Diederich, F.; Valentine, J. S. J. Am. Chem. Soc. 1990, 112, 7826–7828. doi:10.1021/ja00177a071
Return to citation in text: [1] -
Engelmann, X.; Malik, D. D.; Corona, T.; Warm, K.; Farquhar, E. R.; Swart, M.; Nam, W.; Ray, K. Angew. Chem., Int. Ed. 2019, 58, 4012–4016. doi:10.1002/anie.201812758
Return to citation in text: [1] -
Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. ACS Catal. 2018, 8, 4528–4538. doi:10.1021/acscatal.8b00874
Return to citation in text: [1] -
Wei, Y.; Zheng, X.; Xie, X. Chem. Res. Chin. Univ. 2019, 35, 26–32. doi:10.1007/s40242-019-8264-9
Return to citation in text: [1] -
Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328–337. doi:10.1002/1615-4169(200206)344:3/4<328::aid-adsc328>3.0.co;2-s
Return to citation in text: [1] -
Adam, W.; Herold, M.; Hill, C. L.; Saha-Möller, C. R. Eur. J. Org. Chem. 2002, 941–946. doi:10.1002/1099-0690(200203)2002:5<941::aid-ejoc941>3.0.co;2-a
Return to citation in text: [1] -
Moriarty, R. M.; Hu, H.; Gupta, S. C. Tetrahedron Lett. 1981, 22, 1283–1286. doi:10.1016/s0040-4039(01)90297-7
Return to citation in text: [1] -
Zhen, X.; Wan, X.; Zhang, W.; Li, Q.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2019, 21, 890–894. doi:10.1021/acs.orglett.8b03741
Return to citation in text: [1] -
Sun, X.; Lyu, Y.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Org. Lett. 2013, 15, 6222–6225. doi:10.1021/ol4030716
Return to citation in text: [1] -
Liu, X.; Cheng, R.; Zhao, F.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Org. Lett. 2012, 14, 5480–5483. doi:10.1021/ol3025583
Return to citation in text: [1] -
We also investigated the substrate with R2 = R3 = Me but failed to achieve any successful result.
Return to citation in text: [1] -
Cui, L.-Q.; Dong, Z.-L.; Liu, K.; Zhang, C. Org. Lett. 2011, 13, 6488–6491. doi:10.1021/ol202777h
Return to citation in text: [1] -
We tentatively propose that polymeric (PhIO)n would undergo depolymerization to give PhIO species in DMF, as the HRMS study of a solution of (PhIO)n in DMF implied the existence of an m/z signal for [M + Na]+ for PhIO (see Supporting Information File 1 for HRMS spectra).
Return to citation in text: [1] -
Fan, H.; Pan, P.; Zhang, Y.; Wang, W. Org. Lett. 2018, 20, 7929–7932. doi:10.1021/acs.orglett.8b03503
Return to citation in text: [1] -
Sreenithya, A.; Sunoj, R. B. Org. Lett. 2014, 16, 6224–6227. doi:10.1021/ol503161g
Return to citation in text: [1] -
Shin, Y.; Yoo, C.; Moon, Y.; Lee, Y.; Hong, S. Chem. – Asian J. 2015, 10, 878–881. doi:10.1002/asia.201402876
Return to citation in text: [1] -
Di Paolo, E. R.; Hamburger, M. O.; Stoeckli-Evans, H.; Rogers, C.; Hostettmann, K. Helv. Chim. Acta 1989, 72, 1455–1462. doi:10.1002/hlca.19890720705
Return to citation in text: [1] -
Thelingwani, R. S.; Dhansay, K.; Smith, P.; Chibale, K.; Masimirembwa, C. M. Xenobiotica 2012, 42, 989–1000. doi:10.3109/00498254.2012.681077
Return to citation in text: [1] -
Boonsombat, J.; Thongnest, S.; Ruchirawat, S. Eur. J. Org. Chem. 2019, 2971–2983. doi:10.1002/ejoc.201900078
Return to citation in text: [1]
| 63. | Nicolaou, K. C.; Montagnon, T.; Baran, P. S.; Zhong, Y.-L. J. Am. Chem. Soc. 2002, 124, 2245–2258. doi:10.1021/ja012127+ |
| 64. | Mashraqui, S.; Keehn, P. Synth. Commun. 2006, 12, 637–645. doi:10.1080/13504620601054090 |
| 65. | Singh, O. V.; Muthukrishnan, M.; Raj, G. Synth. Commun. 2005, 35, 2723–2728. doi:10.1080/00397910500214490 |
| 66. | Gayen, K. S.; Chatterjee, N.; Khamarui, S.; Tarafdar, P. K. Eur. J. Org. Chem. 2018, 425–439. doi:10.1002/ejoc.201701306 |
| 67. | Narayan, R.; Manna, S.; Antonchick, A. P. Synlett 2015, 26, 1785–1803. doi:10.1055/s-0034-1379912 |
| 68. | Minatti, A. Synlett 2003, 140–141. doi:10.1055/s-2003-36242 |
| 69. | Yoshimura, A.; Jarvi, M. E.; Shea, M. T.; Makitalo, C. L.; Rohde, G. T.; Yusubov, M. S.; Saito, A.; Zhdankin, V. V. Eur. J. Org. Chem. 2019, 6682–6689. doi:10.1002/ejoc.201901258 |
| 70. | Xing, Q.; Liang, H.; Bao, M.; Li, X.; Zhang, J.; Bi, T.; Zhang, Y.; Xu, J.; Du, Y.; Zhao, K. Adv. Synth. Catal. 2019, 361, 4669–4673. doi:10.1002/adsc.201900652 |
| 71. | Zhdankin, V. V.; Protasiewicz, J. D. Coord. Chem. Rev. 2014, 275, 54–62. doi:10.1016/j.ccr.2014.04.007 |
| 72. | Reddy Kandimalla, S.; Parvathaneni, S. P.; Sabitha, G.; Subba Reddy, B. V. Eur. J. Org. Chem. 2019, 1687–1714. doi:10.1002/ejoc.201801469 |
| 73. | Kita, Y.; Dohi, T. Chem. Rec. 2015, 15, 886–906. doi:10.1002/tcr.201500020 |
| 1. | Gaspar, A.; Matos, M. J.; Garrido, J.; Uriarte, E.; Borges, F. Chem. Rev. 2014, 114, 4960–4992. doi:10.1021/cr400265z |
| 2. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 3. | Keri, R. S.; Budagumpi, S.; Pai, R. K.; Balakrishna, R. G. Eur. J. Med. Chem. 2014, 78, 340–374. doi:10.1016/j.ejmech.2014.03.047 |
| 8. | Kuroda, M.; Uchida, S.; Watanabe, K.; Mimaki, Y. Phytochemistry 2009, 70, 288–293. doi:10.1016/j.phytochem.2008.12.002 |
| 20. | Shukla, S.; Gupta, S. Pharm. Res. 2010, 27, 962–978. doi:10.1007/s11095-010-0089-7 |
| 21. | Li, Y.; Yang, B.; Bai, J.-y.; Xia, S.; Mao, M.; Li, X.; Li, N.; Chen, L. Int. Immunopharmacol. 2019, 73, 362–369. doi:10.1016/j.intimp.2019.05.024 |
| 22. | Erdogan, S.; Turkekul, K.; Dibirdik, I.; Doganlar, Z. B.; Doganlar, O.; Bilir, A. Invest. New Drugs 2019. doi:10.1007/s10637-019-00774-8 |
| 83. | Sun, X.; Lyu, Y.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Org. Lett. 2013, 15, 6222–6225. doi:10.1021/ol4030716 |
| 7. | Martens, S.; Mithöfer, A. Phytochemistry 2005, 66, 2399–2407. doi:10.1016/j.phytochem.2005.07.013 |
| 23. | Zhou, Q.; Cheng, K.-W.; Gong, J.; Li, E. T. S.; Wang, M. Biochem. Pharmacol. 2019, 166, 231–241. doi:10.1016/j.bcp.2019.05.027 |
| 84. | Liu, X.; Cheng, R.; Zhao, F.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Org. Lett. 2012, 14, 5480–5483. doi:10.1021/ol3025583 |
| 5. | Jovanovic, S. V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M. G. J. Am. Chem. Soc. 1994, 116, 4846–4851. doi:10.1021/ja00090a032 |
| 6. | Grindlay, D.; Reynolds, T. J. Ethnopharmacol. 1986, 16, 117–151. doi:10.1016/0378-8741(86)90085-1 |
| 3. | Keri, R. S.; Budagumpi, S.; Pai, R. K.; Balakrishna, R. G. Eur. J. Med. Chem. 2014, 78, 340–374. doi:10.1016/j.ejmech.2014.03.047 |
| 17. | Tang, H. C.; Lam, W. P.; Zhang, X.; Leung, P. C.; Yew, D. T.; Liang, W. LUTS: Lower Urinary Tract Symptoms 2015, 7, 149–154. doi:10.1111/luts.12063 |
| 81. | Moriarty, R. M.; Hu, H.; Gupta, S. C. Tetrahedron Lett. 1981, 22, 1283–1286. doi:10.1016/s0040-4039(01)90297-7 |
| 4. | Li, X.; Xu, L.; Cui, Y. Q.; Pang, M.; Wang, F.; Qi, J. IOP Conf. Ser.: Mater. Sci. Eng. 2017, 275, No. 012013. doi:10.1088/1757-899x/275/1/012013 |
| 18. | Chinsembu, K. C. Braz. J. Pharm. 2019, 572–596. |
| 19. | Li, H.-M.; Zhou, C.; Chen, C.-H.; Li, R.-T.; Lee, K.-H. Helv. Chim. Acta 2015, 98, 1177–1187. doi:10.1002/hlca.201500123 |
| 82. | Zhen, X.; Wan, X.; Zhang, W.; Li, Q.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2019, 21, 890–894. doi:10.1021/acs.orglett.8b03741 |
| 12. | Binbuga, N.; Ruhs, C.; Hasty, J. K.; Henry, W. P.; Schultz, T. P. Holzforschung 2008, 62, 264–269. doi:10.1515/hf.2008.038 |
| 14. | Fu, Z.; Sun, W. C.; Luan, X. C.; Gao, J. X.; Zhang, Z. H. J. J. Med. Univ. 2018, 39, 88–92. |
| 78. | Wei, Y.; Zheng, X.; Xie, X. Chem. Res. Chin. Univ. 2019, 35, 26–32. doi:10.1007/s40242-019-8264-9 |
| 15. | Asimakopoulos, A. D.; Cerruto, M. A.; Del Popolo, G.; La Martina, M.; Artibani, W.; Carone, R.; Finazzi-Agrò, E. Urol. Int. 2012, 89, 259–269. doi:10.1159/000339600 |
| 16. | Yamaguchi, O.; Nishizawa, O.; Takeda, M.; Yokoyama, O.; Homma, Y.; Kakizaki, H.; Obara, K.; Gotoh, M.; Igawa, Y.; Seki, N.; Yoshida, M. Int. J. Urol. 2009, 16, 126–142. doi:10.1111/j.1442-2042.2008.02177.x |
| 79. | Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328–337. doi:10.1002/1615-4169(200206)344:3/4<328::aid-adsc328>3.0.co;2-s |
| 80. | Adam, W.; Herold, M.; Hill, C. L.; Saha-Möller, C. R. Eur. J. Org. Chem. 2002, 941–946. doi:10.1002/1099-0690(200203)2002:5<941::aid-ejoc941>3.0.co;2-a |
| 10. | Gamal-Eldeen, A. M.; Djemgou, P. C.; Tchuendem, M.; Ngadjui, B. T.; Tane, P.; Toshifumi, H. Z. Naturforsch., C: J. Biosci. 2007, 62, 331–338. doi:10.1515/znc-2007-5-622 |
| 74. | Saltzman, H.; Sharefkin, J. G. Org. Synth. 1963, 43, 62–64. doi:10.15227/orgsyn.043.0062 |
| 9. | Zhou, T.; Shi, Q.; Lee, K. H. Tetrahedron Lett. 2010, 51, 4382–4386. doi:10.1016/j.tetlet.2010.06.058 |
| 13. | Sumiyoshi, M.; Kimura, Y. Phytomedicine 2010, 17, 820–829. doi:10.1016/j.phymed.2010.01.010 |
| 75. | Yang, Y.; Diederich, F.; Valentine, J. S. J. Am. Chem. Soc. 1990, 112, 7826–7828. doi:10.1021/ja00177a071 |
| 76. | Engelmann, X.; Malik, D. D.; Corona, T.; Warm, K.; Farquhar, E. R.; Swart, M.; Nam, W.; Ray, K. Angew. Chem., Int. Ed. 2019, 58, 4012–4016. doi:10.1002/anie.201812758 |
| 77. | Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. ACS Catal. 2018, 8, 4528–4538. doi:10.1021/acscatal.8b00874 |
| 26. | Darmani, N. A.; Chebolu, S.; Zhong, W.; Kim, W. D.; Narlesky, M.; Adams, J.; Dong, F. Eur. J. Pharmacol. 2017, 809, 20–31. doi:10.1016/j.ejphar.2017.05.014 |
| 24. | Ha, E.-S.; Kim, J.-S.; Kuk, D.-H.; Ha, D.-H.; Baek, I.-h.; Kim, M.-S. J. Mol. Liq. 2017, 225, 231–234. doi:10.1016/j.molliq.2016.11.082 |
| 85. | We also investigated the substrate with R2 = R3 = Me but failed to achieve any successful result. |
| 25. | Huang, F. C.; Galemmo, R. A.; Poli, G. B.; Learn, K. S.; Morrissette, M. M.; Johnson, W. H.; Dankulich, W. P.; Campbell, H. F.; Carnathan, G. W.; Van Inwegen, R. G. J. Med. Chem. 1991, 34, 1704–1707. doi:10.1021/jm00109a025 |
| 63. | Nicolaou, K. C.; Montagnon, T.; Baran, P. S.; Zhong, Y.-L. J. Am. Chem. Soc. 2002, 124, 2245–2258. doi:10.1021/ja012127+ |
| 86. | Cui, L.-Q.; Dong, Z.-L.; Liu, K.; Zhang, C. Org. Lett. 2011, 13, 6488–6491. doi:10.1021/ol202777h |
| 87. | We tentatively propose that polymeric (PhIO)n would undergo depolymerization to give PhIO species in DMF, as the HRMS study of a solution of (PhIO)n in DMF implied the existence of an m/z signal for [M + Na]+ for PhIO (see Supporting Information File 1 for HRMS spectra). |
| 3. | Keri, R. S.; Budagumpi, S.; Pai, R. K.; Balakrishna, R. G. Eur. J. Med. Chem. 2014, 78, 340–374. doi:10.1016/j.ejmech.2014.03.047 |
| 59. | Shanker, C. G.; Mallaiah, B. V.; Srimannarayana, G. Synthesis 1983, 310–311. doi:10.1055/s-1983-30317 |
| 60. | Hoshino, Y.; Takeno, N. Bull. Chem. Soc. Jpn. 1987, 60, 4468–4470. doi:10.1246/bcsj.60.4468 |
| 61. | Zhou, Z.; Zhao, P.; Huang, W.; Yang, G. Adv. Synth. Catal. 2006, 348, 63–67. doi:10.1002/adsc.200505223 |
| 62. | Vaz, P. A. A. M.; Pinto, D. C. G. A.; Rocha, D. H. A.; Silva, A. M. S.; Cavaleiro, J. A. S. Synlett 2012, 23, 2353–2356. doi:10.1055/s-0032-1317159 |
| 55. | Pan, G.-F.; Zhu, X.-Q.; Guo, R.-L.; Gao, Y.-R.; Wang, Y.-Q. Adv. Synth. Catal. 2018, 360, 4774–4783. doi:10.1002/adsc.201801058 |
| 56. | Chen, M.; Rago, A. J.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 16205–16209. doi:10.1002/anie.201811197 |
| 57. | Diao, T.; Stahl, S. S. J. Am. Chem. Soc. 2011, 133, 14566–14569. doi:10.1021/ja206575j |
| 58. | Lee, J.; Yu, J.; Son, S. H.; Heo, J.; Kim, T.; An, J.-Y.; Inn, K.-S.; Kim, N.-J. Org. Biomol. Chem. 2016, 14, 777–784. doi:10.1039/c5ob01911g |
| 2. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 90. | Shin, Y.; Yoo, C.; Moon, Y.; Lee, Y.; Hong, S. Chem. – Asian J. 2015, 10, 878–881. doi:10.1002/asia.201402876 |
| 91. | Di Paolo, E. R.; Hamburger, M. O.; Stoeckli-Evans, H.; Rogers, C.; Hostettmann, K. Helv. Chim. Acta 1989, 72, 1455–1462. doi:10.1002/hlca.19890720705 |
| 92. | Thelingwani, R. S.; Dhansay, K.; Smith, P.; Chibale, K.; Masimirembwa, C. M. Xenobiotica 2012, 42, 989–1000. doi:10.3109/00498254.2012.681077 |
| 29. | Ellis, G. P. General Methods of Preparing Chromones. In Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; Ellis, G. P., Ed.; John Wiley & Sons: Hoboken, NJ, 1977; Vol. 31, pp 495–555. doi:10.1002/9780470187012.ch9 |
| 30. | Singh, O. V.; Kapoor, R. P. Tetrahedron Lett. 1990, 31, 1459–1462. doi:10.1016/s0040-4039(00)88832-2 |
| 31. | Ciattini, P. G.; Morera, E.; Ortar, G. J. Heterocycl. Chem. 1982, 19, 395–400. doi:10.1002/jhet.5570190236 |
| 32. | Löwe, W.; Matzanke, N. J. Heterocycl. Chem. 1996, 33, 943–948. doi:10.1002/jhet.5570330364 |
| 33. | Turcas, R.; Kripli, B.; Attia, A. A. A.; Lakk-Bogáth, D.; Speier, G.; Giorgi, M.; Silaghi-Dumitrescu, R.; Kaizer, J. Dalton Trans. 2018, 47, 14416–14420. doi:10.1039/c8dt03519a |
| 34. | Yoshii, D.; Jin, X.; Yatabe, T.; Hasegawa, J.-y.; Yamaguchi, K.; Mizuno, N. Chem. Commun. 2016, 52, 14314–14317. doi:10.1039/c6cc07846j |
| 35. | Patonay, T.; Vasas, A.; Kiss-Szikszai, A.; Silva, A. M. S.; Cavaleiro, J. A. S. Aust. J. Chem. 2010, 63, 1582–1593. doi:10.1071/ch10295 |
| 36. | Bovicelli, P.; D'Angelo, V.; Collalto, D.; Verzina, A.; D'Antona, N.; Lambusta, D. J. Pharm. Pharmacol. 2007, 59, 1697–1701. doi:10.1211/jpp.59.12.0012 |
| 37. | Zacuto, M. J.; Cai, D. Tetrahedron Lett. 2005, 46, 447–450. doi:10.1016/j.tetlet.2004.11.092 |
| 38. | Muthukrishnan, M.; Patil, P. S.; More, S. V.; Joshi, R. A. Mendeleev Commun. 2005, 15, 100–101. doi:10.1070/mc2005v015n03abeh001997 |
| 39. | Prakash, O.; Pahuja, S.; Moriarty, R. M. Synth. Commun. 1990, 20, 1417–1422. doi:10.1080/00397919008052857 |
| 40. | Harvey, R. G.; Hahn, J. T.; Bukowska, M.; Jackson, H. J. Org. Chem. 1990, 55, 6161–6166. doi:10.1021/jo00312a023 |
| 41. | Takeno, N.; Fukushima, T.; Takeda, S.-i.; Kishimoto, K. Bull. Chem. Soc. Jpn. 1985, 58, 1599–1600. doi:10.1246/bcsj.58.1599 |
| 42. | Wu, L.-L.; Tang, L.; Zhou, S.-G.; Peng, Y.-J.; He, X.-D.; Guan, Z.; He, Y.-H. Tetrahedron 2017, 73, 6471–6478. doi:10.1016/j.tet.2017.09.050 |
| 43. | Patonay, T.; Lévai, A.; Nemes, C.; Timár, T.; Tóth, G.; Adam, W. J. Org. Chem. 1996, 61, 5375–5383. doi:10.1021/jo960163z |
| 44. | Mal, K.; Kaur, A.; Haque, F.; Das, I. J. Org. Chem. 2015, 80, 6400–6410. doi:10.1021/acs.joc.5b00846 |
| 45. | Thomsen, I.; Torssell, K. B. G. Acta Chem. Scand., Ser. B 1988, 42, 303–308. doi:10.3891/acta.chem.scand.42b-0303 |
| 46. | Litkei, G.; Patonay, T.; Szilágyi, L.; Dinya, Z. Org. Prep. Proced. Int. 1991, 23, 741–747. doi:10.1080/00304949109458249 |
| 47. | Zanarotti, A. Heterocycles 1982, 19, 1585–1586. doi:10.3987/r-1982-09-1585 |
| 48. | Prakash, O.; Mendiratta, S. Synth. Commun. 1992, 22, 327–334. doi:10.1080/00397919208021310 |
| 49. | Somogyi, L. Synth. Commun. 1999, 29, 1857–1872. doi:10.1080/00397919908086175 |
| 50. | Marié, J.-C.; Xiong, Y.; Min, G. K.; Yeager, A. R.; Taniguchi, T.; Berova, N.; Schaus, S. E.; Porco, J. A., Jr. J. Org. Chem. 2010, 75, 4584–4590. doi:10.1021/jo100889c |
| 51. | Kumar, D.; Singh, O. V.; Prakash, O.; Singh, S. P. Synth. Commun. 1994, 24, 2637–2644. doi:10.1080/00397919408010576 |
| 52. | Prakash, O.; Tanwar, M. P. Bull. Chem. Soc. Jpn. 1995, 68, 1168–1171. doi:10.1246/bcsj.68.1168 |
| 53. | Lambaa, M.; Makrandi, J. K. J. Chem. Res. 2008, 225–226. doi:10.3184/030823408x313591 |
| 54. | Varma, R. S.; Varma, M. Synth. Commun. 1982, 12, 927–930. doi:10.1080/00397918208061928 |
| 93. | Boonsombat, J.; Thongnest, S.; Ruchirawat, S. Eur. J. Org. Chem. 2019, 2971–2983. doi:10.1002/ejoc.201900078 |
| 27. | Sharma, R.; Williams, I. S.; Gatchie, L.; Sonawane, V. R.; Chaudhuri, B.; Bharate, S. B. ACS Omega 2018, 3, 8553–8566. doi:10.1021/acsomega.8b01088 |
| 88. | Fan, H.; Pan, P.; Zhang, Y.; Wang, W. Org. Lett. 2018, 20, 7929–7932. doi:10.1021/acs.orglett.8b03503 |
| 28. | Machado, N. F. L.; Marques, M. P. M. Curr. Bioact. Compd. 2010, 6, 76–89. doi:10.2174/157340710791184859 |
| 89. | Sreenithya, A.; Sunoj, R. B. Org. Lett. 2014, 16, 6224–6227. doi:10.1021/ol503161g |
© 2019 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)