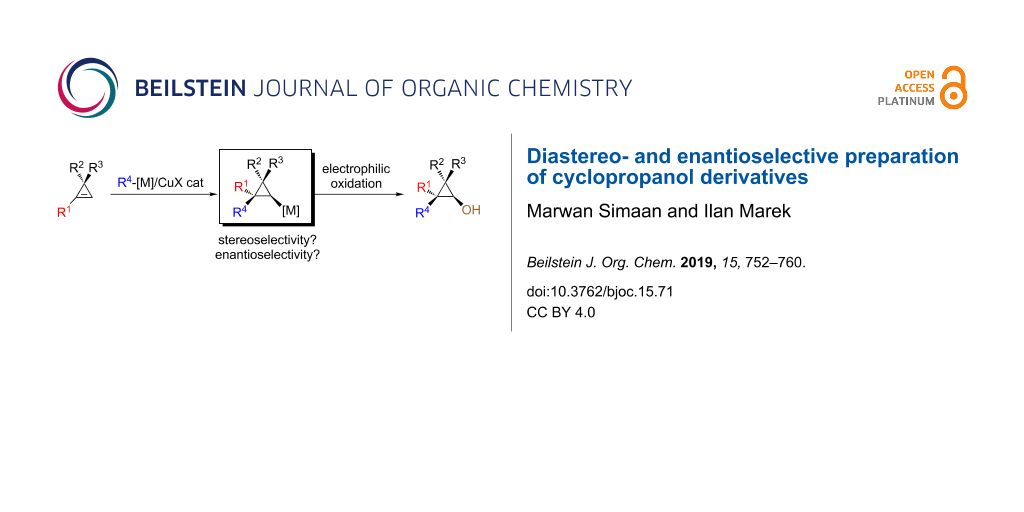
Abstract
The diastereoselective carbocupration reaction of alkoxy-functionalized cyclopropene derivatives, followed by a subsequent trapping of the resulting cyclopropylmetal species with an electrophilic source of oxygen (oxenoid) afforded various tetrasubstituted cyclopropanol derivatives in high diastereo- and enantiomeric ratios. Similarly, the enantioselective copper-catalyzed carbomagnesiation/oxidation (or amination) sequence on achiral nonfunctionalized cyclopropenes provided the desired cyclopropanol (and cyclopropylamine) derivatives in excellent diastereo- and enantiomeric excesses.

Graphical Abstract
Introduction
The highly strained structure and bonding properties of cyclopropyl rings have constantly fascinated successive generations of chemists. These small carbocycles are known to have high ring strain (27.5 kcal/mol) and limited degrees of freedom, making them very attractive substrates for various chemical transformations [1]. The cyclopropane subunit is also present in many biologically important compounds such as pheromones, fatty acid metabolites, unusual amino acids and possess interesting herbicidal, insecticidal, antibiotic, antibacterial, antifungal, antiviral and antitumor activities [2]. For these reasons, cyclopropanes have been extensively studied and numerous approaches have been described for their preparation [3]. Among all possible three-membered ring subunits that have been reported, cyclopropanols have recently attracted renewed attention as they are not only contained in many natural products but they are also important precursors in the synthesis of various biologically active molecules and pharmaceuticals such as antidepressants, antiviral and antibacterial drugs [4]. Cyclopropanols and their derivatives are considered to be carbocyclic homologues of enols presenting similar chemical properties which are caused by the unsaturated character of the cyclopropyl ring. Although cyclopropanols are usually less reactive than enols and enolates, their chemical properties are somehow more diverse as they undergo useful transformations either with preservation or rupture of the cyclic structure [5]. Several reliable approaches to produce cyclopropanols have been reported in the literature (Scheme 1) [6] but a number of challenges still exist particularly for the stereoselective preparation of cyclopropanols of high structural complexity and substitution pattern. Since the first synthesis of cyclopropanol by Cottle [7], the most popular methods for the preparation of cyclopropanols rely on the transformation of enolates [8,9], silyl enol ether [10-12], vinyl borane [13-17], Fischer carbene addition [18], addition of nucleophiles to carbonyl groups [19-25], elimination [26,27], oxidation [28] or on the Kulinkovich reaction [29-41] as summarized in Scheme 1. However, and despite the increasing and justified popularity of all of these methods, the short summary described in Scheme 1 emphasizes an intrinsic problem: a different strategy is required for every cyclopropanol and cyclopropylamine that one needs to prepare, limiting the rapid structural diversification which is usually essential for biological studies. From this rapid tour d’horizon, it is clear that if one could design a stereoselective synthetic pathway to afford polysubstituted cyclopropanols (or cyclopropylamines) potentially bearing several diastereo- and enantiomerically enriched adjacent stereogenic centers, including quaternary carbon stereocenters, as single diastereo- and enantiomer from a simple precursor, it would certainly provide an additional and useful entry to the synthesis of these heterosubstituted three-membered rings.
![[1860-5397-15-71-i1]](/bjoc/content/inline/1860-5397-15-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Various strategies leading to the formation of cyclopropanols.
Scheme 1: Various strategies leading to the formation of cyclopropanols.
Results and Discussion
To reach these goals, we are describing herein the diastereo- and enantioselective carbometalation reaction of cyclopropenes to provide cyclopropylmetal species. By subsequent stereoselective reactions of the cyclopropylmetal species with an electrophilic source of oxygen (or nitrogen), cyclopropanols (and cyclopropylamines) should be easily accessible (Scheme 2).
![[1860-5397-15-71-i2]](/bjoc/content/inline/1860-5397-15-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General approach to the preparation of cyclopropanol and cyclopropylamine derivatives.
Scheme 2: General approach to the preparation of cyclopropanol and cyclopropylamine derivatives.
In order to assemble both cyclopropanols and cyclopropylamines from a single and unique precursor in a single-pot reaction double facial selection by the catalyst is required: (i) regioselectivity when R1 is different to H and enantioselectivity when R1 is equal to H (left or right) and (ii) diastereotopic face selection (top or bottom) as described in Scheme 2. Since the pioneering addition of a carbon–metal bond (carbometalation) across the double bond of cyclopropenes [42], a very large number of groups have reported the addition of organometallic species demonstrating the generality of this approach for the preparation of cyclopropanes [43-63]. To achieve good diastereoselectivity during the carbometalation reaction, few conditions needed to be fulfilled in the design of the starting cyclopropenyl ring (Figure 1):
- The presence of a coordinating group (such as an oxygen) at the C3 position is crucial for the selective facial addition of the incoming organometallic species.
- The presence of a bulky substituent (alkyl or aryl) at the opposite face at the C3 position might equally be important as it might induce an additional steric parameter leading to a potentially more selective carbometalation reaction.
- The substitution pattern on the double bond needs to be addressed carefully as it plays an important role in the control of the regioselectivity of the reaction pathway. An alkyl group on the C1 position of the cyclopropene should control the regioselectivity of the addition of the organometallic species to give the more stable secondary cyclopropylmetal species (metal at C2).
![[1860-5397-15-71-1]](/bjoc/content/figures/1860-5397-15-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Prerequisite for a regio- and diastereoselective carbometalation.
Figure 1: Prerequisite for a regio- and diastereoselective carbometalation.
We therefore decided to start our research with cyclopropanes bearing an electron-rich methoxy group on one side of the ring and a phenyl or methyl substituent on the opposite side, respectively. Following reported methods from the literature [64-67], cyclopropenes 3 were easily prepared through the well-known RhII-catalyzed decomposition of diazo esters in the presence of alkynes to give cyclopropenyl esters 1. Reduction of 1 using DIBAL-H afforded cyclopropenyl alcohols 2 and subsequent protection of the primary alcohols gave 3 in good yields (Scheme 3).
![[1860-5397-15-71-i3]](/bjoc/content/inline/1860-5397-15-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of cyclopropenyl methyl ethers 3a–d.
Scheme 3: Preparation of cyclopropenyl methyl ethers 3a–d.
First, we checked that the carbometalation reaction was regio- and diastereoselective by addition of lower order cyanocuprate, easily obtained from the corresponding organolithium and a stoichiometric amount of CuCN (Scheme 4) [68-70].
![[1860-5397-15-71-i4]](/bjoc/content/inline/1860-5397-15-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Regio- and diastereoselective carbocupration of cyclopropenyl methyl ethers 3a,c.
Scheme 4: Regio- and diastereoselective carbocupration of cyclopropenyl methyl ethers 3a,c.
The addition reaction proceeds similarly for the addition of alkyl- or arylcuprate (4a,b and 4c) as well as for the addition on cyclopropene possessing either a primary or secondary alkyl group at the vinylic carbon center (4a–c and 4d,e). Having in hand diastereoisomerically pure and configurationally stable cyclopropylcopper species 3Cu, we next turned our attention to their stereoselective oxidation reaction (Scheme 5). Considering electrophilic oxidation processes of organometallic species, molecular oxygen seems to be the most obvious choice due to its abundancy and low cost. Nevertheless, the reaction of molecular oxygen with a organocopper species usually proceeds through single-electron transfer to dioxygen, leading to either a loss of stereoselectivity, degradation of the organocopper or to the formation of dimer as major products [71]. Therefore, it was clear that a different approach for the oxidation process was needed. Oxenoid, possessing the general structure M–O–LG, with a metal and a leaving group connected to an oxygen atom, have been shown to be an excellent electrophilic oxygen source for nucleophilic organometallic species [72]. Since the original discovery of Müller and Töpel of lithiated peroxides [73], several studies have been reported on the reactivity of oxenoids [74-77], indicating that the reaction of a nucleophile with oxenoid proceeds through an SN2 process [74]. Following the carbocupration of cyclopropene 3 into cyclopropylcopper species, the subsequent oxidation with the amphiphilic lithiated hydroperoxide t-BuOOLi (oxenoid), simply generated by deprotonation of t-BuOOH with n-BuLi, led to the copper alkoxide, as anticipated, without the formation of free radical intermediates. As already reported [78], the expected 2,2,3,3-tetrasubstituted cyclopropanols 5 were obtained as single diastereoisomers (Scheme 5).
![[1860-5397-15-71-i5]](/bjoc/content/inline/1860-5397-15-71-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Diastereoselective formation of cyclopropanols.
Scheme 5: Diastereoselective formation of cyclopropanols.
The reaction proceeded for all R1 and R2 groups tested and determination of the stereochemistry confirmed that the oxidation reaction proceeds with pure retention of configuration at the metalated center (intramolecular SN2 reaction or 1,2-metalate rearrangement) [78-80]. It should be noted that when the same sequence of diastereoselective carbometalation/oxidation was performed on cyclopropenyl ester 1, the in situ-formed donor–acceptor cyclopropanol undergoes a selective ring-opening to provide the acyclic product possessing a quaternary carbon stereocenter [57]. As the enantioselective synthesis of the cyclopropenylmethyl ether 3 was easily achieved in high enantiomeric ratio (er 93:7, Scheme 5 [78,81,82]), the subsequent combined diastereoselective carbometalation reaction and oxidation gave the enantiomerically enriched cyclopropanols 5 as unique diastereoisomer with the same enantiomeric ratio as the starting material (Scheme 5, dr 98:2:0:0, er 93:7). Having established the optimized reaction conditions for the preparation of diastereomerically pure 2,2,3,3-tetrasubstituted cyclopropanol derivatives 5, we were interested to expand the scope of this transformation and include different types of cyclopropene precursors. We concentrated our efforts on the reaction of diversely substituted 3-methyl-3-arylcyclopropenes 6. In this case, as there is no coordinating functionality to dictate the facial selectivity, the control of the diastereoselectivity may be more challenging. Performing the same carbocupration/oxidation sequence on 3-methyl-3-phenylcyclopropene 6a,b (R1 = H), we were pleased to observe that trisubstituted cyclopropanols 7a–h were obtained in good yields with excellent diastereoselectivities (Scheme 6).
![[1860-5397-15-71-i6]](/bjoc/content/inline/1860-5397-15-71-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Diastereoselective carbometalation/oxidation of nonfunctionalized cyclopropenes 6.
Scheme 6: Diastereoselective carbometalation/oxidation of nonfunctionalized cyclopropenes 6.
The addition of primary, secondary and tertiary alkylcuprates or arylcuprates proceeded smoothly to give the desired cyclopropanols (7a–h) after hydrolysis. In all cases, the easily prepared cyclopropanol derivatives were obtained with the methyl, the incoming organometallic and the alcohol in the syn-orientation which was determined through comparison of the hydrolyzed carbometalated products with compounds already described in the literature [83]. To further increase the structural complexity of the final cyclopropanols, we also tested the reaction on nonfunctionalized trisubstituted cyclopropenes (6c–g). Addition of primary alkyl- or arylcuprates followed by the oxidation of the cyclopropylcopper species proceeded equally well and gave the corresponding cyclopropanols possessing two adjacent quaternary carbon stereocenters (7i–r) in good yields and excellent diastereomeric ratios. Here again, the methyl, the alkyl group from the organometallic and the alcohol in the resulting cyclopropanols are syn-oriented as previously observed. As reported in a different context, the nature of the two substituents on the cyclopropene rings could be changed without drastically altering the selectivity of the reaction [83,91].
We were also interested to develop the access to non-racemic unfunctionalized cyclopropanols. Based on the pioneering work of Lautens [62], Nakamura [60], Fox [61], Gevorgyan [51] and Tortosa [84], we anticipated that an enantioselective copper-catalyzed carbometalation reaction [83,85] would be an ideal solution to access the desired polysubstituted enantioenriched cyclopropanols. As reported, the copper-catalyzed diastereo- and enantioselective carbomagnesiation reaction of cyclopropenes 6 was easily achieved in the presence of (R,S)-Josiphos (2.2 mol %, Scheme 7). Having in hand, diastereoisomerically pure and enantiomerically enriched cyclopropylmagnesium species 6MgBr, the selective oxidation reaction of the copper species, resulting from a transmetalation reaction, was similarly achieved by reaction with oxenoid [71,79,80,86-89]. In all cases, cyclopropanols 7 were obtained as single diastereoisomer (dr 98:2:0:0) with excellent enantiomeric ratios (er up to 99:1, Scheme 7). Following the same concept of copper-catalyzed diastereo- and enantioselective carbomagnesiation reaction of cyclopropenes 6 followed now by a selective electrophilic amination reaction, a powerful entry to cyclopropylamines as single diastereoisomer and in excellent enantiomeric ratios could also be achieved (Scheme 7) [89]. However, the enantioselective and catalytic copper-catalyzed carbomagnesiation reaction gave poor enantiomeric ratios for the addition of vinyl, aryl and allyl groups and alternative strategies have been recently developed in our research group [90-92].
![[1860-5397-15-71-i7]](/bjoc/content/inline/1860-5397-15-71-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Preparation of diastereoisomerically pure and enantioenriched cyclopropanols and cyclopropylamines.
Scheme 7: Preparation of diastereoisomerically pure and enantioenriched cyclopropanols and cyclopropylamines.
Conclusion
In conclusion, we have successfully merged the regio- and diastereoselective carbocupration reaction of alkoxy-functionalized cyclopropenes with electrophilic oxidation of the resulting cyclopropylcopper species to afford 2,2,3,3-polysubstituted cyclopropanol derivatives bearing two adjacent quaternary stereogenic centers in a single pot operation. The simple preparation of enantiomerically enriched cyclopropene afforded the corresponding cyclopropanols in high enantiomeric excess. This transformation was then applied to unfunctionalized diversely substituted cyclopropenes. Using the catalytic and enantioselective carbometalation reaction of unfunctionalized cyclopropenes followed by an electrophilic oxidation reaction, polysubstituted cyclopropanols were obtained as single diastereoisomer with high enantiomeric ratios. In all cases, the configurationally stable cyclopropylmetal species reacted with retention of configuration with those electrophiles opening a new approach to O-heterosubstituted cyclopropyl rings.
Experimental
General procedure for the carbocupration reaction of 3a,c with RCuCNLi
To a suspension of CuCN (1.5 equiv) in 8 mL of Et2O was added alkyllithium dropwise at −35 °C (2 equiv). The resulting mixture (pale yellow in case of MeLi and PhLi and dark brown in case of n-BuLi and n-HexLi) was allowed to stir for 30 min. Cyclopropene 3a,c (1 equiv in 2 mL/mmol of Et2O) was added at that temperature and the reaction mixture was stirred until TLC shows complete consumption of the starting material (eluent hexane/EtOAc 9:1, ca. 30 min). The reaction was then quenched with an aqueous solution of NH4Cl/NH4OH (2:1). The aqueous layer was extracted twice with EtOAc and the combined organic phases were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude mixtures were then purified by flash chromatography using pentane/diethyl ether as eluent.
General procedure for the combined carbocupration/oxidation sequence
The reaction was performed on a 1 mmol scale. To a suspension of CuCN (2 equiv) in 8 mL of Et2O was added alkyllithium dropwise at −35 °C (2 equiv/2 mmol). The resulting mixture (pale yellow in case of MeLi and PhLi and dark brown in case of n-BuLi and n-HexLi) was allowed to stir for 30 min. Cyclopropene 6a–g (1 equiv/1 mmol in 2 mL of Et2O) was added at that temperature and the reaction mixture was stirred until TLC shows complete consumption of the starting material (eluent hexane/EtOAc 9:1, ca. 30 min). The oxenoid was prepared in a different flask by slowly adding n-BuLi (1.2 equiv) to a solution of tert-butyl hydroperoxide (2 equiv) in THF (5 mL/2 mmol) at −80 °C. After 30 min at −80 °C, the resulting t-BuOOLi was transferred to the organocopper dropwise at −78 °C via a cannula. The mixture (orange to brown) was stirred at this temperature until disappearance of the cyclopropylcopper species (followed by TLC, eluent hexane/EtOAc 9:1, ca. 30 min). The reaction was then quenched with an aqueous solution of NH4Cl/NH4OH (2:1). The aqueous layer was extracted twice with Et2O and the combined organic phases were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Crude mixtures were then purified by flash chromatography using pentane/diethyl ether as eluent.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 308.7 KB | Download |
References
-
D’yakonov, V. A.; Trapeznikova, O. A.; de Meijere, A.; Dzhemilev, U. M. Chem. Rev. 2014, 114, 5775–5814. doi:10.1021/cr400291c
Return to citation in text: [1] -
Salaün, J.; Bairds, M. S. Curr. Med. Chem. 1995, 2, 511–542.
Return to citation in text: [1] -
Wong, H. N. C.; Hon, M.-Y.; Tse, C.-W.; Yip, Y.-C.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165–198. doi:10.1021/cr00091a005
Return to citation in text: [1] -
Haym, I.; Brimble, M. A. Org. Biomol. Chem. 2012, 10, 7649–7665. doi:10.1039/c2ob26082d
Return to citation in text: [1] -
Orellana, A.; Nikolaev, A. Synthesis 2016, 48, 1741–1768. doi:10.1055/s-0035-1560442
Return to citation in text: [1] -
Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597–2632. doi:10.1021/cr010012i
Return to citation in text: [1] -
Magrane, J. K., Jr.; Cottle, D. L. J. Am. Chem. Soc. 1942, 64, 484–487. doi:10.1021/ja01255a004
Return to citation in text: [1] -
Imamoto, T.; Kamiya, Y.; Hatajima, T.; Takahashi, H. Tetrahedron Lett. 1989, 30, 5149–5152. doi:10.1016/s0040-4039(01)93471-9
Return to citation in text: [1] -
Ito, S.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 1998, 39, 5253–5256. doi:10.1016/s0040-4039(98)01036-3
Return to citation in text: [1] -
Rubottom, G. M.; Lopez, M. I. J. Org. Chem. 1973, 38, 2097–2099. doi:10.1021/jo00951a032
Return to citation in text: [1] -
Murai, S.; Aya, T.; Sonoda, N. J. Org. Chem. 1973, 38, 4354–4356. doi:10.1021/jo00964a037
Return to citation in text: [1] -
Du, H.; Long, J.; Shi, Y. Org. Lett. 2006, 8, 2827–2829. doi:10.1021/ol0609659
Return to citation in text: [1] -
Fontani, P.; Carboni, B.; Vaultier, M.; Maas, G. Synthesis 1991, 605–609. doi:10.1055/s-1991-26524
Return to citation in text: [1] -
Pietruszka, J.; Widenmeyer, M. Synlett 1997, 977–979. doi:10.1055/s-1997-5790
Return to citation in text: [1] -
Hussain, M. M.; Li, H.; Hussain, N.; Ureña, M.; Carroll, P. J.; Walsh, P. J. J. Am. Chem. Soc. 2009, 131, 6516–6524. doi:10.1021/ja900147s
Return to citation in text: [1] -
Imai, T.; Mineta, H.; Nishida, S. J. Org. Chem. 1990, 55, 4986–4988. doi:10.1021/jo00304a004
Return to citation in text: [1] -
Benoit, G.; Charette, A. B. J. Am. Chem. Soc. 2017, 139, 1364–1367. doi:10.1021/jacs.6b09090
Return to citation in text: [1] -
Barluenga, J.; Suero, M. G.; Pérez-Sánchez, I.; Flórez, J. J. Am. Chem. Soc. 2008, 130, 2708–2709. doi:10.1021/ja074363b
Return to citation in text: [1] -
Ukai, K.; Oshima, K.; Matsubara, S. J. Am. Chem. Soc. 2000, 122, 12047–12048. doi:10.1021/ja003360v
Return to citation in text: [1] -
Matsubara, S.; Ukai, K.; Fushimi, H.; Yokota, Y.; Yoshino, H.; Oshima, K.; Omoto, K.; Ogawa, A.; Hioki, Y.; Fujimoto, H. Tetrahedron 2002, 58, 8255–8262. doi:10.1016/s0040-4020(02)00975-4
Return to citation in text: [1] -
Cheng, K.; Carroll, P. J.; Walsh, P. J. Org. Lett. 2011, 13, 2346–2349. doi:10.1021/ol200597h
Return to citation in text: [1] -
Turro, N. J. Acc. Chem. Res. 1969, 2, 25–32. doi:10.1021/ar50013a004
Return to citation in text: [1] -
Salaun, J. Chem. Rev. 1983, 83, 619–632. doi:10.1021/cr00058a002
Return to citation in text: [1] -
Salaün, J.; Bennani, F.; Compain, J. C.; Fadel, A.; Ollivier, J. J. Org. Chem. 1980, 45, 4129–4135. doi:10.1021/jo01309a012
Return to citation in text: [1] -
Stolle, A.; Ollivier, J.; Piras, P. P.; Salaün, J.; de Meijere, A. J. Am. Chem. Soc. 1992, 114, 4051–4067. doi:10.1021/ja00037a006
Return to citation in text: [1] -
Trost, B. M.; Bogdanowicz, M. J. J. Am. Chem. Soc. 1973, 95, 289–290. doi:10.1021/ja00782a077
Return to citation in text: [1] -
Trost, B. M.; Bogdanowicz, M. J. J. Am. Chem. Soc. 1973, 95, 5311–5321. doi:10.1021/ja00797a036
Return to citation in text: [1] -
Longone, D. T.; Wright, W. D. Tetrahedron Lett. 1969, 10, 2859–2862. doi:10.1016/s0040-4039(01)88292-7
Return to citation in text: [1] -
Kulinkovich, O. G.; Sviridov, S. V.; Vasilevskii, D. A.; Pritytskaya, T. S. Zh. Org. Khim. 1989, 25, 2244.
J. Org. Chem. USSR 1989, 25, 2027.
Return to citation in text: [1] -
Kulinkovich, O. G.; Sviridov, S. V.; Vasilevski, D. A. Synthesis 1991, 234. doi:10.1055/s-1991-26431
Return to citation in text: [1] -
Sviridov, S. V.; Vasilevskii, D. A.; Savchenko, A. I.; Pritytskaya, T. S.; Kulinkovich, O. G. Zh. Org. Khim. 1991, 27, 294.
J. Org. Chem. USSR 1991, 27, 250.
Return to citation in text: [1] -
Vasilevskii, D. A.; Savchenko, A. I.; Sviridov, S. V.; Kulinkovich, O. G. Zh. Org. Khim. 1991, 27, 1428.
J. Org. Chem. USSR 1991, 27, 1249.
Return to citation in text: [1] -
Savchenko, I. A.; Kulinkovich, O. G. Zh. Org. Khim. 1997, 33, 913.
Russ. J. Org. Chem. 1997, 33, 846.
Return to citation in text: [1] -
Savchenko, A. I.; Shevchuk, T. A.; Kulinkovich, O. G. Zh. Org. Khim. 1999, 35, 244.
Russ. J. Org. Chem. 1999, 35, 225.
Return to citation in text: [1] -
Lee, J.; Kim, H.; Cha, J. J. Am. Chem. Soc. 1996, 118, 4198–4199. doi:10.1021/ja954147f
Return to citation in text: [1] -
Epstein, O. L.; Savchenko, A. I.; Kulinkovich, O. G. Tetrahedron Lett. 1999, 40, 5935–5938. doi:10.1016/s0040-4039(99)01177-6
Return to citation in text: [1] -
Kananovich, D. G.; Kulinkovich, O. G. Tetrahedron 2008, 64, 1536–1547. doi:10.1016/j.tet.2007.10.075
Return to citation in text: [1] -
Lee, J.; Kang, C. H.; Kim, H.; Cha, J. K. J. Am. Chem. Soc. 1996, 118, 291–292. doi:10.1021/ja953055n
Return to citation in text: [1] -
Kasatkin, A.; Sato, F. Tetrahedron Lett. 1995, 36, 6079–6082. doi:10.1016/0040-4039(95)01208-y
Return to citation in text: [1] -
Corey, E. J.; Rao, S. A.; Noe, M. C. J. Am. Chem. Soc. 1994, 116, 9345–9346. doi:10.1021/ja00099a068
Return to citation in text: [1] -
Kulinkovich, O. G.; Kananovich, D. G.; Lopp, M.; Snieckus, V. Adv. Synth. Catal. 2014, 356, 3615–3626. doi:10.1002/adsc.201400480
Return to citation in text: [1] -
Welch, J. G.; Magid, R. M. J. Am. Chem. Soc. 1967, 89, 5300–5301. doi:10.1021/ja00996a047
Return to citation in text: [1] -
Lukina, M. Y.; Rudavshevskaya, T. Y.; Nesmeyanova, O. A. Dokl. Akad. Nauk. SSSR 1970, 190, 133.
Return to citation in text: [1] -
Avezov, I. B.; Bolesov, I. G.; Levina, R. Y. J. Org. Chem. USSR 1975, 10, 2129.
Return to citation in text: [1] -
Moiseenkov, A. M.; Czeskis, B. A.; Semenovsky, A. V. J. Chem. Soc., Chem. Commun. 1982, 109–110. doi:10.1039/c39820000109
Return to citation in text: [1] -
Lehmkuhl, H.; Mehler, K. Justus Liebigs Ann. Chem. 1978, 1841–1853. doi:10.1002/jlac.197819781117
Return to citation in text: [1] -
Lehmkuhl, H.; Mehler, K. Liebigs Ann. Chem. 1982, 2244–2246. doi:10.1002/jlac.198219821215
Return to citation in text: [1] -
Richey, H. G.; Watkins, E. K. J. Chem. Soc., Chem. Commun. 1984, 772–773. doi:10.1039/c39840000772
Return to citation in text: [1] -
Watkins, E. K.; Richey, H. G. Organometallics 1992, 11, 3785–3794. doi:10.1021/om00059a048
Return to citation in text: [1] -
Stoll, A. T.; Negishi, E.-i. Tetrahedron Lett. 1985, 26, 5671–5674. doi:10.1016/s0040-4039(01)80915-1
Return to citation in text: [1] -
Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l
Return to citation in text: [1] [2] -
Lautens, M.; Klute, W.; Tam, W. Chem. Rev. 1996, 96, 49–92. doi:10.1021/cr950016l
Return to citation in text: [1] -
Marek, I.; Simaan, S.; Masarwa, A. Angew. Chem., Int. Ed. 2007, 46, 7364–7376. doi:10.1002/anie.200604774
Return to citation in text: [1] -
Miege, F.; Meyer, C.; Cossy, J. Beilstein J. Org. Chem. 2011, 7, 717–734. doi:10.3762/bjoc.7.82
Return to citation in text: [1] -
Rubin, M.; Rubina, M.; Gevorgyan, V. Synthesis 2006, 1221–1245. doi:10.1055/s-2006-926404
Return to citation in text: [1] -
Isaka, M.; Nakamura, E. J. Am. Chem. Soc. 1990, 112, 7428–7430. doi:10.1021/ja00176a072
Return to citation in text: [1] -
Delaye, P.-O.; Didier, D.; Marek, I. Angew. Chem., Int. Ed. 2013, 52, 5333–5337. doi:10.1002/anie.201300664
Return to citation in text: [1] [2] -
Roy, S. R.; Didier, D.; Kleiner, A.; Marek, I. Chem. Sci. 2016, 7, 5989–5994. doi:10.1039/c6sc02191c
Return to citation in text: [1] -
Liao, L.-a.; Fox, J. M. J. Am. Chem. Soc. 2002, 124, 14322–14323. doi:10.1021/ja0278234
Return to citation in text: [1] -
Nakamura, M.; Hirai, A.; Nakamura, E. J. Am. Chem. Soc. 2000, 122, 978–979. doi:10.1021/ja983066r
Return to citation in text: [1] [2] -
Liu, X.; Fox, J. M. J. Am. Chem. Soc. 2006, 128, 5600–5601. doi:10.1021/ja058101q
Return to citation in text: [1] [2] -
Krämer, K.; Leong, P.; Lautens, M. Org. Lett. 2011, 13, 819–821. doi:10.1021/ol1029904
Return to citation in text: [1] [2] -
Rubin, M.; Gevorgyan, V. Synthesis 2004, 796–800. doi:10.1055/s-2003-44368
Return to citation in text: [1] -
Baird, M. S. In Carbocyclic Three-Membered Ring Compounds, 4th ed.; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart; Germany, 1996; Vol. 17, pp 2695 ff.
Return to citation in text: [1] -
Petiniot, N.; Anciaux, A. J.; Noels, A. F.; Hubert, A. J.; Teyssié, P. Tetrahedron Lett. 1978, 19, 1239–1242. doi:10.1016/s0040-4039(01)94511-3
Return to citation in text: [1] -
Panne, P.; Fox, J. M. J. Am. Chem. Soc. 2007, 129, 22–23. doi:10.1021/ja0660195
Return to citation in text: [1] -
Díaz-Requejo, M. M.; Mairena, M. A.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. Chem. Commun. 2001, 1804–1805. doi:10.1039/b105020f
Return to citation in text: [1] -
Lipshutz, B. H.; Wilhelm, R. S. J. Am. Chem. Soc. 1982, 104, 4696–4698. doi:10.1021/ja00381a041
Return to citation in text: [1] -
Lipshutz, B. H.; Kozlowski, J.; Wilhelm, R. S. J. Am. Chem. Soc. 1982, 104, 2305–2307. doi:10.1021/ja00372a032
Return to citation in text: [1] -
Lipshutz, B. H.; Wilhelm, R. S.; Floyd, D. M. J. Am. Chem. Soc. 1981, 103, 7672–7674. doi:10.1021/ja00415a055
Return to citation in text: [1] -
Minko, Y.; Marek, I. Org. Biomol. Chem. 2014, 12, 1535–1546. doi:10.1039/c3ob42349b
Return to citation in text: [1] [2] -
Boche, G.; Lohrenz, J. C. W. Chem. Rev. 2001, 101, 697–756. doi:10.1021/cr940260x
Return to citation in text: [1] -
Müller, E.; Töpel, T. Ber. Dtsch. Chem. Ges. B 1939, 72, 273–290. doi:10.1002/cber.19390720208
Return to citation in text: [1] -
Boche, G.; Möbus, K.; Harms, K.; Lohrenz, J. C. W.; Marsch, M. Chem. – Eur. J. 1996, 2, 604–607. doi:10.1002/chem.19960020521
Return to citation in text: [1] [2] -
Möller, M.; Husemann, M.; Boche, G. J. Organomet. Chem. 2001, 624, 47–52. doi:10.1016/s0022-328x(00)00596-9
Return to citation in text: [1] -
Warner, P.; Lu, S.-L. J. Org. Chem. 1976, 41, 1459–1461. doi:10.1021/jo00870a037
Return to citation in text: [1] -
Surry, D. S.; Spring, D. R. Chem. Soc. Rev. 2006, 35, 218–225. doi:10.1039/b508391p
Return to citation in text: [1] -
Simaan, M.; Delaye, P.-O.; Shi, M.; Marek, I. Angew. Chem., Int. Ed. 2015, 54, 12345–12348. doi:10.1002/anie.201412440
Return to citation in text: [1] [2] [3] -
Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569
Return to citation in text: [1] [2] -
Minko, Y.; Pasco, M.; Lercher, L.; Marek, I. Nat. Protoc. 2013, 8, 749–754. doi:10.1038/nprot.2013.036
Return to citation in text: [1] [2] -
Davies, H. M. L.; Lee, G. H. Org. Lett. 2004, 6, 1233–1236. doi:10.1021/ol049928c
Return to citation in text: [1] -
Marek, I.; Simaan, S.; Masarwa, A. Angew. Chem., Int. Ed. 2008, 47, 1982. doi:10.1002/anie.200890046
Return to citation in text: [1] -
Dian, L.; Müller, D. S.; Marek, I. Angew. Chem., Int. Ed. 2017, 56, 6783–6787. doi:10.1002/anie.201701094
Return to citation in text: [1] [2] [3] -
Parra, A.; Amenós, L.; Guisán-Ceinos, M.; López, A.; García Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833–15836. doi:10.1021/ja510419z
Return to citation in text: [1] -
Müller, D. S.; Marek, I. J. Am. Chem. Soc. 2015, 137, 15414–15417. doi:10.1021/jacs.5b11220
Return to citation in text: [1] -
Panek, E. J.; Kaiser, L. R.; Whitesides, G. M. J. Am. Chem. Soc. 1977, 99, 3708–3713. doi:10.1021/ja00453a032
Return to citation in text: [1] -
DeBergh, J. R.; Spivey, K. M.; Ready, J. M. J. Am. Chem. Soc. 2008, 130, 7828–7829. doi:10.1021/ja803480b
Return to citation in text: [1] -
Zhang, D.; Ready, J. M. Org. Lett. 2005, 7, 5681–5683. doi:10.1021/ol052413g
Return to citation in text: [1] -
Simaan, M.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 1543–1546. doi:10.1002/anie.201710707
Return to citation in text: [1] [2] -
Sommer, H.; Marek, I. Chem. Sci. 2018, 9, 6503–6508. doi:10.1039/c8sc02085j
Return to citation in text: [1] -
Dian, L.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 3682–3686. doi:10.1002/anie.201713324
Return to citation in text: [1] [2] -
Müller, D. S.; Werner, V.; Akyol, S.; Schmalz, H.-G.; Marek, I. Org. Lett. 2017, 19, 3970–3973. doi:10.1021/acs.orglett.7b01661
Return to citation in text: [1]
| 78. | Simaan, M.; Delaye, P.-O.; Shi, M.; Marek, I. Angew. Chem., Int. Ed. 2015, 54, 12345–12348. doi:10.1002/anie.201412440 |
| 81. | Davies, H. M. L.; Lee, G. H. Org. Lett. 2004, 6, 1233–1236. doi:10.1021/ol049928c |
| 82. | Marek, I.; Simaan, S.; Masarwa, A. Angew. Chem., Int. Ed. 2008, 47, 1982. doi:10.1002/anie.200890046 |
| 83. | Dian, L.; Müller, D. S.; Marek, I. Angew. Chem., Int. Ed. 2017, 56, 6783–6787. doi:10.1002/anie.201701094 |
| 83. | Dian, L.; Müller, D. S.; Marek, I. Angew. Chem., Int. Ed. 2017, 56, 6783–6787. doi:10.1002/anie.201701094 |
| 91. | Dian, L.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 3682–3686. doi:10.1002/anie.201713324 |
| 1. | D’yakonov, V. A.; Trapeznikova, O. A.; de Meijere, A.; Dzhemilev, U. M. Chem. Rev. 2014, 114, 5775–5814. doi:10.1021/cr400291c |
| 5. | Orellana, A.; Nikolaev, A. Synthesis 2016, 48, 1741–1768. doi:10.1055/s-0035-1560442 |
| 29. |
Kulinkovich, O. G.; Sviridov, S. V.; Vasilevskii, D. A.; Pritytskaya, T. S. Zh. Org. Khim. 1989, 25, 2244.
J. Org. Chem. USSR 1989, 25, 2027. |
| 30. | Kulinkovich, O. G.; Sviridov, S. V.; Vasilevski, D. A. Synthesis 1991, 234. doi:10.1055/s-1991-26431 |
| 31. |
Sviridov, S. V.; Vasilevskii, D. A.; Savchenko, A. I.; Pritytskaya, T. S.; Kulinkovich, O. G. Zh. Org. Khim. 1991, 27, 294.
J. Org. Chem. USSR 1991, 27, 250. |
| 32. |
Vasilevskii, D. A.; Savchenko, A. I.; Sviridov, S. V.; Kulinkovich, O. G. Zh. Org. Khim. 1991, 27, 1428.
J. Org. Chem. USSR 1991, 27, 1249. |
| 33. |
Savchenko, I. A.; Kulinkovich, O. G. Zh. Org. Khim. 1997, 33, 913.
Russ. J. Org. Chem. 1997, 33, 846. |
| 34. |
Savchenko, A. I.; Shevchuk, T. A.; Kulinkovich, O. G. Zh. Org. Khim. 1999, 35, 244.
Russ. J. Org. Chem. 1999, 35, 225. |
| 35. | Lee, J.; Kim, H.; Cha, J. J. Am. Chem. Soc. 1996, 118, 4198–4199. doi:10.1021/ja954147f |
| 36. | Epstein, O. L.; Savchenko, A. I.; Kulinkovich, O. G. Tetrahedron Lett. 1999, 40, 5935–5938. doi:10.1016/s0040-4039(99)01177-6 |
| 37. | Kananovich, D. G.; Kulinkovich, O. G. Tetrahedron 2008, 64, 1536–1547. doi:10.1016/j.tet.2007.10.075 |
| 38. | Lee, J.; Kang, C. H.; Kim, H.; Cha, J. K. J. Am. Chem. Soc. 1996, 118, 291–292. doi:10.1021/ja953055n |
| 39. | Kasatkin, A.; Sato, F. Tetrahedron Lett. 1995, 36, 6079–6082. doi:10.1016/0040-4039(95)01208-y |
| 40. | Corey, E. J.; Rao, S. A.; Noe, M. C. J. Am. Chem. Soc. 1994, 116, 9345–9346. doi:10.1021/ja00099a068 |
| 41. | Kulinkovich, O. G.; Kananovich, D. G.; Lopp, M.; Snieckus, V. Adv. Synth. Catal. 2014, 356, 3615–3626. doi:10.1002/adsc.201400480 |
| 71. | Minko, Y.; Marek, I. Org. Biomol. Chem. 2014, 12, 1535–1546. doi:10.1039/c3ob42349b |
| 79. | Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569 |
| 80. | Minko, Y.; Pasco, M.; Lercher, L.; Marek, I. Nat. Protoc. 2013, 8, 749–754. doi:10.1038/nprot.2013.036 |
| 86. | Panek, E. J.; Kaiser, L. R.; Whitesides, G. M. J. Am. Chem. Soc. 1977, 99, 3708–3713. doi:10.1021/ja00453a032 |
| 87. | DeBergh, J. R.; Spivey, K. M.; Ready, J. M. J. Am. Chem. Soc. 2008, 130, 7828–7829. doi:10.1021/ja803480b |
| 88. | Zhang, D.; Ready, J. M. Org. Lett. 2005, 7, 5681–5683. doi:10.1021/ol052413g |
| 89. | Simaan, M.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 1543–1546. doi:10.1002/anie.201710707 |
| 4. | Haym, I.; Brimble, M. A. Org. Biomol. Chem. 2012, 10, 7649–7665. doi:10.1039/c2ob26082d |
| 42. | Welch, J. G.; Magid, R. M. J. Am. Chem. Soc. 1967, 89, 5300–5301. doi:10.1021/ja00996a047 |
| 89. | Simaan, M.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 1543–1546. doi:10.1002/anie.201710707 |
| 3. | Wong, H. N. C.; Hon, M.-Y.; Tse, C.-W.; Yip, Y.-C.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165–198. doi:10.1021/cr00091a005 |
| 26. | Trost, B. M.; Bogdanowicz, M. J. J. Am. Chem. Soc. 1973, 95, 289–290. doi:10.1021/ja00782a077 |
| 27. | Trost, B. M.; Bogdanowicz, M. J. J. Am. Chem. Soc. 1973, 95, 5311–5321. doi:10.1021/ja00797a036 |
| 84. | Parra, A.; Amenós, L.; Guisán-Ceinos, M.; López, A.; García Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833–15836. doi:10.1021/ja510419z |
| 28. | Longone, D. T.; Wright, W. D. Tetrahedron Lett. 1969, 10, 2859–2862. doi:10.1016/s0040-4039(01)88292-7 |
| 83. | Dian, L.; Müller, D. S.; Marek, I. Angew. Chem., Int. Ed. 2017, 56, 6783–6787. doi:10.1002/anie.201701094 |
| 85. | Müller, D. S.; Marek, I. J. Am. Chem. Soc. 2015, 137, 15414–15417. doi:10.1021/jacs.5b11220 |
| 10. | Rubottom, G. M.; Lopez, M. I. J. Org. Chem. 1973, 38, 2097–2099. doi:10.1021/jo00951a032 |
| 11. | Murai, S.; Aya, T.; Sonoda, N. J. Org. Chem. 1973, 38, 4354–4356. doi:10.1021/jo00964a037 |
| 12. | Du, H.; Long, J.; Shi, Y. Org. Lett. 2006, 8, 2827–2829. doi:10.1021/ol0609659 |
| 18. | Barluenga, J.; Suero, M. G.; Pérez-Sánchez, I.; Flórez, J. J. Am. Chem. Soc. 2008, 130, 2708–2709. doi:10.1021/ja074363b |
| 61. | Liu, X.; Fox, J. M. J. Am. Chem. Soc. 2006, 128, 5600–5601. doi:10.1021/ja058101q |
| 8. | Imamoto, T.; Kamiya, Y.; Hatajima, T.; Takahashi, H. Tetrahedron Lett. 1989, 30, 5149–5152. doi:10.1016/s0040-4039(01)93471-9 |
| 9. | Ito, S.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 1998, 39, 5253–5256. doi:10.1016/s0040-4039(98)01036-3 |
| 19. | Ukai, K.; Oshima, K.; Matsubara, S. J. Am. Chem. Soc. 2000, 122, 12047–12048. doi:10.1021/ja003360v |
| 20. | Matsubara, S.; Ukai, K.; Fushimi, H.; Yokota, Y.; Yoshino, H.; Oshima, K.; Omoto, K.; Ogawa, A.; Hioki, Y.; Fujimoto, H. Tetrahedron 2002, 58, 8255–8262. doi:10.1016/s0040-4020(02)00975-4 |
| 21. | Cheng, K.; Carroll, P. J.; Walsh, P. J. Org. Lett. 2011, 13, 2346–2349. doi:10.1021/ol200597h |
| 22. | Turro, N. J. Acc. Chem. Res. 1969, 2, 25–32. doi:10.1021/ar50013a004 |
| 23. | Salaun, J. Chem. Rev. 1983, 83, 619–632. doi:10.1021/cr00058a002 |
| 24. | Salaün, J.; Bennani, F.; Compain, J. C.; Fadel, A.; Ollivier, J. J. Org. Chem. 1980, 45, 4129–4135. doi:10.1021/jo01309a012 |
| 25. | Stolle, A.; Ollivier, J.; Piras, P. P.; Salaün, J.; de Meijere, A. J. Am. Chem. Soc. 1992, 114, 4051–4067. doi:10.1021/ja00037a006 |
| 51. | Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l |
| 7. | Magrane, J. K., Jr.; Cottle, D. L. J. Am. Chem. Soc. 1942, 64, 484–487. doi:10.1021/ja01255a004 |
| 62. | Krämer, K.; Leong, P.; Lautens, M. Org. Lett. 2011, 13, 819–821. doi:10.1021/ol1029904 |
| 13. | Fontani, P.; Carboni, B.; Vaultier, M.; Maas, G. Synthesis 1991, 605–609. doi:10.1055/s-1991-26524 |
| 14. | Pietruszka, J.; Widenmeyer, M. Synlett 1997, 977–979. doi:10.1055/s-1997-5790 |
| 15. | Hussain, M. M.; Li, H.; Hussain, N.; Ureña, M.; Carroll, P. J.; Walsh, P. J. J. Am. Chem. Soc. 2009, 131, 6516–6524. doi:10.1021/ja900147s |
| 16. | Imai, T.; Mineta, H.; Nishida, S. J. Org. Chem. 1990, 55, 4986–4988. doi:10.1021/jo00304a004 |
| 17. | Benoit, G.; Charette, A. B. J. Am. Chem. Soc. 2017, 139, 1364–1367. doi:10.1021/jacs.6b09090 |
| 60. | Nakamura, M.; Hirai, A.; Nakamura, E. J. Am. Chem. Soc. 2000, 122, 978–979. doi:10.1021/ja983066r |
| 68. | Lipshutz, B. H.; Wilhelm, R. S. J. Am. Chem. Soc. 1982, 104, 4696–4698. doi:10.1021/ja00381a041 |
| 69. | Lipshutz, B. H.; Kozlowski, J.; Wilhelm, R. S. J. Am. Chem. Soc. 1982, 104, 2305–2307. doi:10.1021/ja00372a032 |
| 70. | Lipshutz, B. H.; Wilhelm, R. S.; Floyd, D. M. J. Am. Chem. Soc. 1981, 103, 7672–7674. doi:10.1021/ja00415a055 |
| 43. | Lukina, M. Y.; Rudavshevskaya, T. Y.; Nesmeyanova, O. A. Dokl. Akad. Nauk. SSSR 1970, 190, 133. |
| 44. | Avezov, I. B.; Bolesov, I. G.; Levina, R. Y. J. Org. Chem. USSR 1975, 10, 2129. |
| 45. | Moiseenkov, A. M.; Czeskis, B. A.; Semenovsky, A. V. J. Chem. Soc., Chem. Commun. 1982, 109–110. doi:10.1039/c39820000109 |
| 46. | Lehmkuhl, H.; Mehler, K. Justus Liebigs Ann. Chem. 1978, 1841–1853. doi:10.1002/jlac.197819781117 |
| 47. | Lehmkuhl, H.; Mehler, K. Liebigs Ann. Chem. 1982, 2244–2246. doi:10.1002/jlac.198219821215 |
| 48. | Richey, H. G.; Watkins, E. K. J. Chem. Soc., Chem. Commun. 1984, 772–773. doi:10.1039/c39840000772 |
| 49. | Watkins, E. K.; Richey, H. G. Organometallics 1992, 11, 3785–3794. doi:10.1021/om00059a048 |
| 50. | Stoll, A. T.; Negishi, E.-i. Tetrahedron Lett. 1985, 26, 5671–5674. doi:10.1016/s0040-4039(01)80915-1 |
| 51. | Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l |
| 52. | Lautens, M.; Klute, W.; Tam, W. Chem. Rev. 1996, 96, 49–92. doi:10.1021/cr950016l |
| 53. | Marek, I.; Simaan, S.; Masarwa, A. Angew. Chem., Int. Ed. 2007, 46, 7364–7376. doi:10.1002/anie.200604774 |
| 54. | Miege, F.; Meyer, C.; Cossy, J. Beilstein J. Org. Chem. 2011, 7, 717–734. doi:10.3762/bjoc.7.82 |
| 55. | Rubin, M.; Rubina, M.; Gevorgyan, V. Synthesis 2006, 1221–1245. doi:10.1055/s-2006-926404 |
| 56. | Isaka, M.; Nakamura, E. J. Am. Chem. Soc. 1990, 112, 7428–7430. doi:10.1021/ja00176a072 |
| 57. | Delaye, P.-O.; Didier, D.; Marek, I. Angew. Chem., Int. Ed. 2013, 52, 5333–5337. doi:10.1002/anie.201300664 |
| 58. | Roy, S. R.; Didier, D.; Kleiner, A.; Marek, I. Chem. Sci. 2016, 7, 5989–5994. doi:10.1039/c6sc02191c |
| 59. | Liao, L.-a.; Fox, J. M. J. Am. Chem. Soc. 2002, 124, 14322–14323. doi:10.1021/ja0278234 |
| 60. | Nakamura, M.; Hirai, A.; Nakamura, E. J. Am. Chem. Soc. 2000, 122, 978–979. doi:10.1021/ja983066r |
| 61. | Liu, X.; Fox, J. M. J. Am. Chem. Soc. 2006, 128, 5600–5601. doi:10.1021/ja058101q |
| 62. | Krämer, K.; Leong, P.; Lautens, M. Org. Lett. 2011, 13, 819–821. doi:10.1021/ol1029904 |
| 63. | Rubin, M.; Gevorgyan, V. Synthesis 2004, 796–800. doi:10.1055/s-2003-44368 |
| 90. | Sommer, H.; Marek, I. Chem. Sci. 2018, 9, 6503–6508. doi:10.1039/c8sc02085j |
| 91. | Dian, L.; Marek, I. Angew. Chem., Int. Ed. 2018, 57, 3682–3686. doi:10.1002/anie.201713324 |
| 92. | Müller, D. S.; Werner, V.; Akyol, S.; Schmalz, H.-G.; Marek, I. Org. Lett. 2017, 19, 3970–3973. doi:10.1021/acs.orglett.7b01661 |
| 64. | Baird, M. S. In Carbocyclic Three-Membered Ring Compounds, 4th ed.; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart; Germany, 1996; Vol. 17, pp 2695 ff. |
| 65. | Petiniot, N.; Anciaux, A. J.; Noels, A. F.; Hubert, A. J.; Teyssié, P. Tetrahedron Lett. 1978, 19, 1239–1242. doi:10.1016/s0040-4039(01)94511-3 |
| 66. | Panne, P.; Fox, J. M. J. Am. Chem. Soc. 2007, 129, 22–23. doi:10.1021/ja0660195 |
| 67. | Díaz-Requejo, M. M.; Mairena, M. A.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. Chem. Commun. 2001, 1804–1805. doi:10.1039/b105020f |
| 78. | Simaan, M.; Delaye, P.-O.; Shi, M.; Marek, I. Angew. Chem., Int. Ed. 2015, 54, 12345–12348. doi:10.1002/anie.201412440 |
| 79. | Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569 |
| 80. | Minko, Y.; Pasco, M.; Lercher, L.; Marek, I. Nat. Protoc. 2013, 8, 749–754. doi:10.1038/nprot.2013.036 |
| 57. | Delaye, P.-O.; Didier, D.; Marek, I. Angew. Chem., Int. Ed. 2013, 52, 5333–5337. doi:10.1002/anie.201300664 |
| 74. | Boche, G.; Möbus, K.; Harms, K.; Lohrenz, J. C. W.; Marsch, M. Chem. – Eur. J. 1996, 2, 604–607. doi:10.1002/chem.19960020521 |
| 78. | Simaan, M.; Delaye, P.-O.; Shi, M.; Marek, I. Angew. Chem., Int. Ed. 2015, 54, 12345–12348. doi:10.1002/anie.201412440 |
| 73. | Müller, E.; Töpel, T. Ber. Dtsch. Chem. Ges. B 1939, 72, 273–290. doi:10.1002/cber.19390720208 |
| 74. | Boche, G.; Möbus, K.; Harms, K.; Lohrenz, J. C. W.; Marsch, M. Chem. – Eur. J. 1996, 2, 604–607. doi:10.1002/chem.19960020521 |
| 75. | Möller, M.; Husemann, M.; Boche, G. J. Organomet. Chem. 2001, 624, 47–52. doi:10.1016/s0022-328x(00)00596-9 |
| 76. | Warner, P.; Lu, S.-L. J. Org. Chem. 1976, 41, 1459–1461. doi:10.1021/jo00870a037 |
| 77. | Surry, D. S.; Spring, D. R. Chem. Soc. Rev. 2006, 35, 218–225. doi:10.1039/b508391p |
| 71. | Minko, Y.; Marek, I. Org. Biomol. Chem. 2014, 12, 1535–1546. doi:10.1039/c3ob42349b |
| 72. | Boche, G.; Lohrenz, J. C. W. Chem. Rev. 2001, 101, 697–756. doi:10.1021/cr940260x |
© 2019 Simaan and Marek; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)