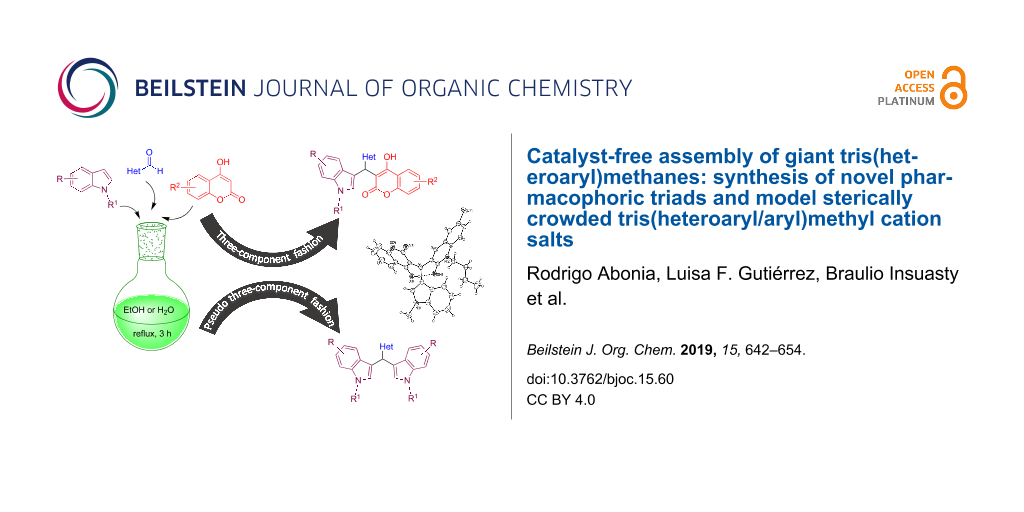
Abstract
A series of giant tris(heteroaryl)methanes are easily assembled by one-pot three-component synthesis by simple reflux in ethanol without catalyst or additives. Diversely substituted indoles (Ar1) react with quinoline aldehydes, quinolone aldehydes, chromone aldehydes, and fluorene aldehydes (Ar2CHO) and coumarins (Ar3) in 1:1:1 ratio to form the corresponding tris(heteroaryl)methanes (Ar1Ar2Ar3)CH along with (Ar1Ar1Ar2)CH triads. A series of new 2:1 triads were also synthesized by coupling substituted indoles with Ar2CHO. The coupling reactions could also be carried out in water (at circa 80 °C) but with chemoselectivity favoring (Ar1Ar1Ar2)CH over (Ar1Ar2Ar3)CH. The molecular structure of a representative (Ar1Ar2Ar3)CH triad was confirmed by X-ray analysis. Model tris(heteroaryl/aryl)methylium salts were generated by reaction with DDQ/HPF6 and studied by NMR and by DFT and GIAO-DFT.

Graphical Abstract
Introduction
During the last few decades multicomponent reactions (MCRs) have gained importance as a suitable strategy for the synthesis of diverse synthetic and naturally occurring compounds of biological and practical interest. This approach offers several advantages including simplicity, high reaction rates, and high bond-forming efficiency [1-5]. Furthermore, it is highly desirable to perform these reactions in environmentally friendly solvents such as water, ethanol, and PEG [6,7].
Motifs bearing triarylmethane (Ar3CH) [8-10] and their heterocyclic variants (Het-Ar)3CH [8-12], constitute an integral part of a number of bioactive compounds [13-16]. Due to their valuable properties, they are also well exploited by the chemical industry as dyes and photochromic agents [17,18], protective groups in organic synthesis [19] and as building blocks for dendrimers [20] and nonlinear optical (NLO) properties [21] (Scheme 1). Numerous methods for the construction of triarylmethane frameworks have been developed, with the majority of them bearing simple diaryl or triaryl moieties in their structures [22], and many are performed in multistep processes or require harsh reaction conditions [1-22]. Although, “Yonemitsu-type” three-component reactions have been employed for the synthesis of indole-based triarylmethanes [23-26] (Scheme 2), there still exists a need for the development of new approaches for easy access to libraries of triarylmethanes of higher complexity by employing simpler, more efficient, catalytic methods that are also environmentally friendly.
![[1860-5397-15-60-i1]](/bjoc/content/inline/1860-5397-15-60-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative examples of tris(hetero/aryl)methanes, molecular hybrids and bis(indolyl)methanes with useful properties.
Scheme 1: Representative examples of tris(hetero/aryl)methanes, molecular hybrids and bis(indolyl)methanes wi...
Molecular hybridization has emerged as an interesting strategy for the synthesis of bioactive molecules with improved properties by combining two or more pharmacophore fragments in a new structure. This concept has recently received attention by the pharmaceutical industry because it provides new options to develop more specific drugs for the treatment of persistent and challenging pathologies [27,28] (Scheme 1).
The indole, coumarin, quinoline, chromone and fluorene moieties are a set of “privileged structural motifs” that are present in both synthetic and naturally occurring compounds of practical and biological interest [29-36]. Consequently, there have been many attempts to produce hybrid structures with interesting properties by combining two such pharmacophores in one molecule, using both catalytic and non-catalytic reactions [37-48].
However, a remaining challenge is to discover methods to construct asymmetric triads consisting of three different pharmacophores (i.e., heterodimeric entities) via a simple synthetic step. According to the literature, most attempts in this direction have resulted in isolation of symmetric and asymmetric bis(indolyl)methane derivatives as the main components [39-41,49-52] (Scheme 1). Some exceptions to this tendency have been reported by Appendino et al. [52] and by Mousavizadeh et al. [53] through the three-component reactions of indole and coumarin, but in all cases, ordinary aliphatic and aromatic aldehydes as the third partner, mediated by a catalyst or by a biphasic system as solvent, respectively, were used. The lack of structural diversity in the indole and coumarin partners also characterizes these approaches, Scheme 2.
![[1860-5397-15-60-i2]](/bjoc/content/inline/1860-5397-15-60-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Previous synthetic approaches for the synthesis of triarylmethane analogues in comparison to the present study.
Scheme 2: Previous synthetic approaches for the synthesis of triarylmethane analogues in comparison to the pr...
Continuing our current program on the synthesis of quinoline-based heterocyclic compounds of biological interest [54-57], we describe here a Yonemitsu-based direct and reproducible three-component synthesis of ternary heteroarylmethane-inspired hybrids by coupling diversely substituted indoles (Ar1) with quinoline aldehydes, quinolone aldehydes, chromone aldehydes, and fluorene aldehydes (Ar2CHO) and coumarins (Ar3) in 1:1:1 ratio by simple reflux in ethanol solvent to form the corresponding highly crowded tris(heteroaryl)methanes (Ar1Ar2Ar3)CH (Scheme 2). Formation of (Ar1Ar1Ar2)CH triads is a competing process, whose relative proportion varies depending on the choice of the substituents. The efficacy to perform these remarkable reactions in water as solvent, and to generate highly crowded triarylmethylium salts by hydride abstraction from (Ar1Ar1Ar2)CH are also demonstrated.
Results and Discussion
At the onset a series of non-commercial N-alkylindoles 1{4–10} and quinoline-/quinolone aldehydes 6{1-7} were prepared (Scheme 3 and Scheme 4). The N-methyl-, N-butyl- and N-benzylindoles 1{4–10} were synthesized in 80–98% yield by N-alkylation of commercially available N–H indoles 1{1–3} ({1} R = H; {2} R = F and {3} R = OMe), by adopting a procedure similar to that described by Kong et al. [58] (Scheme 3).
![[1860-5397-15-60-i3]](/bjoc/content/inline/1860-5397-15-60-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the starting N-alkylindoles 1{4–10}.
Scheme 3: Synthesis of the starting N-alkylindoles 1{4–10}.
The quinolone aldehydes 5 were synthesized via 2-chloroquinoline-3-carbaldehydes 4 mediated by a Meth-Cohn type methodology through the Vilsmeier–Haack (DMF + POCl3) reagent [59,60]. A subsequent sequence of hydrolysis and N/O-alkylation processes, respectively, afforded the starting quinoline-/quinolone aldehydes 6{1–7} in 70–85% yield as described previously [54] (Scheme 4).
![[1860-5397-15-60-i4]](/bjoc/content/inline/1860-5397-15-60-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: General procedure for the synthesis of the starting quinoline-/quinolone aldehydes 6{1–7}.
Scheme 4: General procedure for the synthesis of the starting quinoline-/quinolone aldehydes 6{1–7}.
Additionally, a chemset of hydroxycoumarins 7{1–4} (Scheme 5) was chosen as the second source of nucleophilic partners for elaboration in our MCR experiments.
![[1860-5397-15-60-i5]](/bjoc/content/inline/1860-5397-15-60-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Chemset of coumarins 7{1–4} for elaboration in the MCR experiments.
Scheme 5: Chemset of coumarins 7{1–4} for elaboration in the MCR experiments.
With these building blocks at hand, an initial three-component assay was performed starting with indole 1{1} (1.0 equiv), quinoline aldehyde 6{1} (1.0 equiv) and coumarin 7{1} (1.0 equiv) in ethanol as solvent with no catalyst. The mixture was subjected to stirring at ambient temperature, and the reaction progress was monitored by TLC. After 24 h, the starting materials 1{1} and 6{1} were almost totally consumed, but several spots were observed (including unreacted 7{1}), with two of them as main components. A white solid fell out of solution, which was collected by filtration and washed with cold ethanol. NMR and HRMS analysis showed that it corresponded to the bisindole derivative 8{1,1,1}. The remaining crude reaction mixture was purified by column chromatography, and led to isolation of a second major component corresponding to the desired three-component product 9{1,1,1}. The relative weight ratios of the two isolated products 8{1,1,1} and 9{1,1,1} were circa 1:1 (Scheme 6).
![[1860-5397-15-60-i6]](/bjoc/content/inline/1860-5397-15-60-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Exploratory reaction leading to isolation of products 8{1,1,1} and 9{1,1,1}.
Scheme 6: Exploratory reaction leading to isolation of products 8{1,1,1} and 9{1,1,1}.
These initial findings encouraged us to perform an in-depth study aimed at optimizing chemoselectivity. As a model reaction, an equimolar three-component mixture of precursors 1{1}, 6{2} and 7{1} was subjected to various catalyzed and uncatalyzed conditions and the results are summarized in Table 1.
Table 1: Optimization of the reaction conditions for the three-component synthesis of triads 8{1,1,2} and 9{1,2,1}.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-60-i9.svg?max-width=637&scale=1.0)
|
||||||
| entrya |
solvent
(2 mL) |
catalyst
(mol %) |
temp.
(°C) |
time
(h) |
bisindole triad 8{1,1,2}
(% w/w) |
tris-triad 9{1,2,1}
(% w/w) |
| 1 | EtOH | – | rt | 24 | ≈50 | ≈50 |
| 2 | EtOH | – | reflux | 3 | ≈50 | ≈50 |
| 3 | ACN | Yb(OTf)3 (5) | rt | 6 | 100 | – |
| 4 | ACN | Sc(OTf)3 (5) | rt | 3 | 100 | – |
| 5 | ACN | Al(OTf)3 (5) | rt | 3 | 100 | – |
| 6 | ACN | Bi(OTf)3 (5) | rt | 3 | 100 | – |
| 7 | ACN | I2 (5) | rt | 1 | 100 | – |
| 8 | ACN | BF3·OEt (5) | rt | 2 | 100 | – |
| 9 | ACN | – | rt | 48 | ≈50 | ≈50 |
| 10 | EtOHb | AcOH (0.5 mL) | rt | 8 | ≈50 | ≈50 |
| 11 | H2O | – | reflux | 3 | ≈67 | ≈33 |
aAll reactions were performed starting with compound 1{1} (10 mg), 6{2} (20 mg) and 7{1} (13 mg) corresponding to a 1:1:1 mmolar ratio. b1.5 mL of EtOH was used.
Further studies showed that the Lewis acid-catalyzed reactions (Table 1, entries 3–8) greatly favored the formation of bisindole triad 8{1,1,2}, while EtOH at room temperature produced an optimal (circa 1:1 w/w) mixture of 8{1,1,2} and 9{1,2,1} (Table 1, entry 1), and reflux accelerated the process without affecting the w/w ratio (Table 1, entry 2). The reaction time was notably shorter in EtOH at rt in the presence of AcOH as catalyst (Table 1, entry 10), while longer reaction times were noted when MeCN was used as solvent at rt (compare entry 9 and entry 1). Finally, performing the reaction in hot water instead of EtOH resulted in a 2:1 mixture of 8 and 9.
Using the outcomes in Table 1 as a guide, an adaptation of entry 3 was chosen to obtain a library of diversely substituted bisindole triads 8. Since coumarin 7 remained unreacted in this approach the examples described in Figure 1 were performed by employing a 2:1 ratio of precursors 1 and 6, respectively, in the absence of coumarin 7.
![[1860-5397-15-60-1]](/bjoc/content/figures/1860-5397-15-60-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Pseudo-three-component synthesis of bisindole triads 8 employing quinoline-/quinolone-CHO 6{1–6}, chromone-CHO 6{8–9} and fluorene-CHO 6{10} as coupling partners. Although entries 4 and 7 (Table 1) were satisfactory, reactions of Figure 1 were performed by following an adaptation of entry 3 (using Yb(OTf)3 with a 2:1 ratio of 1 and 6, respectively) due to lower catalyst cost (in comparison with Sc(OTf)3) and/or easier work-up (in comparison with I2) (see experimental section). aThis product was obtained as an inseparable mixture along compound 9{9,6,2} from the approach described in entry 2 of Table 1 (see also Supporting Information File 1).
Figure 1: Pseudo-three-component synthesis of bisindole triads 8 employing quinoline-/quinolone-CHO 6{1–6}, c...
For a broader scope of this approach, bisindole triads 8{4,4,8}, 8{4,4,9}, 8{10,10,9} and 8{1,1,10} were also synthesized in good yields by replacing the corresponding quinoline-/quinolone aldehydes 6{1–6} with 4-oxo-4H-chromene-3-carbaldehyde (6{8}), 6-fluoro-4-oxo-4H-chromene-3-carbaldehyde (6{9}), and 9H-fluorene-2-carbaldehyde (6{10}), respectively (Scheme 7 and Figure 1).
![[1860-5397-15-60-i7]](/bjoc/content/inline/1860-5397-15-60-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Chemset of further aldehydes 6{8–10} for elaboration in the MCR experiments.
Scheme 7: Chemset of further aldehydes 6{8–10} for elaboration in the MCR experiments.
Focusing our attention on the synthesis of diversely substituted tris(heteroaryl)methane triads of type 9 via a three-component procedure, the approach described in Table 1, entry 2 was adopted, and the method was extended to a variety of indoles 1, quinoline-/quinolone- and chromene aldehydes 6, and hydroxycoumarins 7 as illustrated in Schemes 3–5, leading to a set of novel tris(heteroaryl)methane triads 9{1,1,1} to 9{6,4,1}, as shown in Figure 2.
![[1860-5397-15-60-2]](/bjoc/content/figures/1860-5397-15-60-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Three-component synthesis of tris(heteroaryl)methane triads 9. aThis product was obtained as an inseparable mixture along compound 8{9,9,6} (see Supporting Information File 1).
Figure 2: Three-component synthesis of tris(heteroaryl)methane triads 9. aThis product was obtained as an ins...
Structures of the newly obtained triads 8 and 9 were ascertained by 1D and 2D NMR spectroscopy and by EIMS, elemental analysis, and HRMS (see experimental section and Supporting Information File 1). Additionally, single crystals of compound 9{4,7,1} suitable for X-ray analysis were grown from ACN at room temperature. Compound 9{4,7,1} crystallizes in the triclinic space group ![[Graphic 2]](/bjoc/content/inline/1860-5397-15-60-i10.svg?max-width=637&scale=1.18182) (Figure 3). The asymmetric unit corresponds to one molecule of 9{4,7,1} and one molecule of ACN. A packing diagram is shown in Figure S1 (Supporting Information File 1). Interestingly, the unit cell consists of a pair of enantiomers. Within the structure of 9{4,7,1}, there is a short distance between the quinolone carbonyl and the OH of hydroxycoumarin (H(1)–O(4) 1.740 Å).
(Figure 3). The asymmetric unit corresponds to one molecule of 9{4,7,1} and one molecule of ACN. A packing diagram is shown in Figure S1 (Supporting Information File 1). Interestingly, the unit cell consists of a pair of enantiomers. Within the structure of 9{4,7,1}, there is a short distance between the quinolone carbonyl and the OH of hydroxycoumarin (H(1)–O(4) 1.740 Å).
![[1860-5397-15-60-3]](/bjoc/content/figures/1860-5397-15-60-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Thermal ellipsoid plot (40% probability level) of the tris(heteroaryl)methane triad 9{4,7,1}.
Figure 3: Thermal ellipsoid plot (40% probability level) of the tris(heteroaryl)methane triad 9{4,7,1}.
The DFT-optimized structure of 9{4,7,1} (Figure 4) confirms the formation of a highly stable hydrogen bond between the quinolone carbonyl and the OH of hydroxycoumarin, with a O···H bond distance of 1.603 Å. It should be noted that the hydrogen-bonded conformation is ca. 15 kcal/mol more stable than other rotamers that do not present this O···H interaction.
![[1860-5397-15-60-4]](/bjoc/content/figures/1860-5397-15-60-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: DFT-optimized structure of 9{4,7,1} triad.
Figure 4: DFT-optimized structure of 9{4,7,1} triad.
In the next phase of the study the possibility to synthesize crowded tris(heteroaryl/aryl)methylium salts from 8{4,4,8} and 8{4,4,11} was examined. Whereas attempts to cleanly generate the salts by hydride abstraction with trityl-BF4 were unsuccessful [61], presumably due to extreme steric crowding, the reaction with DDQ/HPF6 (Scheme 8) [62-65] was successful and the methylium-PF6 salts 10{4,4,8} and 10{4,4,11}, respectively, precipitated from DCM as purple solids. Both salts were studied in detail by 1D and 2D (COSY, DEPT, HSQC, and HMBC) NMR. Restricted rotation of the N-methylindole moiety is clearly deduced from 1H NMR for both methylium salts by broadening the pair of protons at δ 8.82/7.43 and 8.62/6.85 ppm, respectively (Figure 5, Figure 6 and Supporting Information File 1). Assignments of the quaternary carbons including the formal carbocation centers were made by HMBC correlations.
![[1860-5397-15-60-i8]](/bjoc/content/inline/1860-5397-15-60-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of crowed (Het12Het2/Ar2)C+PF6− salts 10.
Scheme 8: Synthesis of crowed (Het12Het2/Ar2)C+PF6− salts 10.
![[1860-5397-15-60-5]](/bjoc/content/figures/1860-5397-15-60-5.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 5: 1D- and 2D-based NMR assignments for methylium-PF6 salt 10{4,4,8}.
Figure 5: 1D- and 2D-based NMR assignments for methylium-PF6 salt 10{4,4,8}.
![[1860-5397-15-60-6]](/bjoc/content/figures/1860-5397-15-60-6.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 6: 1D- and 2D-based NMR assignments for methylium-PF6 salt 10{4,4,11}.
Figure 6: 1D- and 2D-based NMR assignments for methylium-PF6 salt 10{4,4,11}.
NMR data suggest that the positive charge is more effectively delocalized into the indole rings. The GIAO-NMR data show the same general trend, as evidenced by the 13C Δδ chemical shifts, with largest charge locations at the conjugated carbon of the indole ring (Figure S2, Supporting Information File 1). The DFT-optimized structures of methylium-PF6 salts 10{4,4,8} and 10{4,4,11} are shown in Figure 7 and Figure 8, where close cation–anion contacts are observed despite significant steric crowding. Steric congestion restricts the conjugation of the carbocationic center with the aromatic/heteroaromatic substituents, as evidenced by the bond length shortenings from only 0.052 Å to 0.111 Å observed upon hydride abstraction. The optimized geometries confirm the restricted rotation of the N-methylindole moiety deduced from experimental 1H NMR for both methylium salts as described above (broadening of pair of protons at δ 8.82/7.43 and 8.62/6.85 ppm), as this moiety is anchored by the position of the PF6− anion (Figure 7 and Figure 8). The distance between the formal carbocationic center and the closest fluorine atom was 3.084 Å in the methylium-PF6 salt 10{4,4,8}, and 3.275 Å in case of the 10{4,4,11} salt. Moreover, C–H···F interactions where also observed, with H···F bond distances between 2.094 Å and 2.575 Å.
![[1860-5397-15-60-7]](/bjoc/content/figures/1860-5397-15-60-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Optimized geometry of methylium-PF6 salts 10{4,4,8}.
Figure 7: Optimized geometry of methylium-PF6 salts 10{4,4,8}.
![[1860-5397-15-60-8]](/bjoc/content/figures/1860-5397-15-60-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Optimized geometry of methylium-PF6 salt 10{4,4,11}.
Figure 8: Optimized geometry of methylium-PF6 salt 10{4,4,11}.
Conclusion
A facile one-pot method for the three-component synthesis of ternary heteroarylmethane-inspired hybrids is presented, by coupling quinoline aldehydes, quinolone aldehydes, chromone aldehydes, and fluorene aldehydes with substituted indoles and coumarins. The method enabled the synthesis of novel libraries of giant (Ar1Ar1Ar2)CH and (Ar1Ar2Ar3)CH triads 8 and 9, respectively, packed with up to three different pharmacophors in a single molecule. The ability to perform these reactions in ethanol and even in water, with no catalysts is noteworthy. Representative methylium salts generated by ionization with DDQ/HPF6 exhibited 1H NMR signal broadening reflecting restricted rotation of the N-methylindole moieties at room temperature.
Experimental
General. Melting points were measured using a Stuart SMP3 melting point apparatus and are uncorrected. IR spectra were recorded on a Shimadzu IRAffinity-1 spectrophotometer by ATR method. 1H and 13C NMR spectra were recorded on Bruker Avance 400 and Varian INOVA 500 MHz instruments using DMSO-d6 and CDCl3 as solvents with and without added TMS as internal standard. Mass spectra were run on a SHIMADZU-GCMS 2010-DI-2010 spectrometer (equipped with a direct inlet probe) operating at 70 eV. HRMS analyses were performed on a Finnigan Quantum ultra-AM in electrospray mode using methanol as solvent. Single-crystal X-ray data for compound 9{4,7,1} was collected at 200 K on a Bruker AXS diffractometer upgraded with an APEX II CCD detector. Crystallographic data for the structure has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no: CCDC 1864804. TLC analyses were performed on silica gel aluminum plates (Merck 60 F254) and spots visualized under UV light. The starting precursors and reagents for the synthesis of indoles 1{4–9} and quinoline-/quinolone aldehydes 6, and the required solvents were purchased from Sigma-Aldrich, Fluka and Merck (analytical grade reagent), and were used without further purification.
Catalyzed general procedure for the direct synthesis of bisindoles 8. A mixture of indole 1 (2.0 equiv), aldehyde 6 (1.0 equiv), Yb(OTf)3 (5 mol %) and ACN (2 mL), was stirred at ambient temperature for 6 h until the starting materials 1 and 6 were no longer detected by TLC. The white precipitate formed was collected by filtration and washed with cold EtOH (2 × 0.5 mL). No further purification of product 8 was required. Alternatively, the more expensive Lewis acid Sc(OTf)3 was used instead of Yb(OTf)3 with similar behavior and results, although, reactions just took about 3 h. In the case of I2, although, the reaction worked quite well, the isolation of products 8 required filtering the colored solid formed and treatment of the re-dissolved solid in ethyl acetate with sodium thiosulfate to destroy the excess iodine. Finally, purification of the crude reaction mixtures by column chromatography was required in all cases.
Uncatalyzed general procedure for the synthesis of products 9. An equimolar mixture of the appropriate indole 1 (1.0 equiv), aldehyde 6 (1.0 equiv), and 4-hydroxycoumarin 7 (1.0 equiv) was dissolved in ethanol (2 mL). The solution was heated to reflux for 3 h until the starting materials 1 and 6 were no longer detected by TLC. After the solvent was removed under reduced pressure, the crude reaction mixture was purified by column chromatography on silica gel, using hexane/EtOAc (7:3) as eluent. The desired products 9 along with the side-products 8 were isolated and quantified.
General procedure for the synthesis of carbocation salts 10{4,4,8} and 10{4,4,11}. DDQ (2 equiv) was added to a solution of 3,3'-(arylmethylene)bis(1-methyl-1H-indoles) 8{4,4,8} or 8{4,4,11} (50 mg, 1 equiv) in CH2Cl2 (8 mL) at room temperature. After the solution was stirred at the same temperature for 30 min, 60% HPF6 (1 mL) and water (10 mL) were added to the mixture. The resulting suspension was filtered with suction. The organic layer was washed with water, dried over MgSO4, and concentrated under reduced pressure. Finally, the crystals were obtained after simple trituration with Et2O.
Computational methods. Density functional theory (DFT) calculations were carried out with the Gaussian 09 program suite [66]. Geometries were fully optimized at the B3LYP [67-69]/6-311+G(d,p) level. Stationary points were characterized as minima by harmonic vibrational frequency calculations (no imaginary frequencies). NMR chemical shifts were computed by the GIAO (gauge independent atomic orbitals) [70,71] method at the B3LYP/6-311+G(d,p) level. The 1H and 13C NMR chemical shifts were referenced to TMS. GIAO magnetic shielding tensors were 31.88 for 1H, 182.5 for 13C, values related to the GIAO isotropic magnetic susceptibility.
X-ray crystallography. Colorless crystals were isolated for 9{4,7,1} from acetonitrile and used for the following X-ray diffraction studies. A crystal was mounted onto a fiber from FluorolubeTM and was placed under a liquid N2 cooled stream, on a Bruker AXS diffractometer updated with an APEX II CCD detector. The radiation used was graphite monochromatized Mo Kα radiation (λ = 0.7107 Å). Lattice determination, data collection, structure refinement, scaling, and data reduction were carried out using the APEX2 Version 2014.11 software package [72,73]. The data were corrected for absorption using the SCALE program within the APEX2 software suite [72,73]. The structure was solved using SHELXT [74]. This procedure yielded a number of the C, N and O atoms. Subsequent Fourier synthesis yielded the remaining atom positions. The hydrogen atoms are fixed in positions of ideal geometry (riding model) and refined within the XSHELL software package [75]. The final refinement of the compound included anisotropic thermal parameters on all non-hydrogen atoms was performed using OLEX2-1.2 [76]. The crystal data for compound 9{4,7,1} is given in Table S1, and a packing diagram is shown in Figure S1 [77] (Supporting Information File 1). Crystallographic data for the structure has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no: CCDC 1864804. Copies of the data can be obtained on application to the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax, +44-(0)1223-336033; or email, deposit@ccdc.cam.ac.uk).
Acknowledgements
Authors thank COLCIENCIAS and Universidad del Valle for financial support—Project Numbers 110665842661 and CI-71111. L.G. specially thanks COLCIENCIAS for her “Joven Investigador” fellowship award. K.K.L. thanks the University of North Florida for the outstanding faculty scholarship and presidential professorship awards, as well as faculty scholarship and UNF Foundation Board grants. G.L.B. acknowledges funding from CONICET and Secyt-UNC.
References
-
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] [2] -
Shiri, M. Chem. Rev. 2012, 112, 3508–3549. doi:10.1021/cr2003954
Return to citation in text: [1] [2] -
da Silveira Pinto, L. S.; Couri, M. R. C.; de Souza, M. V. N. Curr. Org. Synth. 2018, 15, 21–37. doi:10.2174/1570179414666170614124053
Return to citation in text: [1] [2] -
Mohammadi Ziarani, G.; Moradi, R.; Ahmadi, T.; Lashgari, N. RSC Adv. 2018, 8, 12069–12103. doi:10.1039/c7ra13321a
Return to citation in text: [1] [2] -
Vroemans, R.; Verhaegen, Y.; Dieu, M. T. T.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 2689–2697. doi:10.3762/bjoc.14.246
Return to citation in text: [1] [2] -
Shanab, K.; Neudorfer, C.; Schirmer, E.; Spreitzer, H. Curr. Org. Chem. 2013, 17, 1179–1187. doi:10.2174/1385272811317110005
Return to citation in text: [1] [2] -
Gu, Y. Green Chem. 2012, 14, 2091–2128. doi:10.1039/c2gc35635j
Return to citation in text: [1] [2] -
Nair, V.; Thomas, S.; Mathew, S. C.; Abhilash, K. G. Tetrahedron 2006, 62, 6731–6747. doi:10.1016/j.tet.2006.04.081
Return to citation in text: [1] [2] [3] -
Podder, S.; Choudhury, J.; Roy, U. K.; Roy, S. J. Org. Chem. 2007, 72, 3100–3103. doi:10.1021/jo062633n
Return to citation in text: [1] [2] [3] -
Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875
Return to citation in text: [1] [2] [3] -
Khaksar, S.; Vahdat, S. M.; Gholizadeh, M.; Talesh, S. M. J. Fluorine Chem. 2012, 136, 8–11. doi:10.1016/j.jfluchem.2012.01.002
Return to citation in text: [1] [2] -
Kumar Das, S.; Shagufta; Panda, G. Tetrahedron Lett. 2005, 46, 3097–3102. doi:10.1016/j.tetlet.2005.03.001
Return to citation in text: [1] [2] -
Bushby, S. R. M. Toxicity in Chemotherapy. In Experimental Chemotherapy; Schnitzer, R. J.; Hawking, F., Eds.; Academic Press: New York, NY, 1963; Vol. 1, pp 25–53. doi:10.1016/c2013-0-12425-7
Return to citation in text: [1] [2] -
Yamato, M.; Hashigaki, K.; Yasumoto, Y.; Sakai, J.; Luduena, R. F.; Banerjee, A.; Tsukagoshi, S.; Tashiro, T.; Tsuruo, T. J. Med. Chem. 1987, 30, 1897–1900. doi:10.1021/jm00393a035
Return to citation in text: [1] [2] -
Wulff, H.; Miller, M. J.; Hänsel, W.; Grissmer, S.; Cahalan, M. D.; Chandy, K. G. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8151–8156. doi:10.1073/pnas.97.14.8151
Return to citation in text: [1] [2] -
Mibu, N.; Yokomizo, K.; Uyeda, M.; Sumoto, K. Chem. Pharm. Bull. 2003, 51, 1325–1327. doi:10.1248/cpb.51.1325
Return to citation in text: [1] [2] -
Rys, P.; Zollinger, H. Fundamentals of the Chemistry and Application of Dyes; Wiley-Interscience: New York, NY, 1972.
Return to citation in text: [1] [2] -
Aldag, R. Photochromism based on dissociation processes. In Photochromism: Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: London, 1990.
Return to citation in text: [1] [2] -
Shchepinov, M. S.; Korshun, V. A. Chem. Soc. Rev. 2003, 32, 170–180. doi:10.1039/b008900l
Return to citation in text: [1] [2] -
Baker, L. A.; Sun, L.; Crooks, R. M. Bull. Korean Chem. Soc. 2002, 23, 647–654. doi:10.5012/bkcs.2002.23.5.647
Return to citation in text: [1] [2] -
Sanguinet, L.; Twieg, R. J.; Wiggers, G.; Mao, G.; Singer, K. D.; Petschek, R. G. Tetrahedron Lett. 2005, 46, 5121–5125. doi:10.1016/j.tetlet.2005.05.138
Return to citation in text: [1] [2] -
Yu, L.; Li, S.-S.; Li, W.; Yu, S.; Liu, Q.; Xiao, J. J. Org. Chem. 2018, 83, 15277–15283. doi:10.1021/acs.joc.8b02549
Return to citation in text: [1] [2] -
Oikawa, Y.; Hirasawa, H.; Yonemitsu, O. Tetrahedron Lett. 1978, 19, 1759–1762. doi:10.1016/0040-4039(78)80037-9
Return to citation in text: [1] -
Lü, C.-W.; Wang, J.-J.; Liu, Y.-H.; Shan, W.-J.; Sun, Q.; Shi, L. Res. Chem. Intermed. 2017, 43, 943–949. doi:10.1007/s11164-016-2675-8
Return to citation in text: [1] -
Renzetti, A.; Boffa, E.; Colazzo, M.; Gérard, S.; Sapi, J.; Chan, T.-H.; Nakazawa, H.; Villani, C.; Fontana, A. RSC Adv. 2014, 4, 47992–47999. doi:10.1039/c4ra08853k
Return to citation in text: [1] -
Gérard, S.; Renzetti, A.; Lefevre, B.; Fontana, A.; de Maria, P.; Sapi, J. Tetrahedron 2010, 66, 3065–3069. doi:10.1016/j.tet.2010.02.025
Return to citation in text: [1] -
Viegas-Junior, C.; Danuello, A.; da Silva-Bolzani, V.; Barreiro, E. J.; Fraga, C. A. M. Curr. Med. Chem. 2007, 14, 1829–1852. doi:10.2174/092986707781058805
Return to citation in text: [1] -
Zhang, S.; Saathoff, J. M.; He, L. Molecular Hybridization: An Emerging Tool for the Design of Novel Therapeutics for Alzheimer’s Disease. In Design of Hybrid Molecules for Drug Development; Decker, M., Ed.; Elsevier: Amsterdam, 2017; pp 219–237. doi:10.1016/b978-0-08-101011-2.00008-8
Return to citation in text: [1] -
Rad-Moghadam, K.; Mohseni, M. Monatsh. Chem. 2004, 135, 817–821. doi:10.1007/s00706-003-0138-6
Return to citation in text: [1] -
Alizadeh, A.; Ghanbaripour, R.; Zhu, L.-G. Tetrahedron 2014, 70, 2048–2053. doi:10.1016/j.tet.2014.01.038
Return to citation in text: [1] -
El-Mekabaty, A.; Mesbah, A.; Fadda, A. A. J. Heterocycl. Chem. 2017, 54, 916–922. doi:10.1002/jhet.2654
Return to citation in text: [1] -
Joule, J. A. In Houben-Weyl Methods of Molecular Transformations; Thomas, E. J., Ed.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany , 2000; p 10.
Return to citation in text: [1] -
Mugnaini, C.; Nocerino, S.; Pedani, V.; Pasquini, S.; Tafi, A.; De Chiaro, M.; Bellucci, L.; Valoti, M.; Guida, F.; Luongo, L.; Dragoni, S.; Ligresti, A.; Rosenberg, A.; Bolognini, D.; Cascio, M. G.; Pertwee, R. G.; Moaddel, R.; Maione, S.; Di Marzo, V.; Corelli, F. ChemMedChem 2012, 7, 920–934. doi:10.1002/cmdc.201100573
Return to citation in text: [1] -
Chung, H. J.; Kamli, M. R.; Lee, H. J.; Ha, J. D.; Cho, S. Y.; Lee, J.; Kong, J. Y.; Han, S.-Y. Biochem. Biophys. Res. Commun. 2014, 445, 561–565. doi:10.1016/j.bbrc.2014.02.029
Return to citation in text: [1] -
Rezayan, A. H.; Hariri, S.; Azerang, P.; Ghavami, G.; Portugal, I.; Sardari, S. Iran. J. Pharm. Res. 2017, 16, 745–755.
Return to citation in text: [1] -
Hranjec, M.; Lučić, B.; Ratkaj, I.; Kraljević Pavelić, S.; Piantanida, I.; Pavelić, K.; Karminski-Zamola, G. Eur. J. Med. Chem. 2011, 46, 2748–2758. doi:10.1016/j.ejmech.2011.03.062
Return to citation in text: [1] -
Desyatkin, V. G.; Beletskaya, I. P. Synthesis 2017, 49, 4327–4334. doi:10.1055/s-0036-1589024
Return to citation in text: [1] -
Karthik, G.; Rajasekaran, T.; Sridhar, B.; Subba Reddy, B. V. Tetrahedron 2014, 70, 8148–8154. doi:10.1016/j.tet.2014.07.093
Return to citation in text: [1] -
Jadhav, S. D.; Bakshi, D.; Singh, A. J. Org. Chem. 2015, 80, 10187–10196. doi:10.1021/acs.joc.5b01736
Return to citation in text: [1] [2] -
Shelke, G. M.; Kumar, A. Synthesis 2017, 49, 4321–4326. doi:10.1055/s-0036-1588181
Return to citation in text: [1] [2] -
Deb, M. L.; Deka, B.; Saikia, P. J.; Baruah, P. K. Tetrahedron Lett. 2017, 58, 1999–2003. doi:10.1016/j.tetlet.2017.04.032
Return to citation in text: [1] [2] -
Teguh, S. C.; Klonis, N.; Duffy, S.; Lucantoni, L.; Avery, V. M.; Hutton, C. A.; Baell, J. B.; Tilley, L. J. Med. Chem. 2013, 56, 6200–6215. doi:10.1021/jm400656s
Return to citation in text: [1] -
Lin, W.; Yuan, L.; Cao, Z.; Feng, J.; Feng, Y. Dyes Pigm. 2009, 83, 14–20. doi:10.1016/j.dyepig.2009.03.006
Return to citation in text: [1] -
Khan, M. N.; Pal, S.; Karamthulla, S.; Choudhury, L. H. New J. Chem. 2014, 38, 4722–4729. doi:10.1039/c4nj00630e
Return to citation in text: [1] -
Oelerich, J.; Roelfes, G. Org. Biomol. Chem. 2015, 13, 2793–2799. doi:10.1039/c4ob02487g
Return to citation in text: [1] -
Shao, Z.; Xu, L.; Wang, L.; Wei, H.; Xiao, J. Org. Biomol. Chem. 2014, 12, 2185–2188. doi:10.1039/c3ob42582g
Return to citation in text: [1] -
Jennings, J. J.; Bhatt, C. P.; Franz, A. K. J. Org. Chem. 2016, 81, 6211–6222. doi:10.1021/acs.joc.6b00541
Return to citation in text: [1] -
Prabhakar, M.; Reddy, G. N.; Srinu, G.; Manjulatha, K.; Prasad, J. V.; Kumar, S. P.; Srinivas, O.; Iqbal, J.; Kumar, K. A. Synlett 2010, 947–951. doi:10.1055/s-0029-1219532
Return to citation in text: [1] -
Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a
Return to citation in text: [1] -
Kottawar, S. S.; Siddiqui, S. A.; Pendalwar, S. S.; Jadhav, W. N.; Bhusare, S. R. Res. Chem. Intermed. 2014, 40, 2929–2934. doi:10.1007/s11164-013-1139-7
Return to citation in text: [1] -
Shelke, G. M.; Rao, V. K.; Tiwari, R. K.; Chhikara, B. S.; Parang, K.; Kumar, A. RSC Adv. 2013, 3, 22346–22352. doi:10.1039/c3ra44693j
Return to citation in text: [1] -
Appendino, G.; Cicione, L.; Minassi, A. Tetrahedron Lett. 2009, 50, 5559–5561. doi:10.1016/j.tetlet.2009.05.033
Return to citation in text: [1] [2] -
Mousavizadeh, F.; Hekmatshoar, R.; Beheshtiha, S. Y. S.; Rahnamafar, R. Synth. Commun. 2014, 44, 1483–1491. doi:10.1080/00397911.2013.862723
Return to citation in text: [1] -
Laali, K. K.; Insuasty, D.; Abonia, R.; Insuasty, B.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 4395–4399. doi:10.1016/j.tetlet.2014.05.094
Return to citation in text: [1] [2] -
Ramírez-Prada, J.; Robledo, S. M.; Vélez, I. D.; del Pilar Crespo, M.; Quiroga, J.; Abonia, R.; Montoya, A.; Svetaz, L.; Zacchino, S.; Insuasty, B. Eur. J. Med. Chem. 2017, 131, 237–254. doi:10.1016/j.ejmech.2017.03.016
Return to citation in text: [1] -
Abonia, R.; Insuasty, D.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Eur. J. Med. Chem. 2012, 57, 29–40. doi:10.1016/j.ejmech.2012.08.039
Return to citation in text: [1] -
Insuasty, D.; Abonia, R.; Insuasty, B.; Quiroga, J.; Laali, K. K.; Nogueras, M.; Cobo, J. ACS Comb. Sci. 2017, 19, 555–563. doi:10.1021/acscombsci.7b00091
Return to citation in text: [1] -
Kong, A.; Han, X.; Lu, X. Org. Lett. 2006, 8, 1339–1342. doi:10.1021/ol060039u
Return to citation in text: [1] -
Meth-Cohn, O.; Narine, B. Tetrahedron Lett. 1978, 19, 2045–2048. doi:10.1016/s0040-4039(01)94745-8
Return to citation in text: [1] -
Meth-Cohn, O.; Narine, B.; Tarnowski, B. J. Chem. Soc., Perkin Trans. 1 1981, 1520–1530. doi:10.1039/p19810001520
Return to citation in text: [1] -
Oda, M.; Fukuta, A.; Kajioka, T.; Uchiyama, T.; Kainuma, H.; Miyatake, R.; Kuroda, S. Tetrahedron 2000, 56, 9917–9925. doi:10.1016/s0040-4020(00)00964-9
See for an example.
Return to citation in text: [1] -
Ito, S.; Akimoto, K.; Morita, N. In Recent Developments in Carbocation and Onium Ion Chemistry; Laali, K. K., Ed.; ACS Symposium Series 965; ACS: Washington, DC, 2007; pp 160–183.
See for DDQ/HPF6 previously employed for generation of tri(1-azulenyl)methyl cation salts.
Return to citation in text: [1] -
Ito, S.; Morita, N.; Asao, T. Bull. Chem. Soc. Jpn. 1995, 68, 1409–1436. doi:10.1246/bcsj.68.1409
Return to citation in text: [1] -
Pfeuffer, L.; Mueller, J.; Pindur, U. Chimia 1985, 39, 141–142.
See for 3-indolylmethylethoxycarbenium BF4 salt.
Return to citation in text: [1] -
Pindur, U.; Mueller, J. Chem.-Ztg. 1986, 110, 333–335.
See for a UV–vis study of diindolylmethylium.
Return to citation in text: [1] -
Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785
Return to citation in text: [1] -
Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200–206. doi:10.1016/0009-2614(89)87234-3
Return to citation in text: [1] -
Wolinski, K.; Hinton, J. F.; Pulay, P. J. Am. Chem. Soc. 1990, 112, 8251–8260. doi:10.1021/ja00179a005
Return to citation in text: [1] -
Ditchfield, R. Mol. Phys. 1974, 27, 789–807. doi:10.1080/00268977400100711
Return to citation in text: [1] -
APEX II; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2005.
Return to citation in text: [1] [2] -
SAINT, Version 6.45A; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2003.
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
SHELXTL, V6.12; Bruker AXS, Inc., Analytical X-ray Systems: East Cheryl Parkway, Madison WI 53711–5373, USA, 2002.
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
DIAMOND, Bonn, Germany; Crystal Impact GbR, 1999.
Return to citation in text: [1]
| 72. | APEX II; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2005. |
| 73. | SAINT, Version 6.45A; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2003. |
| 72. | APEX II; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2005. |
| 73. | SAINT, Version 6.45A; Bruker AXS, Inc., Analytical X-ray Systems: 5465 East Cheryl Parkway, Madison WI 53711–5373, USA, 2003. |
| 74. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 1. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 2. | Shiri, M. Chem. Rev. 2012, 112, 3508–3549. doi:10.1021/cr2003954 |
| 3. | da Silveira Pinto, L. S.; Couri, M. R. C.; de Souza, M. V. N. Curr. Org. Synth. 2018, 15, 21–37. doi:10.2174/1570179414666170614124053 |
| 4. | Mohammadi Ziarani, G.; Moradi, R.; Ahmadi, T.; Lashgari, N. RSC Adv. 2018, 8, 12069–12103. doi:10.1039/c7ra13321a |
| 5. | Vroemans, R.; Verhaegen, Y.; Dieu, M. T. T.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 2689–2697. doi:10.3762/bjoc.14.246 |
| 13. | Bushby, S. R. M. Toxicity in Chemotherapy. In Experimental Chemotherapy; Schnitzer, R. J.; Hawking, F., Eds.; Academic Press: New York, NY, 1963; Vol. 1, pp 25–53. doi:10.1016/c2013-0-12425-7 |
| 14. | Yamato, M.; Hashigaki, K.; Yasumoto, Y.; Sakai, J.; Luduena, R. F.; Banerjee, A.; Tsukagoshi, S.; Tashiro, T.; Tsuruo, T. J. Med. Chem. 1987, 30, 1897–1900. doi:10.1021/jm00393a035 |
| 15. | Wulff, H.; Miller, M. J.; Hänsel, W.; Grissmer, S.; Cahalan, M. D.; Chandy, K. G. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8151–8156. doi:10.1073/pnas.97.14.8151 |
| 16. | Mibu, N.; Yokomizo, K.; Uyeda, M.; Sumoto, K. Chem. Pharm. Bull. 2003, 51, 1325–1327. doi:10.1248/cpb.51.1325 |
| 37. | Desyatkin, V. G.; Beletskaya, I. P. Synthesis 2017, 49, 4327–4334. doi:10.1055/s-0036-1589024 |
| 38. | Karthik, G.; Rajasekaran, T.; Sridhar, B.; Subba Reddy, B. V. Tetrahedron 2014, 70, 8148–8154. doi:10.1016/j.tet.2014.07.093 |
| 39. | Jadhav, S. D.; Bakshi, D.; Singh, A. J. Org. Chem. 2015, 80, 10187–10196. doi:10.1021/acs.joc.5b01736 |
| 40. | Shelke, G. M.; Kumar, A. Synthesis 2017, 49, 4321–4326. doi:10.1055/s-0036-1588181 |
| 41. | Deb, M. L.; Deka, B.; Saikia, P. J.; Baruah, P. K. Tetrahedron Lett. 2017, 58, 1999–2003. doi:10.1016/j.tetlet.2017.04.032 |
| 42. | Teguh, S. C.; Klonis, N.; Duffy, S.; Lucantoni, L.; Avery, V. M.; Hutton, C. A.; Baell, J. B.; Tilley, L. J. Med. Chem. 2013, 56, 6200–6215. doi:10.1021/jm400656s |
| 43. | Lin, W.; Yuan, L.; Cao, Z.; Feng, J.; Feng, Y. Dyes Pigm. 2009, 83, 14–20. doi:10.1016/j.dyepig.2009.03.006 |
| 44. | Khan, M. N.; Pal, S.; Karamthulla, S.; Choudhury, L. H. New J. Chem. 2014, 38, 4722–4729. doi:10.1039/c4nj00630e |
| 45. | Oelerich, J.; Roelfes, G. Org. Biomol. Chem. 2015, 13, 2793–2799. doi:10.1039/c4ob02487g |
| 46. | Shao, Z.; Xu, L.; Wang, L.; Wei, H.; Xiao, J. Org. Biomol. Chem. 2014, 12, 2185–2188. doi:10.1039/c3ob42582g |
| 47. | Jennings, J. J.; Bhatt, C. P.; Franz, A. K. J. Org. Chem. 2016, 81, 6211–6222. doi:10.1021/acs.joc.6b00541 |
| 48. | Prabhakar, M.; Reddy, G. N.; Srinu, G.; Manjulatha, K.; Prasad, J. V.; Kumar, S. P.; Srinivas, O.; Iqbal, J.; Kumar, K. A. Synlett 2010, 947–951. doi:10.1055/s-0029-1219532 |
| 8. | Nair, V.; Thomas, S.; Mathew, S. C.; Abhilash, K. G. Tetrahedron 2006, 62, 6731–6747. doi:10.1016/j.tet.2006.04.081 |
| 9. | Podder, S.; Choudhury, J.; Roy, U. K.; Roy, S. J. Org. Chem. 2007, 72, 3100–3103. doi:10.1021/jo062633n |
| 10. | Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875 |
| 11. | Khaksar, S.; Vahdat, S. M.; Gholizadeh, M.; Talesh, S. M. J. Fluorine Chem. 2012, 136, 8–11. doi:10.1016/j.jfluchem.2012.01.002 |
| 12. | Kumar Das, S.; Shagufta; Panda, G. Tetrahedron Lett. 2005, 46, 3097–3102. doi:10.1016/j.tetlet.2005.03.001 |
| 39. | Jadhav, S. D.; Bakshi, D.; Singh, A. J. Org. Chem. 2015, 80, 10187–10196. doi:10.1021/acs.joc.5b01736 |
| 40. | Shelke, G. M.; Kumar, A. Synthesis 2017, 49, 4321–4326. doi:10.1055/s-0036-1588181 |
| 41. | Deb, M. L.; Deka, B.; Saikia, P. J.; Baruah, P. K. Tetrahedron Lett. 2017, 58, 1999–2003. doi:10.1016/j.tetlet.2017.04.032 |
| 49. | Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a |
| 50. | Kottawar, S. S.; Siddiqui, S. A.; Pendalwar, S. S.; Jadhav, W. N.; Bhusare, S. R. Res. Chem. Intermed. 2014, 40, 2929–2934. doi:10.1007/s11164-013-1139-7 |
| 51. | Shelke, G. M.; Rao, V. K.; Tiwari, R. K.; Chhikara, B. S.; Parang, K.; Kumar, A. RSC Adv. 2013, 3, 22346–22352. doi:10.1039/c3ra44693j |
| 52. | Appendino, G.; Cicione, L.; Minassi, A. Tetrahedron Lett. 2009, 50, 5559–5561. doi:10.1016/j.tetlet.2009.05.033 |
| 8. | Nair, V.; Thomas, S.; Mathew, S. C.; Abhilash, K. G. Tetrahedron 2006, 62, 6731–6747. doi:10.1016/j.tet.2006.04.081 |
| 9. | Podder, S.; Choudhury, J.; Roy, U. K.; Roy, S. J. Org. Chem. 2007, 72, 3100–3103. doi:10.1021/jo062633n |
| 10. | Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875 |
| 27. | Viegas-Junior, C.; Danuello, A.; da Silva-Bolzani, V.; Barreiro, E. J.; Fraga, C. A. M. Curr. Med. Chem. 2007, 14, 1829–1852. doi:10.2174/092986707781058805 |
| 28. | Zhang, S.; Saathoff, J. M.; He, L. Molecular Hybridization: An Emerging Tool for the Design of Novel Therapeutics for Alzheimer’s Disease. In Design of Hybrid Molecules for Drug Development; Decker, M., Ed.; Elsevier: Amsterdam, 2017; pp 219–237. doi:10.1016/b978-0-08-101011-2.00008-8 |
| 6. | Shanab, K.; Neudorfer, C.; Schirmer, E.; Spreitzer, H. Curr. Org. Chem. 2013, 17, 1179–1187. doi:10.2174/1385272811317110005 |
| 7. | Gu, Y. Green Chem. 2012, 14, 2091–2128. doi:10.1039/c2gc35635j |
| 29. | Rad-Moghadam, K.; Mohseni, M. Monatsh. Chem. 2004, 135, 817–821. doi:10.1007/s00706-003-0138-6 |
| 30. | Alizadeh, A.; Ghanbaripour, R.; Zhu, L.-G. Tetrahedron 2014, 70, 2048–2053. doi:10.1016/j.tet.2014.01.038 |
| 31. | El-Mekabaty, A.; Mesbah, A.; Fadda, A. A. J. Heterocycl. Chem. 2017, 54, 916–922. doi:10.1002/jhet.2654 |
| 32. | Joule, J. A. In Houben-Weyl Methods of Molecular Transformations; Thomas, E. J., Ed.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany , 2000; p 10. |
| 33. | Mugnaini, C.; Nocerino, S.; Pedani, V.; Pasquini, S.; Tafi, A.; De Chiaro, M.; Bellucci, L.; Valoti, M.; Guida, F.; Luongo, L.; Dragoni, S.; Ligresti, A.; Rosenberg, A.; Bolognini, D.; Cascio, M. G.; Pertwee, R. G.; Moaddel, R.; Maione, S.; Di Marzo, V.; Corelli, F. ChemMedChem 2012, 7, 920–934. doi:10.1002/cmdc.201100573 |
| 34. | Chung, H. J.; Kamli, M. R.; Lee, H. J.; Ha, J. D.; Cho, S. Y.; Lee, J.; Kong, J. Y.; Han, S.-Y. Biochem. Biophys. Res. Commun. 2014, 445, 561–565. doi:10.1016/j.bbrc.2014.02.029 |
| 35. | Rezayan, A. H.; Hariri, S.; Azerang, P.; Ghavami, G.; Portugal, I.; Sardari, S. Iran. J. Pharm. Res. 2017, 16, 745–755. |
| 36. | Hranjec, M.; Lučić, B.; Ratkaj, I.; Kraljević Pavelić, S.; Piantanida, I.; Pavelić, K.; Karminski-Zamola, G. Eur. J. Med. Chem. 2011, 46, 2748–2758. doi:10.1016/j.ejmech.2011.03.062 |
| 21. | Sanguinet, L.; Twieg, R. J.; Wiggers, G.; Mao, G.; Singer, K. D.; Petschek, R. G. Tetrahedron Lett. 2005, 46, 5121–5125. doi:10.1016/j.tetlet.2005.05.138 |
| 1. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 2. | Shiri, M. Chem. Rev. 2012, 112, 3508–3549. doi:10.1021/cr2003954 |
| 3. | da Silveira Pinto, L. S.; Couri, M. R. C.; de Souza, M. V. N. Curr. Org. Synth. 2018, 15, 21–37. doi:10.2174/1570179414666170614124053 |
| 4. | Mohammadi Ziarani, G.; Moradi, R.; Ahmadi, T.; Lashgari, N. RSC Adv. 2018, 8, 12069–12103. doi:10.1039/c7ra13321a |
| 5. | Vroemans, R.; Verhaegen, Y.; Dieu, M. T. T.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 2689–2697. doi:10.3762/bjoc.14.246 |
| 6. | Shanab, K.; Neudorfer, C.; Schirmer, E.; Spreitzer, H. Curr. Org. Chem. 2013, 17, 1179–1187. doi:10.2174/1385272811317110005 |
| 7. | Gu, Y. Green Chem. 2012, 14, 2091–2128. doi:10.1039/c2gc35635j |
| 8. | Nair, V.; Thomas, S.; Mathew, S. C.; Abhilash, K. G. Tetrahedron 2006, 62, 6731–6747. doi:10.1016/j.tet.2006.04.081 |
| 9. | Podder, S.; Choudhury, J.; Roy, U. K.; Roy, S. J. Org. Chem. 2007, 72, 3100–3103. doi:10.1021/jo062633n |
| 10. | Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875 |
| 11. | Khaksar, S.; Vahdat, S. M.; Gholizadeh, M.; Talesh, S. M. J. Fluorine Chem. 2012, 136, 8–11. doi:10.1016/j.jfluchem.2012.01.002 |
| 12. | Kumar Das, S.; Shagufta; Panda, G. Tetrahedron Lett. 2005, 46, 3097–3102. doi:10.1016/j.tetlet.2005.03.001 |
| 13. | Bushby, S. R. M. Toxicity in Chemotherapy. In Experimental Chemotherapy; Schnitzer, R. J.; Hawking, F., Eds.; Academic Press: New York, NY, 1963; Vol. 1, pp 25–53. doi:10.1016/c2013-0-12425-7 |
| 14. | Yamato, M.; Hashigaki, K.; Yasumoto, Y.; Sakai, J.; Luduena, R. F.; Banerjee, A.; Tsukagoshi, S.; Tashiro, T.; Tsuruo, T. J. Med. Chem. 1987, 30, 1897–1900. doi:10.1021/jm00393a035 |
| 15. | Wulff, H.; Miller, M. J.; Hänsel, W.; Grissmer, S.; Cahalan, M. D.; Chandy, K. G. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8151–8156. doi:10.1073/pnas.97.14.8151 |
| 16. | Mibu, N.; Yokomizo, K.; Uyeda, M.; Sumoto, K. Chem. Pharm. Bull. 2003, 51, 1325–1327. doi:10.1248/cpb.51.1325 |
| 17. | Rys, P.; Zollinger, H. Fundamentals of the Chemistry and Application of Dyes; Wiley-Interscience: New York, NY, 1972. |
| 18. | Aldag, R. Photochromism based on dissociation processes. In Photochromism: Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: London, 1990. |
| 19. | Shchepinov, M. S.; Korshun, V. A. Chem. Soc. Rev. 2003, 32, 170–180. doi:10.1039/b008900l |
| 20. | Baker, L. A.; Sun, L.; Crooks, R. M. Bull. Korean Chem. Soc. 2002, 23, 647–654. doi:10.5012/bkcs.2002.23.5.647 |
| 21. | Sanguinet, L.; Twieg, R. J.; Wiggers, G.; Mao, G.; Singer, K. D.; Petschek, R. G. Tetrahedron Lett. 2005, 46, 5121–5125. doi:10.1016/j.tetlet.2005.05.138 |
| 22. | Yu, L.; Li, S.-S.; Li, W.; Yu, S.; Liu, Q.; Xiao, J. J. Org. Chem. 2018, 83, 15277–15283. doi:10.1021/acs.joc.8b02549 |
| 20. | Baker, L. A.; Sun, L.; Crooks, R. M. Bull. Korean Chem. Soc. 2002, 23, 647–654. doi:10.5012/bkcs.2002.23.5.647 |
| 23. | Oikawa, Y.; Hirasawa, H.; Yonemitsu, O. Tetrahedron Lett. 1978, 19, 1759–1762. doi:10.1016/0040-4039(78)80037-9 |
| 24. | Lü, C.-W.; Wang, J.-J.; Liu, Y.-H.; Shan, W.-J.; Sun, Q.; Shi, L. Res. Chem. Intermed. 2017, 43, 943–949. doi:10.1007/s11164-016-2675-8 |
| 25. | Renzetti, A.; Boffa, E.; Colazzo, M.; Gérard, S.; Sapi, J.; Chan, T.-H.; Nakazawa, H.; Villani, C.; Fontana, A. RSC Adv. 2014, 4, 47992–47999. doi:10.1039/c4ra08853k |
| 26. | Gérard, S.; Renzetti, A.; Lefevre, B.; Fontana, A.; de Maria, P.; Sapi, J. Tetrahedron 2010, 66, 3065–3069. doi:10.1016/j.tet.2010.02.025 |
| 19. | Shchepinov, M. S.; Korshun, V. A. Chem. Soc. Rev. 2003, 32, 170–180. doi:10.1039/b008900l |
| 75. | SHELXTL, V6.12; Bruker AXS, Inc., Analytical X-ray Systems: East Cheryl Parkway, Madison WI 53711–5373, USA, 2002. |
| 17. | Rys, P.; Zollinger, H. Fundamentals of the Chemistry and Application of Dyes; Wiley-Interscience: New York, NY, 1972. |
| 18. | Aldag, R. Photochromism based on dissociation processes. In Photochromism: Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: London, 1990. |
| 22. | Yu, L.; Li, S.-S.; Li, W.; Yu, S.; Liu, Q.; Xiao, J. J. Org. Chem. 2018, 83, 15277–15283. doi:10.1021/acs.joc.8b02549 |
| 76. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 54. | Laali, K. K.; Insuasty, D.; Abonia, R.; Insuasty, B.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 4395–4399. doi:10.1016/j.tetlet.2014.05.094 |
| 55. | Ramírez-Prada, J.; Robledo, S. M.; Vélez, I. D.; del Pilar Crespo, M.; Quiroga, J.; Abonia, R.; Montoya, A.; Svetaz, L.; Zacchino, S.; Insuasty, B. Eur. J. Med. Chem. 2017, 131, 237–254. doi:10.1016/j.ejmech.2017.03.016 |
| 56. | Abonia, R.; Insuasty, D.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Eur. J. Med. Chem. 2012, 57, 29–40. doi:10.1016/j.ejmech.2012.08.039 |
| 57. | Insuasty, D.; Abonia, R.; Insuasty, B.; Quiroga, J.; Laali, K. K.; Nogueras, M.; Cobo, J. ACS Comb. Sci. 2017, 19, 555–563. doi:10.1021/acscombsci.7b00091 |
| 52. | Appendino, G.; Cicione, L.; Minassi, A. Tetrahedron Lett. 2009, 50, 5559–5561. doi:10.1016/j.tetlet.2009.05.033 |
| 53. | Mousavizadeh, F.; Hekmatshoar, R.; Beheshtiha, S. Y. S.; Rahnamafar, R. Synth. Commun. 2014, 44, 1483–1491. doi:10.1080/00397911.2013.862723 |
| 67. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 68. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785 |
| 69. | Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200–206. doi:10.1016/0009-2614(89)87234-3 |
| 70. | Wolinski, K.; Hinton, J. F.; Pulay, P. J. Am. Chem. Soc. 1990, 112, 8251–8260. doi:10.1021/ja00179a005 |
| 71. | Ditchfield, R. Mol. Phys. 1974, 27, 789–807. doi:10.1080/00268977400100711 |
| 62. |
Ito, S.; Akimoto, K.; Morita, N. In Recent Developments in Carbocation and Onium Ion Chemistry; Laali, K. K., Ed.; ACS Symposium Series 965; ACS: Washington, DC, 2007; pp 160–183.
See for DDQ/HPF6 previously employed for generation of tri(1-azulenyl)methyl cation salts. |
| 63. | Ito, S.; Morita, N.; Asao, T. Bull. Chem. Soc. Jpn. 1995, 68, 1409–1436. doi:10.1246/bcsj.68.1409 |
| 64. |
Pfeuffer, L.; Mueller, J.; Pindur, U. Chimia 1985, 39, 141–142.
See for 3-indolylmethylethoxycarbenium BF4 salt. |
| 65. |
Pindur, U.; Mueller, J. Chem.-Ztg. 1986, 110, 333–335.
See for a UV–vis study of diindolylmethylium. |
| 54. | Laali, K. K.; Insuasty, D.; Abonia, R.; Insuasty, B.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 4395–4399. doi:10.1016/j.tetlet.2014.05.094 |
| 61. |
Oda, M.; Fukuta, A.; Kajioka, T.; Uchiyama, T.; Kainuma, H.; Miyatake, R.; Kuroda, S. Tetrahedron 2000, 56, 9917–9925. doi:10.1016/s0040-4020(00)00964-9
See for an example. |
| 58. | Kong, A.; Han, X.; Lu, X. Org. Lett. 2006, 8, 1339–1342. doi:10.1021/ol060039u |
| 59. | Meth-Cohn, O.; Narine, B. Tetrahedron Lett. 1978, 19, 2045–2048. doi:10.1016/s0040-4039(01)94745-8 |
| 60. | Meth-Cohn, O.; Narine, B.; Tarnowski, B. J. Chem. Soc., Perkin Trans. 1 1981, 1520–1530. doi:10.1039/p19810001520 |
© 2019 Abonia et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)