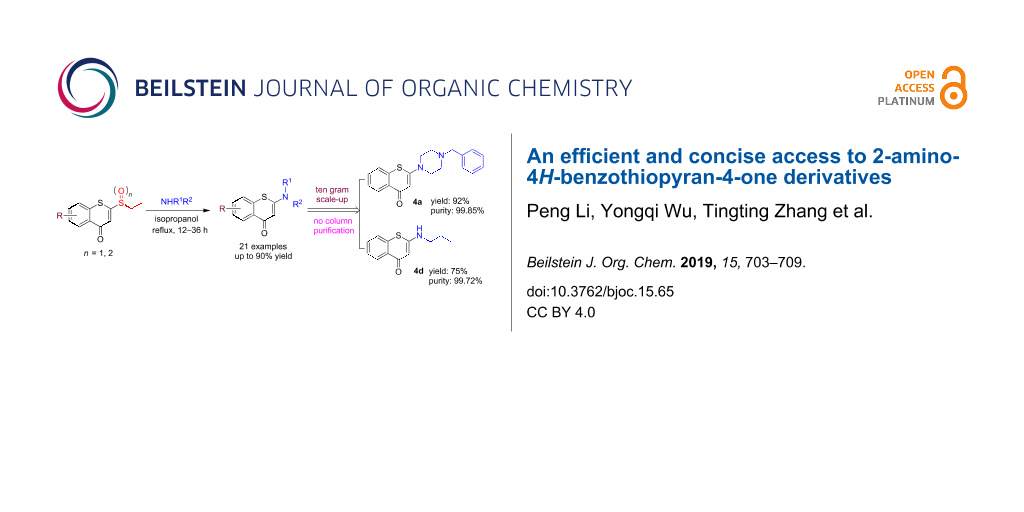
Abstract
A highly efficient and convenient protocol was developed to access 2-amino-4H-benzothiopyran-4-ones through a process of conjugated addition–elimination. The sulfinyl group was proved to be the optimum leaving group by thorough investigations on the elimination of sulfide, sulfinyl, and sulfonyl groups at the 2-position of benzothiopyranone. Most 2-aminobenzothiopyranones were obtained in good to excellent yields under refluxing in isopropanol within 36 h. This method is base-free and the substrate scope in terms of electronic properties of the substituents of the benzothiopyranone is broad. The ten grams scale-up synthesis of the representative compounds 4a and 4d was implemented to show the practical application of this reaction, which afforded the corresponding compounds in good yields and excellent chemical purity without requiring column chromatographical purification.

Graphical Abstract
Introduction
Benzothiopyranones are a class of molecules displaying biological activities in part due to their structural relationship with benzopyranones, which are known as one privileged scaffold in medicinal chemistry [1]. The corresponding 2-aminobenzothiopyranones are molecules of high interest as they have remarkable anticancer, antifungal and antitubercular activities [2-5].
Compared to the versatile synthetic methods leading to 2-aminobenzopyranone derivatives, the approaches to 2-aminobenzothiopyranones are somewhat limited. Some simple 2-aminobenzothiopyranones have been obtained by the cyclization of 2-thiobenzoylacetonitrile treated with a strong acid (method A, Scheme 1) [6,7], or through the reduction of 2-nitrobenzothiopyranones with sodium dithionite (method B, Scheme 1) [8]. Apparently, synthetic methods to afford N-substituted 2-aminobenzothiopyranones are less well developed. The first example of an interchange of a 2-methylthio substituent for a nitrogen substituent leading to 2-methylamino-3-cyanobenzothiopyranones was disclosed by Rudorf and co-workers (method C, Scheme 1) [9]. Recently, a similar strategy was successfully utilized to afford N-substituted 2-aminobenzothiopyranones through the replacement of sulfide, sulfinyl or sulfonyl groups with amines in the presence of bases such as NaH, K2CO3, etc. However, for this reaction an adjacent electron-withdrawing group such as a carbonyl or triazole group is required at the 3-position of the thiochromone ring (method D and E, Scheme 1) [3,4]. Very recently, our group obtained a series of 2-aminobenzothiopyranones containing 8-nitro and 6-trifluoromethyl substituents in low to moderate yields through the transformation of a 2-methylthio substituent under harsh conditions (method F, Scheme 1) [5]. In addition, the nucleophile imidazole could also react with 3-bromobenzothiopyranones to afford 2-imidazolylbenzothiopyranones [10].
![[1860-5397-15-65-i1]](/bjoc/content/inline/1860-5397-15-65-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative strategies for the synthesis of N-substituted 2-aminobenzothiopyranones.
Scheme 1: Representative strategies for the synthesis of N-substituted 2-aminobenzothiopyranones.
It is well known that the sulfide, sulfinyl and sulfonyl groups are generally used as the leaving group for the synthesis of 2-substituted 4H-chromen-4-ones [11-22]. Due to the higher electronegativity of the oxygen atom compared to the sulfur atom, the 2-position of the benzopyranone scaffold is easier to be attacked by nucleophiles than that the corresponding benzothiopyranone. We anticipate that the electron-withdrawing effect of a cyano, carbonyl, nitro or trifluoromethyl group, especially at the 3-position could render the sulfide, sulfinyl or sulfonyl moiety with higher reactivity at the 2-position of the benzothiopyranone ring.
In our continuing efforts to discover novel 2-amino-4H-benzothiopyran-4-ones with activity against Mycobacterium tuberculosis (Mtb) [5], we aim to develop an improved and efficient synthetic route by thoroughly evaluating the feasibility of the elimination of sulfide, sulfinyl or sulfonyl groups at the 2-position of the benzothiopyranone scaffold without requiring an adjacent electron-withdrawing substituent at the 3-position.
Results and Discussion
Firstly, 2-ethylthio-4H-thiochromen-4-one 1 was readily prepared from 2’-chloroacetophenone as the starting material through treatment with carbon disulfide in the presence of sodium hydride [23], followed by alkylation using iodoethane according to the literature procedures [13,16,17,19]. The oxidation of 1 with 1.2 or 5 equiv of H2O2 yielded ethyl sulfoxide 2 and ethyl sulfone 3, respectively (Scheme 2).
![[1860-5397-15-65-i2]](/bjoc/content/inline/1860-5397-15-65-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of sulfide 1, sulfoxide 2, and sulfone 3.
Scheme 2: The synthesis of sulfide 1, sulfoxide 2, and sulfone 3.
With the substrates including sulfide, sulfoxide and sulfone in hand, we screened various solvents with compounds 1a, 2a, 3a and 1-benzylpiperazine as the model substrates and the results are summarized in Table 1. No target compound 4a was obtained at room temperature in all solvents with 1a bearing an ethylthio group as the starting material according to the method of benzopyranone preparation [13,14,17]. Even under refluxing conditions, substrate 1a could not provide the target compound 4a (Table 1, entries 1, 4, 7, 10 and 13), suggesting that the replacement of an ethylthio group requires even more drastic conditions [21]. To our delight, under refluxing conditions, the substrates with sulfinyl and sulfonyl groups led to 4a in THF and dioxane, however, in low to moderate yields (Table 1, entries 5, 6, 8 and 9). Furthermore, reaction of the substrate 2a having a sulfinyl group in ethanol or isopropanol afforded the target compound in good to excellent yield from 75% to 90% (Table 1, entries 11 and 14), indicating that a polar protic solvent promotes the reaction. When the reactions were performed with 1.2 equiv of 1-benzylpiperazine with or without Et3N in isopropanol under refluxing conditions, the yields decreased from 90% to 26% and 30%, respectively, due to considerable amounts of unreacted substrate 2a (Table 1, entries 14, 16 and 17). Although the sulfonyl group has been used more frequently due to its higher leaving ability reactivity toward nucleophiles, we found that substrate 3a with sulfonyl group easily decomposed at high temperature and led to low yields (Table 1, entries 9, 12 and 15 vs entries 8, 11 and 14). Therefore, we were encouraged to confirm the consistent superiority of ethylsulfinyl over ethylsulfonyl as a leaving group to afford the desired 2-amino-4H-benzothiopyran-4-ones through conjugated addition–elimination by nucleophiles.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-65-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | n | Solvent | Time (h) | Yieldb (%) |
| 1 | 0 | acetonitrile | 24 | – |
| 2 | 1 | acetonitrile | 24 | trace |
| 3 | 2 | acetonitrile | 24 | trace |
| 4 | 0 | THF | 24 | – |
| 5 | 1 | THF | 24 | 21 |
| 6 | 2 | THF | 24 | 18 |
| 7 | 0 | dioxane | 12 | – |
| 8 | 1 | dioxane | 12 | 58 |
| 9 | 2 | dioxane | 12 | 51 |
| 10 | 0 | ethanol | 24 | – |
| 11 | 1 | ethanol | 24 | 75 |
| 12 | 2 | ethanol | 24 | 52 |
| 13 | 0 | isopropanol | 12 | – |
| 14 | 1 | isopropanol | 12 | 90 |
| 15 | 2 | isopropanol | 12 | 37 |
| 16c | 1 | isopropanol | 12 | 30 |
| 17d | 1 | isopropanol | 12 | 26 |
aThe reactions (entries 1–15) were performed with 1.0 mmol of 1a–3a and 2.0 mmol of 1-benzylpiperazine in 5 mL of the indicated solvent under refluxing conditions. bIsolated yields. cThe reaction was performed with 1.0 mmol of 2a and 1.2 mmol of 1-benzylpiperazine in 5 mL of isopropanol under refluxing conditions. dThe reaction was performed with 1.0 mmol of 2a,1.2 mmol of 1-benzylpiperazine and 1.2 mmol Et3N in 5 mL of isopropanol under refluxing conditions.
Subsequently, substrates 2a and 3a were reacted with a variety of alkyl/arylamines in the optimized solvent isopropanol (Table 2). Substrate 2a having an ethylsulfinyl group in general provided a much higher product yield (Table 2, entries 1, 3 and 5 vs entries 2, 4 and 6, respectively) and sometimes required a shorter reaction time (Table 2, entry 3 vs entry 4) than the corresponding substrate 3a. To further verify the advantage of the sulfinyl group as the optimal leaving group, the representative amines including primary and secondary amines were then investigated in reactions with substrate 2a. We were pleased to find that various N-substituted 2-aminobenzothiopyranones 4e–i were afforded in moderate to good yields. In general, aliphatic amines NHR1R2 are stronger nucleophiles to furnish the desired products in high yields in a relatively short reaction time compared to aromatic amines, exemplified by entries 1, 7 vs entries 3, 5, 8–10 in Table 2. Both primary and secondary aliphatic amines could smoothly afford the corresponding products (Table 2, entries 3, 5 and 8–11).
Table 2: Further confirmation of the sulfinyl/sulfonyl groups as optimal leaving groups.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-65-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | n | Product | NHR1R2 | Time (h) | Yieldb (%) |
| 1 | 1 | 4b | aniline | 36 | 71 |
| 2 | 2 | 4b | aniline | 36 | 50 |
| 3 | 1 | 4c | benzylamine | 12 | 75 |
| 4 | 2 | 4c | benzylamine | 36 | 52 |
| 5 | 1 | 4d | propylamine | 12 | 78 |
| 6 | 2 | 4d | propylamine | 12 | 64 |
| 7 | 1 | 4e | p-methoxyaniline | 36 | 55 |
| 8 | 1 | 4f | 3,4-dimethoxybenzylamine | 12 | 79 |
| 9 | 1 | 4g | cyclohexylmethylamine | 12 | 76 |
| 10 | 1 | 4h | cyclohexylamine | 24 | 71 |
| 11 | 1 | 4i | diethylamine | 24 | 67 |
aAll reactions were performed with 1.0 mmol of 2a/3a and 2.0 mmol of NHR1R2 in 5 mL of isopropanol. bIsolated yields.
Having identified the optimal conditions, we next explored the scope of the reaction. Various sulfinyl substrates 2b–h with electron-donating or electron-withdrawing substituents in the benzothiopyranone scaffold were treated with 1-benzylpiperazine as well as other representative amines (Scheme 3). Due to the strong nucleophilicity of piperazine and morpholine, the substrates with electron-donating/withdrawing and halogen groups smoothly delivered the N-substituted 2-aminobenzothiopyranones 4j–o and 4t,u in good to excellent yields. Specifically, the substrates containing a strong electron-withdrawing group such as trifluoromethyl gave the desired products, e.g., 4m,n in high yields. Substrates bearing electron-donating groups also reacted with aliphatic amines giving products 4o–q in good yields. It was noted that the treatment of substrate 2 containing a chloro substituent with weaker nucleophilic amines such as arylamines successfully afforded the targeted compounds 4r,s albeit in only about 50% yield. The results indicated that the nucleophilicity of the amine exerts a more pronounced effect on the reactivity (i.e., aliphatic amines vs aromatic amines) under the above optimal conditions established.
![[1860-5397-15-65-i3]](/bjoc/content/inline/1860-5397-15-65-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the synthesis of versatile 2-aminobenzothiopyranones. All reactions were performed with 1.0 mmol of 2b–h and 2.0 mmol of NHR1R2 in 5 mL of isopropanol under refluxing conditions and yields refer to isolated yields.
Scheme 3: Scope of the synthesis of versatile 2-aminobenzothiopyranones. All reactions were performed with 1....
To further evaluate the efficiency of this methodology, a gram-scale synthesis of the representative compounds 4a and 4d was performed. As depicted in Scheme 4, by treatment of 40 mmol of 2a with 1-benzylpiperazine under the optimized conditions, the product 4a was obtained in an excellent yield (92%, 12.4 g) with >99% chemical purity. Following the same procedure starting from 2b gave product 4d in good yield (75%, 9.8 g) as well as with >99% chemical purity. To our delight, both products 4a and 4d were successfully isolated by directly filtration of the reaction mixture instead of column chromatographical purification that was employed in the milligram-scale synthesis. Overall, this synthetic route is efficient and applicable to scale-up.
![[1860-5397-15-65-i4]](/bjoc/content/inline/1860-5397-15-65-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The gram-scale synthesis of 2-aminobenzothiopyranones 4a and 4d.
Scheme 4: The gram-scale synthesis of 2-aminobenzothiopyranones 4a and 4d.
Conclusion
In summary, we have developed a facile and efficient strategy for the synthesis of 2-aminobenzothiopyranones through a conjugated addition–elimination process. The direct C–N bond formation reaction at the 2-position smoothly took place using ethylsulfinyl as the optimal leaving group and various nucleophiles such as aliphatic and aromatic amines. A variety of 2-aminobenzothiopyranones were obtained in moderate to excellent yields without the assistance of an adjacent electron-withdrawing group or additional base. This practical method could be scaled-up to a ten-gram scale allowing isolation of the products by simple filtration. This synthetic route is complementary to the existing methods for the synthesis of 2-aminobenzothiopyranone derivatives, thereby providing opportunities for discovering more active compounds for medicinal chemistry research.
Supporting Information
| Supporting Information File 1: General methods, general procedures, characterization data and copies of 1H and 13C NMR spectra of 1a–h, 2a–h, 3a and 4a–u. | ||
| Format: PDF | Size: 3.5 MB | Download |
References
-
Vargas, E.; Echeverri, F.; Vélez, I. D.; Robledo, S. M.; Quiñones, W. Molecules 2017, 22, 2041. doi:10.3390/molecules22122041
Return to citation in text: [1] -
Geissler, J. F.; Roesel, J. L.; Meyer, T.; Trinks, U. P.; Traxler, P.; Lydon, N. B. Cancer Res. 1992, 52, 4492–4498.
Return to citation in text: [1] -
Xiao, T.; Feng, Y.; Hu, B.; Cheng, G. C. 2-Triazolyl thiochromone compound and synthesis method thereof, and applications of 2-triazolyl thiochromone compound in preparation of antifungal drugs. Chinese Patent Appl. CN107151242 A, March 2, 2016.
Return to citation in text: [1] [2] -
Xiao, T.; Hu, B.; Cheng, G. C.; Feng, Y.; Wei, F.; Xiao, Z. Y. 2-Substituted-3-azacyclo thiochromone compound and synthetic method and application of 2-substituted-3-azacyclo thiochromone compound in antifungal drug. Chinese Patent Appl. CN107513059 A, June 17, 2016.
Return to citation in text: [1] [2] -
Li, P.; Wang, B.; Zhang, X.; Batt, S. M.; Besra, G. S.; Zhang, T.; Ma, C.; Zhang, D.; Lin, Z.; Li, G.; Huang, H.; Lu, Y. Eur. J. Med. Chem. 2018, 160, 157–170. doi:10.1016/j.ejmech.2018.09.042
Return to citation in text: [1] [2] [3] -
Volovenko, Y. M.; Litenko, V. A.; Dashkovskaya, E. V.; Babichev, F. S. Chem. Heterocycl. Compd. 1982, 18, 796–798. doi:10.1007/bf00506579
Return to citation in text: [1] -
Zhmurenko, L. A.; Glozman, O. M.; Zagorevskii, V. A. Chem. Heterocycl. Compd. 1978, 14, 141–144. doi:10.1007/bf00945325
Return to citation in text: [1] -
Eiden, F.; Felbermeir, G.; Buchborn, H. Arch. Pharm. (Weinheim, Ger.) 1983, 316, 921–933. doi:10.1002/ardp.19833161106
Return to citation in text: [1] -
Rudorf, W.-D.; Augustin, M. J. Prakt. Chem. 1981, 323, 55–62. doi:10.1002/prac.19813230109
Return to citation in text: [1] -
Cozzi, P.; Pillan, A. J. Heterocycl. Chem. 1988, 25, 1613–1616. doi:10.1002/jhet.5570250603
Return to citation in text: [1] -
Savant, M. M.; Gowda, N. S.; Pansuriya, A. M.; Bhuva, C. V.; Kapuriya, N.; Anandalwar, S. M.; Prasad, S. J.; Shah, A.; Naliapara, Y. T. Tetrahedron Lett. 2011, 52, 254–257. doi:10.1016/j.tetlet.2010.11.020
Return to citation in text: [1] -
Lindell, S. D.; Ort, O.; Lümmen, P.; Klein, R. Bioorg. Med. Chem. Lett. 2004, 14, 511–514. doi:10.1016/j.bmcl.2003.10.022
Return to citation in text: [1] -
Hatnapure, G. D.; Keche, A. P.; Rodge, A. H.; Birajdar, S. S.; Tale, R. H.; Kamble, V. M. Bioorg. Med. Chem. Lett. 2012, 22, 6385–6390. doi:10.1016/j.bmcl.2012.08.071
Return to citation in text: [1] [2] [3] -
Lee, G. H.; Ha, S. J.; Pak, C. S. Synth. Commun. 1999, 29, 3155–3164. doi:10.1080/00397919908085940
Return to citation in text: [1] [2] -
Lee, G. H.; Ha, S. J.; Pak, C. S. Synth. Commun. 1999, 29, 2677–2684. doi:10.1080/00397919908086429
Return to citation in text: [1] -
Bantick, J. R.; Suschitzky, J. L. J. Heterocycl. Chem. 1981, 18, 679–684. doi:10.1002/jhet.5570180407
Return to citation in text: [1] [2] -
Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; Pasquet, M.-J.; Péru, A.; Plé, P.; Saleh, T.; Vautier, M.; Walker, M.; Ward, L.; Warin, N. J. Med. Chem. 2015, 58, 943–962. doi:10.1021/jm501629p
Return to citation in text: [1] [2] [3] -
Hardcastle, I. R.; Cockcroft, X.; Curtin, N. J.; El-Murr, M. D.; Leahy, J. J. J.; Stockley, M.; Golding, B. T.; Rigoreau, L.; Richardson, C.; Smith, G. C. M.; Griffin, R. J. J. Med. Chem. 2005, 48, 7829–7846. doi:10.1021/jm050444b
Return to citation in text: [1] -
Obreque-Balboa, J. E.; Sun, Q.; Bernhardt, G.; König, B.; Buschauer, A. Eur. J. Med. Chem. 2016, 109, 124–133. doi:10.1016/j.ejmech.2015.12.010
Return to citation in text: [1] [2] -
Hackett, J. C.; Kim, Y.-W.; Su, B.; Brueggemeier, R. W. Bioorg. Med. Chem. 2005, 13, 4063–4070. doi:10.1016/j.bmc.2005.03.050
Return to citation in text: [1] -
Costantino, L.; Del Corso, A.; Rastelli, G.; Petrash, J. M.; Mura, U. Eur. J. Med. Chem. 2001, 36, 697–703. doi:10.1016/s0223-5234(01)01272-7
Return to citation in text: [1] [2] -
Chisari, A.; Maccarone, E.; Parisi, G.; Perrini, G. J. Chem. Soc., Perkin Trans. 2 1982, 957–959. doi:10.1039/p29820000957
Return to citation in text: [1] -
Andersonmckay, J. E.; Liepa, A. J. Aust. J. Chem. 1987, 40, 1179–1190. doi:10.1071/ch9871179
Return to citation in text: [1]
| 1. | Vargas, E.; Echeverri, F.; Vélez, I. D.; Robledo, S. M.; Quiñones, W. Molecules 2017, 22, 2041. doi:10.3390/molecules22122041 |
| 9. | Rudorf, W.-D.; Augustin, M. J. Prakt. Chem. 1981, 323, 55–62. doi:10.1002/prac.19813230109 |
| 8. | Eiden, F.; Felbermeir, G.; Buchborn, H. Arch. Pharm. (Weinheim, Ger.) 1983, 316, 921–933. doi:10.1002/ardp.19833161106 |
| 6. | Volovenko, Y. M.; Litenko, V. A.; Dashkovskaya, E. V.; Babichev, F. S. Chem. Heterocycl. Compd. 1982, 18, 796–798. doi:10.1007/bf00506579 |
| 7. | Zhmurenko, L. A.; Glozman, O. M.; Zagorevskii, V. A. Chem. Heterocycl. Compd. 1978, 14, 141–144. doi:10.1007/bf00945325 |
| 13. | Hatnapure, G. D.; Keche, A. P.; Rodge, A. H.; Birajdar, S. S.; Tale, R. H.; Kamble, V. M. Bioorg. Med. Chem. Lett. 2012, 22, 6385–6390. doi:10.1016/j.bmcl.2012.08.071 |
| 14. | Lee, G. H.; Ha, S. J.; Pak, C. S. Synth. Commun. 1999, 29, 3155–3164. doi:10.1080/00397919908085940 |
| 17. | Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; Pasquet, M.-J.; Péru, A.; Plé, P.; Saleh, T.; Vautier, M.; Walker, M.; Ward, L.; Warin, N. J. Med. Chem. 2015, 58, 943–962. doi:10.1021/jm501629p |
| 2. | Geissler, J. F.; Roesel, J. L.; Meyer, T.; Trinks, U. P.; Traxler, P.; Lydon, N. B. Cancer Res. 1992, 52, 4492–4498. |
| 3. | Xiao, T.; Feng, Y.; Hu, B.; Cheng, G. C. 2-Triazolyl thiochromone compound and synthesis method thereof, and applications of 2-triazolyl thiochromone compound in preparation of antifungal drugs. Chinese Patent Appl. CN107151242 A, March 2, 2016. |
| 4. | Xiao, T.; Hu, B.; Cheng, G. C.; Feng, Y.; Wei, F.; Xiao, Z. Y. 2-Substituted-3-azacyclo thiochromone compound and synthetic method and application of 2-substituted-3-azacyclo thiochromone compound in antifungal drug. Chinese Patent Appl. CN107513059 A, June 17, 2016. |
| 5. | Li, P.; Wang, B.; Zhang, X.; Batt, S. M.; Besra, G. S.; Zhang, T.; Ma, C.; Zhang, D.; Lin, Z.; Li, G.; Huang, H.; Lu, Y. Eur. J. Med. Chem. 2018, 160, 157–170. doi:10.1016/j.ejmech.2018.09.042 |
| 21. | Costantino, L.; Del Corso, A.; Rastelli, G.; Petrash, J. M.; Mura, U. Eur. J. Med. Chem. 2001, 36, 697–703. doi:10.1016/s0223-5234(01)01272-7 |
| 11. | Savant, M. M.; Gowda, N. S.; Pansuriya, A. M.; Bhuva, C. V.; Kapuriya, N.; Anandalwar, S. M.; Prasad, S. J.; Shah, A.; Naliapara, Y. T. Tetrahedron Lett. 2011, 52, 254–257. doi:10.1016/j.tetlet.2010.11.020 |
| 12. | Lindell, S. D.; Ort, O.; Lümmen, P.; Klein, R. Bioorg. Med. Chem. Lett. 2004, 14, 511–514. doi:10.1016/j.bmcl.2003.10.022 |
| 13. | Hatnapure, G. D.; Keche, A. P.; Rodge, A. H.; Birajdar, S. S.; Tale, R. H.; Kamble, V. M. Bioorg. Med. Chem. Lett. 2012, 22, 6385–6390. doi:10.1016/j.bmcl.2012.08.071 |
| 14. | Lee, G. H.; Ha, S. J.; Pak, C. S. Synth. Commun. 1999, 29, 3155–3164. doi:10.1080/00397919908085940 |
| 15. | Lee, G. H.; Ha, S. J.; Pak, C. S. Synth. Commun. 1999, 29, 2677–2684. doi:10.1080/00397919908086429 |
| 16. | Bantick, J. R.; Suschitzky, J. L. J. Heterocycl. Chem. 1981, 18, 679–684. doi:10.1002/jhet.5570180407 |
| 17. | Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; Pasquet, M.-J.; Péru, A.; Plé, P.; Saleh, T.; Vautier, M.; Walker, M.; Ward, L.; Warin, N. J. Med. Chem. 2015, 58, 943–962. doi:10.1021/jm501629p |
| 18. | Hardcastle, I. R.; Cockcroft, X.; Curtin, N. J.; El-Murr, M. D.; Leahy, J. J. J.; Stockley, M.; Golding, B. T.; Rigoreau, L.; Richardson, C.; Smith, G. C. M.; Griffin, R. J. J. Med. Chem. 2005, 48, 7829–7846. doi:10.1021/jm050444b |
| 19. | Obreque-Balboa, J. E.; Sun, Q.; Bernhardt, G.; König, B.; Buschauer, A. Eur. J. Med. Chem. 2016, 109, 124–133. doi:10.1016/j.ejmech.2015.12.010 |
| 20. | Hackett, J. C.; Kim, Y.-W.; Su, B.; Brueggemeier, R. W. Bioorg. Med. Chem. 2005, 13, 4063–4070. doi:10.1016/j.bmc.2005.03.050 |
| 21. | Costantino, L.; Del Corso, A.; Rastelli, G.; Petrash, J. M.; Mura, U. Eur. J. Med. Chem. 2001, 36, 697–703. doi:10.1016/s0223-5234(01)01272-7 |
| 22. | Chisari, A.; Maccarone, E.; Parisi, G.; Perrini, G. J. Chem. Soc., Perkin Trans. 2 1982, 957–959. doi:10.1039/p29820000957 |
| 23. | Andersonmckay, J. E.; Liepa, A. J. Aust. J. Chem. 1987, 40, 1179–1190. doi:10.1071/ch9871179 |
| 10. | Cozzi, P.; Pillan, A. J. Heterocycl. Chem. 1988, 25, 1613–1616. doi:10.1002/jhet.5570250603 |
| 13. | Hatnapure, G. D.; Keche, A. P.; Rodge, A. H.; Birajdar, S. S.; Tale, R. H.; Kamble, V. M. Bioorg. Med. Chem. Lett. 2012, 22, 6385–6390. doi:10.1016/j.bmcl.2012.08.071 |
| 16. | Bantick, J. R.; Suschitzky, J. L. J. Heterocycl. Chem. 1981, 18, 679–684. doi:10.1002/jhet.5570180407 |
| 17. | Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; Pasquet, M.-J.; Péru, A.; Plé, P.; Saleh, T.; Vautier, M.; Walker, M.; Ward, L.; Warin, N. J. Med. Chem. 2015, 58, 943–962. doi:10.1021/jm501629p |
| 19. | Obreque-Balboa, J. E.; Sun, Q.; Bernhardt, G.; König, B.; Buschauer, A. Eur. J. Med. Chem. 2016, 109, 124–133. doi:10.1016/j.ejmech.2015.12.010 |
| 5. | Li, P.; Wang, B.; Zhang, X.; Batt, S. M.; Besra, G. S.; Zhang, T.; Ma, C.; Zhang, D.; Lin, Z.; Li, G.; Huang, H.; Lu, Y. Eur. J. Med. Chem. 2018, 160, 157–170. doi:10.1016/j.ejmech.2018.09.042 |
| 3. | Xiao, T.; Feng, Y.; Hu, B.; Cheng, G. C. 2-Triazolyl thiochromone compound and synthesis method thereof, and applications of 2-triazolyl thiochromone compound in preparation of antifungal drugs. Chinese Patent Appl. CN107151242 A, March 2, 2016. |
| 4. | Xiao, T.; Hu, B.; Cheng, G. C.; Feng, Y.; Wei, F.; Xiao, Z. Y. 2-Substituted-3-azacyclo thiochromone compound and synthetic method and application of 2-substituted-3-azacyclo thiochromone compound in antifungal drug. Chinese Patent Appl. CN107513059 A, June 17, 2016. |
| 5. | Li, P.; Wang, B.; Zhang, X.; Batt, S. M.; Besra, G. S.; Zhang, T.; Ma, C.; Zhang, D.; Lin, Z.; Li, G.; Huang, H.; Lu, Y. Eur. J. Med. Chem. 2018, 160, 157–170. doi:10.1016/j.ejmech.2018.09.042 |
© 2019 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)