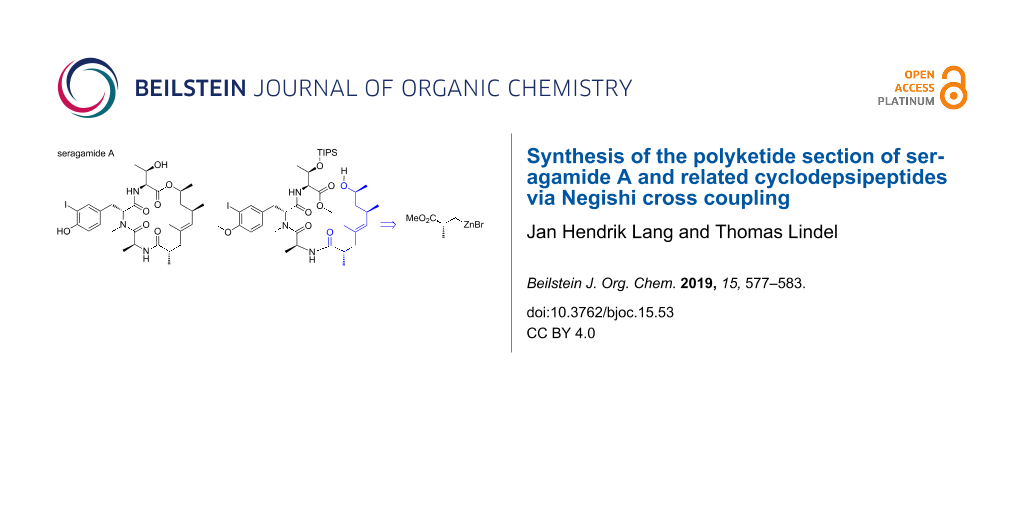
Abstract
The synthesis of the polyketide section present in the potently cytotoxic marine cyclodepsipeptide jasplakinolide and related natural products, geodiamolides and seragamides, is reported. The key step is a Negishi cross coupling of (R)-(3-methoxy-2-methyl-3-oxopropyl)zinc(II) bromide and an (E)-iodoalkene that was synthesized via an aluminium ester enolate attack at (R)-propylene oxide. The overall synthesis comprises nine steps with an overall yield of 21%. It proved to be possible to liberate the free 8-hydroxynonenoic acid and to couple it with a protected tripeptide composed of L-alanine, N,O-dimethyl-D-iodotyrosine, and TIPS-protected L-threonine, which occurs as partial structure of seragamide A. The tripeptide section of seragamide A was assembled by solution-phase synthesis and an open-chain analogue of the natural product was obtained.

Graphical Abstract
Introduction
Our program on the synthesis of biologically active natural products with peptide partial structures is driven by an interest in the chemistry of photoreactive amino acids and heterocycles that may find application in photoaffinity labelling. Knowing the binding of a natural product to a biological target at atomic resolution, as it is the case for the cyclodepsipeptide jasplakinolide A (1, Scheme 1) [1,2], is an ideal situation for the validation of the chemoselectivity and efficiency of photoaffinity labelling. Recently, it has been determined by cryo-electron microscopy how jasplakinolide A (1) binds to F-actin and alters the actin skeleton in vivo, resulting in pronounced cytotoxicity [3]. A photoreactive derivative of the cytotoxic jasplakinolides, geodiamolides [4,5], or seragamides (2–6, seragamides A–E) [6] could also enable the search for additional targets in the cell, including proteins involved in transport or even membrane components. For this purpose, the 2-bromoabrine unit of 1 could be replaced by phototryptophan [7], whereas photo β-phenylalanine [8] could replace the β-tyrosine moiety. For photoaffinity labelling studies with seragamides and geodiamolides, D-photophenylalanine could be incorporated.
![[1860-5397-15-53-i1]](/bjoc/content/inline/1860-5397-15-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Actin-binding cyclodepsipeptides, photo amino acids, retrosynthetic cuts of polyketide 7 leading to organozinc compound 8.
Scheme 1: Actin-binding cyclodepsipeptides, photo amino acids, retrosynthetic cuts of polyketide 7 leading to...
In this paper we describe, as the first step of such an endeavour, a new synthesis of the polyketide section present in seragamides and most of jasplakinolides and geodiamolides. We also include the synthesis of the protected tripeptide section of seragamide A (2), even if the macrocycle still remains to be assembled. Seragamide A (2) from the marine sponge Suberites japonicus (Thiele) has been synthesized only once, with relay ring-closing metathesis being the key step [9]. Characteristically, seragamides A–E exhibit a L-threonine unit at the C-terminus of the peptide moiety.
There is a considerable body of work on the synthesis of the C12 polyketide section. In most cases, the C2–C3 bond was formed by Ireland-, Johnson-, or Eschenmoser–Claisen rearrangement [10-17]. Alternatively, Evans methodology has been used [18-20]. The central C4–C5 double bond has also been constructed by Wittig reaction, cross metathesis, and ring-closing metathesis [9,21,22]. The C3–C4 bond has been assembled by 1,2-cuprate rearrangement [23]. Iterative construction of the polyketide from smaller building blocks has been achieved by Matteson homologation [24], that was also applied to the synthesis of the related polyketide section of lagunamide A [25]. In addition, the shorter C11 polyketide section of the closely related chondramides has been assembled in several ways [26-29].
Results and Discussion
In our approach the polyketide section (7, Scheme 1) of seragamide A (2) was to be assembled via construction of the C3–C4 bond by sp3–sp2 Negishi cross coupling. The required organozinc reagent 8 has been applied occasionally and is accessible from the corresponding commercially available chiral bromide [30-34]. The coupling partner would be an (E)-iodoalkene that was to be constructed from enantiomerically pure (R)-propylene oxide. The C4–C5 double bond would be formed by Corey–Fuchs reaction. Reaction of the aluminium enolate of tert-butyl propionate (9, Scheme 2) with (S)-propylene oxide had provided Taylor et al. with a mixture of diastereomers of α-methylated γ-hydroxyester 11 favouring the (2S,4S)-syn over the (2R,4S)-anti isomer [35,36]. As the configuration at C4 was expected to be invertible in a later step, we aimed for the (2R,4R) configuration by choosing (R)-propylene oxide as electrophile. We obtained an acceptable ratio of diastereomers of 11 (84:16 in favour of (2R,4R), 62% combined) after having treated tert-butyl propionate (9) with LDA/Et2AlCl (Scheme 2). Performing the reaction at −78 °C instead of −40 °C unexpectedly diminished the dr, which is in agreement with results by Liu, Petersen and co-workers [37]. The analogous reaction of adamantyl ester 10 (obtained from adamantan-1-ol and propionyl chloride/pyridine, 85%) instead of 9 gave a quantitative yield of the hydroxy ester 12, but a lower dr (4:1). For both cases, we inverted the carbinol centre by Mitsunobu reaction employing p-methoxyphenol. DEAD was used instead of DIAD because of facilitated purification. It was possible to separate the diastereomers of product 13 by column chromatography. The use of DMEAD (di-2-methoxyethyl azodicarboxylate) [38] was inferior (33%). The PMP protecting group was envisioned to be stable during the following steps and to be selectively cleavable with ceric ammonium nitrate (CAN).
![[1860-5397-15-53-i2]](/bjoc/content/inline/1860-5397-15-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of γ-hydroxy esters 11 and 12, followed by Mitsunobu inversion.
Scheme 2: Synthesis of γ-hydroxy esters 11 and 12, followed by Mitsunobu inversion.
Reduction of tert-butyl ester 13a to the alcohol by portionwise addition of DIBAL and careful temperature control (slow warm-up from −78 to −30 °C) afforded the alcohol without α-epimerization, which was oxidized to aldehyde 15 (IBX, Scheme 3). Adamantyl ester 14 resisted reduction under the same conditions and was abandoned.
![[1860-5397-15-53-i3]](/bjoc/content/inline/1860-5397-15-53-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the polyketide section 7.
Scheme 3: Synthesis of the polyketide section 7.
Dibromoalkene 16 was accessed from aldehyde 15 under Corey–Fuchs conditions and treated with n-hexane to separate triphenylphosphane oxide. For the conversion of 16 to the internal alkyne 17, the use of a large excess of freshly prepared LDA and iodomethane with a slow warm-up from −78 °C to rt [39] proved to be superior to the more commonly employed n-BuLi/MeI reagent combination that led to the formation of a series of side products. We also attempted a Bestmann–Ohira reaction with in situ formation of the diazo phosphonate [40] that gave a slightly inferior yield after methylation of the alkyne terminus. Hydrozirconation/iodination of alkyne 17 required freshly prepared Schwartz reagent (3 equiv) and iodine (3 equiv) to obtain yields of (E)-olefin 18 above 80% with perfect stereo- and regioselectivity. For comparison, as part of a study towards kendomycin, Arimoto and co-workers had published a route to a 1:1 mixture of diastereomers of a TBS-protected analogue of building block 18 [41].
Negishi cross coupling of sp3 organozinc homoenolate 8 (2.75 equiv) and iodoalkene 18 (10 mol % [Pd(dppf)Cl2 × DCM] in DCM) afforded a satisfying 75% yield of the protected nonenoic acid 20 (Scheme 3). For the synthesis of organozinc homoenolate 8, the MnBr2/CuCl-catalyzed reaction of diethylzinc with β-bromopropionic acid ester 19 in DMPU proved to be the best choice [42]. Racemization was avoided.
The PMP protecting group was removed (CAN) affording methyl ester 7 (82%, Scheme 3). For comparison with literature data we converted 7 to the free carboxylic acid carrying a TBS-protected hydroxy group at C8 by silylation with TBSOTf/2,6-lutidine, followed by saponification with LiOH.
To investigate whether polyketide 7 could be coupled in an unprotected form with the peptide fragment of seragamide A (2), we synthesized the tripeptide section. The assembly of the iodotyrosine moiety commenced with D-3-iodotyrosine that was obtained in 63% yield from D-tyrosine upon treatment with iodine (1 equiv, added via syringe pump over 90 min to avoid diiodination) in aqueous ammonia [43] (Scheme 4). This setup also served to avoid the formation of excess amounts of the explosive iodinating reagent NI3. On work-up, the pH had to be adjusted to exactly 4.5 to destroy remaining NI3 and to precipitate the product. Other iodination conditions such as use of NaI/NaOCl [44,45] or I2/CAN [46] did not convince. Boc protection yielded N-Boc-D-3-iodotyrosine (21), which underwent three-fold methylation to N-methyl-O-methyliodotyrosine methyl ester 22 (82% yield, er 4:1, CHIRALPAK® IA, hexane/iPrOH 9:1). Partial racemization of 21 could not be suppressed since a slight excess of base was always necessary, whereas the use of exactly stoichiometric amounts resulted in incomplete N-methylation. Treating the Boc-protected methyl ester of iodotyrosine 21 with a larger excess of base and methyl iodide (10 equiv each) led to the formation of the α-quaternary amino acid 23 (Scheme 4). An alternative route to the N-methylated product via oxazolidinone 24 was hampered by low yield on reaction with paraformaldehyde [47,48]. Although subsequent reduction of 24 with Et3SiH/TFA and reprotection of the free N-methyl amino acid with Boc2O proceeded smoothly and delivered iodotyrosine 25, the overall yield via 24 was not satisfying.
![[1860-5397-15-53-i4]](/bjoc/content/inline/1860-5397-15-53-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Access to methylated D-iodotyrosine derivatives 22, 23, and 25.
Scheme 4: Access to methylated D-iodotyrosine derivatives 22, 23, and 25.
After Boc removal at 22, HATU/HOAt-mediated coupling with Boc-protected L-alanine afforded dipeptide 26, which was saponified and coupled with TIPS-protected threonine methyl ester (obtained with TIPSCl/DBU/MeCN) to provide tripeptide 27 (Scheme 5). Peptides 26 and 27 were obtained in nearly quantitative yield, albeit as mixtures of inseparable diastereomers (4:1), as consequence of the optical impurity of 22. Only when following the coupling order shown in Scheme 5, high yields could be obtained.
![[1860-5397-15-53-i5]](/bjoc/content/inline/1860-5397-15-53-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the doubly protected open chain peptide-polyketide 31.
Scheme 5: Synthesis of the doubly protected open chain peptide-polyketide 31.
The labile carboxylic acid 29 was formed by saponification of methyl ester 7 (LiOH, H2O/MeOH) and underwent BEP-mediated coupling with tripeptide 27 (Scheme 5). Product 31 (60%) was obtained with a higher dr of 9:1 than tripeptide 27 (dr 4:1). We have evidence that the major side product of 31 was a desilylated analogue, for which the 1H NMR spectrum indicated a lower dr of about 7:3. The analogous product 30 carrying an 8-OPMP group was obtained from PMP-protected polyketide 28.
The endgame of a total synthesis of seragamide A (2) will have to address the macrocyclisation, desilylation of the threonine moiety, and O-demethylation of the D-iodotyrosine unit of 31. In orienting reactions on small scale, desilylation of 31 (TBAF) was a spot-to-spot conversion. Saponification of both the silylated and desilylated methyl esters with LiOH was possible, as long as very mild acidic conditions were applied on work-up. However, macrocyclisation of the TIPS-protected or the desilylated acid did not occur. Under Yamaguchi conditions, we observed complete decomposition in both cases. Shiina conditions gave a complex product mixture, if the TIPS group was present, whereas the desilylated acid provided a product that apparently had undergone dehydration, as judged by TLC–MS coupling. In course of their recent synthesis of plusbacin A3, Ichikawa et al. experienced similar difficulties when attempting esterification of a bulky β-OTIPS-substituted amino acid with a secondary alcohol under a variety of conditions [49].
Conclusion
We developed a short synthesis of the polyketide building block present in most of the jasplakinolides, geodiamolides, and seragamides, potently cytotoxic peptide-polyketide cyclodepsipeptides stabilizing filamentous actin. The key step is a Negishi cross coupling, and the carbon atoms stem from (R)-propylene oxide (C7–C9), tert-butyl propionate (9, C5–C6, 6-Me), CBr4 (C4), MeI (4-Me), and (R)-(3-methoxy-2-methyl-3-oxopropyl)zinc(II) bromide (8, C1–C3, 2-Me). We obtained 8-hydroxyester 7 within nine steps and an overall yield of 21% starting from (R)-propylene oxide. The efficiency of our route to polyketide 7 compares well to earlier approaches. It proved to be possible to liberate the free carboxylic acid 29 and to couple it with the protected tripeptide 27 that was obtained by solution phase synthesis. We will now aim at the macrocyclisation of analogues of compound 31, which will probably require replacement of the bulky TIPS group installed at the threonine unit. The synthesis of photoreactive tripeptide analogues incorporating photo amino acids 4–6 will be explored thereafter.
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR spectra. | ||
| Format: PDF | Size: 12.1 MB | Download |
References
-
Crews, P.; Manes, L. V.; Boehler, M. Tetrahedron Lett. 1986, 27, 2797–2800. doi:10.1016/s0040-4039(00)84645-6
Return to citation in text: [1] -
Zabriskie, T. M.; Klocke, J. A.; Ireland, C. M.; Marcus, A. H.; Molinski, T. F.; Faulkner, D. J.; Xu, C.; Clardy, J. C. J. Am. Chem. Soc. 1986, 108, 3123–3124. doi:10.1021/ja00271a062
Return to citation in text: [1] -
Pospich, S.; Kumpula, E.-P.; von der Ecken, J.; Vahokoski, J.; Kursula, I.; Raunser, S. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 10636–10641. doi:10.1073/pnas.1707506114
Return to citation in text: [1] -
Chan, W. R.; Tinto, W. F.; Manchand, P. S.; Todaro, L. J. J. Org. Chem. 1987, 52, 3091–3093. doi:10.1021/jo00390a023
Return to citation in text: [1] -
Coleman, J. E.; Van Soest, R.; Andersen, R. J. J. Nat. Prod. 1999, 62, 1137–1141. doi:10.1021/np990155o
And references cited therein.
Return to citation in text: [1] -
Tanaka, C.; Tanaka, J.; Bolland, R. F.; Marriott, G.; Higa, T. Tetrahedron 2006, 62, 3536–3542. doi:10.1016/j.tet.2006.01.099
Return to citation in text: [1] -
Wartmann, T.; Lindel, T. Eur. J. Org. Chem. 2013, 1649–1652. doi:10.1002/ejoc.201201726
Return to citation in text: [1] -
Schäckermann, J.-N.; Lindel, T. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 1088–1096. doi:10.5560/znb.2014-4152
Return to citation in text: [1] -
Arndt, H.-D.; Rizzo, S.; Nöcker, C.; Wakchaure, V. N.; Milroy, L.-G.; Bieker, V.; Calderon, A.; Tran, T. T. N.; Brand, S.; Dehmelt, L.; Waldmann, H. Chem. – Eur. J. 2015, 21, 5311–5316. doi:10.1002/chem.201500368
Return to citation in text: [1] [2] -
Rao, A. V. R.; Gurjar, M. K.; Nallaganchu, B. R.; Bhandari, A. Tetrahedron Lett. 1993, 34, 7085–7088. doi:10.1016/s0040-4039(00)61605-2
Return to citation in text: [1] -
Grieco, P. A.; Hon, Y. S.; Perez-Medrano, A. J. Am. Chem. Soc. 1988, 110, 1630–1631. doi:10.1021/ja00213a050
Return to citation in text: [1] -
Chu, K. S.; Negrete, G. R.; Konopelski, J. P. J. Org. Chem. 1991, 56, 5196–5202. doi:10.1021/jo00017a037
Return to citation in text: [1] -
Ghosh, A. K.; Moon, D. K. Org. Lett. 2007, 9, 2425–2427. doi:10.1021/ol070855h
Return to citation in text: [1] -
Hirai, Y.; Yokota, K.; Sakai, H.; Yamazaki, T.; Momose, T. Heterocycles 1989, 29, 1865–1869. doi:10.3987/com-89-5123
Return to citation in text: [1] -
White, J. D.; Amedio, J. C., Jr. J. Org. Chem. 1989, 54, 736–738. doi:10.1021/jo00265a003
Return to citation in text: [1] -
Schmidt, U.; Siegel, W.; Mundinger, K. Tetrahedron Lett. 1988, 29, 1269–1270. doi:10.1016/s0040-4039(00)80273-7
Return to citation in text: [1] -
Rao, A. V. R.; Gurjar, M. K.; Nallaganchu, B. R.; Bhandari, A. Tetrahedron Lett. 1993, 34, 7081–7084. doi:10.1016/s0040-4039(00)61604-0
Return to citation in text: [1] -
Imaeda, T.; Hamada, Y.; Shioiri, T. Tetrahedron Lett. 1994, 35, 591–594. doi:10.1016/s0040-4039(00)75846-1
Return to citation in text: [1] -
Kang, S.-K.; Lee, D.-H. Synlett 1991, 175–176. doi:10.1055/s-1991-20668
Return to citation in text: [1] -
Wattanasereekul, S.; Maier, M. E. Adv. Synth. Catal. 2004, 346, 855–861. doi:10.1002/adsc.200404004
Return to citation in text: [1] -
Tannert, R.; Hu, T.-S.; Arndt, H.-D.; Waldmann, H. Chem. Commun. 2009, 1493–1495. doi:10.1039/b900342h
Return to citation in text: [1] -
Tannert, R.; Milroy, L.-G.; Ellinger, B.; Hu, T.-S.; Arndt, H.-D.; Waldmann, H. J. Am. Chem. Soc. 2010, 132, 3063–3077. doi:10.1021/ja9095126
Return to citation in text: [1] -
Ashworth, P.; Broadbelt, B.; Jankowski, P.; Kocienski, P.; Pimm, A.; Bell, R. Synthesis 1995, 199–206. doi:10.1055/s-1995-3870
Return to citation in text: [1] -
Unsworth, P. J.; Leonori, D.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2014, 53, 9846–9850. doi:10.1002/anie.201405700
Return to citation in text: [1] -
Gorges, J.; Kazmaier, U. Org. Lett. 2018, 20, 2033–2036. doi:10.1021/acs.orglett.8b00576
Return to citation in text: [1] -
Eggert, U.; Diestel, R.; Sasse, F.; Jansen, R.; Kunze, B.; Kalesse, M. Angew. Chem., Int. Ed. 2008, 47, 6478–6482. doi:10.1002/anie.200801156
Return to citation in text: [1] -
Waldmann, H.; Hu, T.-S.; Renner, S.; Menninger, S.; Tannert, R.; Oda, T.; Arndt, H.-D. Angew. Chem., Int. Ed. 2008, 47, 6473–6477. doi:10.1002/anie.200801010
Return to citation in text: [1] -
Schmauder, A.; Sibley, L. D.; Maier, M. E. Chem. – Eur. J. 2010, 16, 4328–4336. doi:10.1002/chem.200903500
Return to citation in text: [1] -
Becker, D.; Kazmaier, U. Eur. J. Org. Chem. 2015, 4198–4213. doi:10.1002/ejoc.201500369
Return to citation in text: [1] -
Petri, A. F.; Schneekloth, J. S.; Mandal, A. K.; Crews, C. M. Org. Lett. 2007, 9, 3001–3004. doi:10.1021/ol071024e
Return to citation in text: [1] -
Cui, C.; Dai, W.-M. Org. Lett. 2018, 20, 3358–3361. doi:10.1021/acs.orglett.8b01269
Return to citation in text: [1] -
Dimopoulos, P.; Athlan, A.; Manaviazar, S.; Hale, K. J. Org. Lett. 2005, 7, 5373–5376. doi:10.1021/ol051935t
Return to citation in text: [1] -
Francavilla, C.; Chen, W.; Kinder, F. R., Jr. Org. Lett. 2003, 5, 1233–1236. doi:10.1021/ol034186t
Return to citation in text: [1] -
Corrêa, I. R.; Pilli, R. A. Angew. Chem., Int. Ed. 2003, 42, 3017–3020. doi:10.1002/anie.200351347
Return to citation in text: [1] -
Sturm, T.-J.; Marolewski, A. E.; Rezenka, D. S.; Taylor, S. K. J. Org. Chem. 1989, 54, 2039–2040. doi:10.1021/jo00270a007
Return to citation in text: [1] -
Taylor, S. K.; Fried, J. A.; Grassl, Y. N.; Marolewski, A. E.; Pelton, E. A.; Poel, T.-J.; Rezanka, D. S.; Whittaker, M. R. J. Org. Chem. 1993, 58, 7304–7307. doi:10.1021/jo00077a069
Return to citation in text: [1] -
Qabaja, G.; Benavides, A. R.; Liu, S.; Petersen, K. S. J. Org. Chem. 2015, 80, 133–140. doi:10.1021/jo5022019
Return to citation in text: [1] -
Hagiya, K.; Muramoto, N.; Misaki, T.; Sugimura, T. Tetrahedron 2009, 65, 6109–6114. doi:10.1016/j.tet.2009.05.048
Return to citation in text: [1] -
Handa, M.; Scheidt, K. A.; Bossart, M.; Zheng, N.; Roush, W. R. J. Org. Chem. 2008, 73, 1031–1035. doi:10.1021/jo702250z
Return to citation in text: [1] -
Roth, G. J.; Liepold, B.; Müller, S. G.; Bestmann, H. J. Synthesis 2004, 59–62. doi:10.1055/s-2003-44346
Return to citation in text: [1] -
Sengoku, T.; Uemura, D.; Arimoto, H. Chem. Lett. 2007, 36, 726–727. doi:10.1246/cl.2007.726
Return to citation in text: [1] -
Klement, I.; Knochel, P.; Chau, K.; Cahiez, G. Tetrahedron Lett. 1994, 35, 1177–1180. doi:10.1016/0040-4039(94)88017-4
Return to citation in text: [1] -
Pitt-Rivers, R. Chem. Ind. 1956, 21.
Return to citation in text: [1] -
Edgar, K. J.; Falling, S. N. J. Org. Chem. 1990, 55, 5287–5291. doi:10.1021/jo00305a026
Return to citation in text: [1] -
Schmidt, E. W.; Nelson, J. T.; Fillmore, J. P. Tetrahedron Lett. 2004, 45, 3921–3924. doi:10.1016/j.tetlet.2004.03.112
Return to citation in text: [1] -
Das, B.; Krishnaiah, M.; Venkateswarlu, K.; Reddy, V. S. Tetrahedron Lett. 2007, 48, 81–83. doi:10.1016/j.tetlet.2006.11.009
Return to citation in text: [1] -
Hitotsuyanagi, Y.; Miyazawa, A.; Hinosawa, T.-a.; Nakagawa, Y.; Hasuda, T.; Takeya, K. Bioorg. Med. Chem. Lett. 2013, 23, 6728–6731. doi:10.1016/j.bmcl.2013.10.033
Return to citation in text: [1] -
Moschitto, M. J.; Lewis, C. A. Eur. J. Org. Chem. 2016, 4773–4777. doi:10.1002/ejoc.201600741
Return to citation in text: [1] -
Katsuyama, A.; Yakushiji, F.; Ichikawa, S. J. Org. Chem. 2018, 83, 7085–7101. doi:10.1021/acs.joc.8b00038
Return to citation in text: [1]
| 1. | Crews, P.; Manes, L. V.; Boehler, M. Tetrahedron Lett. 1986, 27, 2797–2800. doi:10.1016/s0040-4039(00)84645-6 |
| 2. | Zabriskie, T. M.; Klocke, J. A.; Ireland, C. M.; Marcus, A. H.; Molinski, T. F.; Faulkner, D. J.; Xu, C.; Clardy, J. C. J. Am. Chem. Soc. 1986, 108, 3123–3124. doi:10.1021/ja00271a062 |
| 7. | Wartmann, T.; Lindel, T. Eur. J. Org. Chem. 2013, 1649–1652. doi:10.1002/ejoc.201201726 |
| 30. | Petri, A. F.; Schneekloth, J. S.; Mandal, A. K.; Crews, C. M. Org. Lett. 2007, 9, 3001–3004. doi:10.1021/ol071024e |
| 31. | Cui, C.; Dai, W.-M. Org. Lett. 2018, 20, 3358–3361. doi:10.1021/acs.orglett.8b01269 |
| 32. | Dimopoulos, P.; Athlan, A.; Manaviazar, S.; Hale, K. J. Org. Lett. 2005, 7, 5373–5376. doi:10.1021/ol051935t |
| 33. | Francavilla, C.; Chen, W.; Kinder, F. R., Jr. Org. Lett. 2003, 5, 1233–1236. doi:10.1021/ol034186t |
| 34. | Corrêa, I. R.; Pilli, R. A. Angew. Chem., Int. Ed. 2003, 42, 3017–3020. doi:10.1002/anie.200351347 |
| 6. | Tanaka, C.; Tanaka, J.; Bolland, R. F.; Marriott, G.; Higa, T. Tetrahedron 2006, 62, 3536–3542. doi:10.1016/j.tet.2006.01.099 |
| 35. | Sturm, T.-J.; Marolewski, A. E.; Rezenka, D. S.; Taylor, S. K. J. Org. Chem. 1989, 54, 2039–2040. doi:10.1021/jo00270a007 |
| 36. | Taylor, S. K.; Fried, J. A.; Grassl, Y. N.; Marolewski, A. E.; Pelton, E. A.; Poel, T.-J.; Rezanka, D. S.; Whittaker, M. R. J. Org. Chem. 1993, 58, 7304–7307. doi:10.1021/jo00077a069 |
| 4. | Chan, W. R.; Tinto, W. F.; Manchand, P. S.; Todaro, L. J. J. Org. Chem. 1987, 52, 3091–3093. doi:10.1021/jo00390a023 |
| 5. |
Coleman, J. E.; Van Soest, R.; Andersen, R. J. J. Nat. Prod. 1999, 62, 1137–1141. doi:10.1021/np990155o
And references cited therein. |
| 25. | Gorges, J.; Kazmaier, U. Org. Lett. 2018, 20, 2033–2036. doi:10.1021/acs.orglett.8b00576 |
| 3. | Pospich, S.; Kumpula, E.-P.; von der Ecken, J.; Vahokoski, J.; Kursula, I.; Raunser, S. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 10636–10641. doi:10.1073/pnas.1707506114 |
| 26. | Eggert, U.; Diestel, R.; Sasse, F.; Jansen, R.; Kunze, B.; Kalesse, M. Angew. Chem., Int. Ed. 2008, 47, 6478–6482. doi:10.1002/anie.200801156 |
| 27. | Waldmann, H.; Hu, T.-S.; Renner, S.; Menninger, S.; Tannert, R.; Oda, T.; Arndt, H.-D. Angew. Chem., Int. Ed. 2008, 47, 6473–6477. doi:10.1002/anie.200801010 |
| 28. | Schmauder, A.; Sibley, L. D.; Maier, M. E. Chem. – Eur. J. 2010, 16, 4328–4336. doi:10.1002/chem.200903500 |
| 29. | Becker, D.; Kazmaier, U. Eur. J. Org. Chem. 2015, 4198–4213. doi:10.1002/ejoc.201500369 |
| 18. | Imaeda, T.; Hamada, Y.; Shioiri, T. Tetrahedron Lett. 1994, 35, 591–594. doi:10.1016/s0040-4039(00)75846-1 |
| 19. | Kang, S.-K.; Lee, D.-H. Synlett 1991, 175–176. doi:10.1055/s-1991-20668 |
| 20. | Wattanasereekul, S.; Maier, M. E. Adv. Synth. Catal. 2004, 346, 855–861. doi:10.1002/adsc.200404004 |
| 23. | Ashworth, P.; Broadbelt, B.; Jankowski, P.; Kocienski, P.; Pimm, A.; Bell, R. Synthesis 1995, 199–206. doi:10.1055/s-1995-3870 |
| 10. | Rao, A. V. R.; Gurjar, M. K.; Nallaganchu, B. R.; Bhandari, A. Tetrahedron Lett. 1993, 34, 7085–7088. doi:10.1016/s0040-4039(00)61605-2 |
| 11. | Grieco, P. A.; Hon, Y. S.; Perez-Medrano, A. J. Am. Chem. Soc. 1988, 110, 1630–1631. doi:10.1021/ja00213a050 |
| 12. | Chu, K. S.; Negrete, G. R.; Konopelski, J. P. J. Org. Chem. 1991, 56, 5196–5202. doi:10.1021/jo00017a037 |
| 13. | Ghosh, A. K.; Moon, D. K. Org. Lett. 2007, 9, 2425–2427. doi:10.1021/ol070855h |
| 14. | Hirai, Y.; Yokota, K.; Sakai, H.; Yamazaki, T.; Momose, T. Heterocycles 1989, 29, 1865–1869. doi:10.3987/com-89-5123 |
| 15. | White, J. D.; Amedio, J. C., Jr. J. Org. Chem. 1989, 54, 736–738. doi:10.1021/jo00265a003 |
| 16. | Schmidt, U.; Siegel, W.; Mundinger, K. Tetrahedron Lett. 1988, 29, 1269–1270. doi:10.1016/s0040-4039(00)80273-7 |
| 17. | Rao, A. V. R.; Gurjar, M. K.; Nallaganchu, B. R.; Bhandari, A. Tetrahedron Lett. 1993, 34, 7081–7084. doi:10.1016/s0040-4039(00)61604-0 |
| 24. | Unsworth, P. J.; Leonori, D.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2014, 53, 9846–9850. doi:10.1002/anie.201405700 |
| 9. | Arndt, H.-D.; Rizzo, S.; Nöcker, C.; Wakchaure, V. N.; Milroy, L.-G.; Bieker, V.; Calderon, A.; Tran, T. T. N.; Brand, S.; Dehmelt, L.; Waldmann, H. Chem. – Eur. J. 2015, 21, 5311–5316. doi:10.1002/chem.201500368 |
| 8. | Schäckermann, J.-N.; Lindel, T. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 1088–1096. doi:10.5560/znb.2014-4152 |
| 9. | Arndt, H.-D.; Rizzo, S.; Nöcker, C.; Wakchaure, V. N.; Milroy, L.-G.; Bieker, V.; Calderon, A.; Tran, T. T. N.; Brand, S.; Dehmelt, L.; Waldmann, H. Chem. – Eur. J. 2015, 21, 5311–5316. doi:10.1002/chem.201500368 |
| 21. | Tannert, R.; Hu, T.-S.; Arndt, H.-D.; Waldmann, H. Chem. Commun. 2009, 1493–1495. doi:10.1039/b900342h |
| 22. | Tannert, R.; Milroy, L.-G.; Ellinger, B.; Hu, T.-S.; Arndt, H.-D.; Waldmann, H. J. Am. Chem. Soc. 2010, 132, 3063–3077. doi:10.1021/ja9095126 |
| 39. | Handa, M.; Scheidt, K. A.; Bossart, M.; Zheng, N.; Roush, W. R. J. Org. Chem. 2008, 73, 1031–1035. doi:10.1021/jo702250z |
| 37. | Qabaja, G.; Benavides, A. R.; Liu, S.; Petersen, K. S. J. Org. Chem. 2015, 80, 133–140. doi:10.1021/jo5022019 |
| 38. | Hagiya, K.; Muramoto, N.; Misaki, T.; Sugimura, T. Tetrahedron 2009, 65, 6109–6114. doi:10.1016/j.tet.2009.05.048 |
| 47. | Hitotsuyanagi, Y.; Miyazawa, A.; Hinosawa, T.-a.; Nakagawa, Y.; Hasuda, T.; Takeya, K. Bioorg. Med. Chem. Lett. 2013, 23, 6728–6731. doi:10.1016/j.bmcl.2013.10.033 |
| 48. | Moschitto, M. J.; Lewis, C. A. Eur. J. Org. Chem. 2016, 4773–4777. doi:10.1002/ejoc.201600741 |
| 49. | Katsuyama, A.; Yakushiji, F.; Ichikawa, S. J. Org. Chem. 2018, 83, 7085–7101. doi:10.1021/acs.joc.8b00038 |
| 44. | Edgar, K. J.; Falling, S. N. J. Org. Chem. 1990, 55, 5287–5291. doi:10.1021/jo00305a026 |
| 45. | Schmidt, E. W.; Nelson, J. T.; Fillmore, J. P. Tetrahedron Lett. 2004, 45, 3921–3924. doi:10.1016/j.tetlet.2004.03.112 |
| 46. | Das, B.; Krishnaiah, M.; Venkateswarlu, K.; Reddy, V. S. Tetrahedron Lett. 2007, 48, 81–83. doi:10.1016/j.tetlet.2006.11.009 |
| 42. | Klement, I.; Knochel, P.; Chau, K.; Cahiez, G. Tetrahedron Lett. 1994, 35, 1177–1180. doi:10.1016/0040-4039(94)88017-4 |
| 40. | Roth, G. J.; Liepold, B.; Müller, S. G.; Bestmann, H. J. Synthesis 2004, 59–62. doi:10.1055/s-2003-44346 |
| 41. | Sengoku, T.; Uemura, D.; Arimoto, H. Chem. Lett. 2007, 36, 726–727. doi:10.1246/cl.2007.726 |
© 2019 Lang and Lindel; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)