Abstract
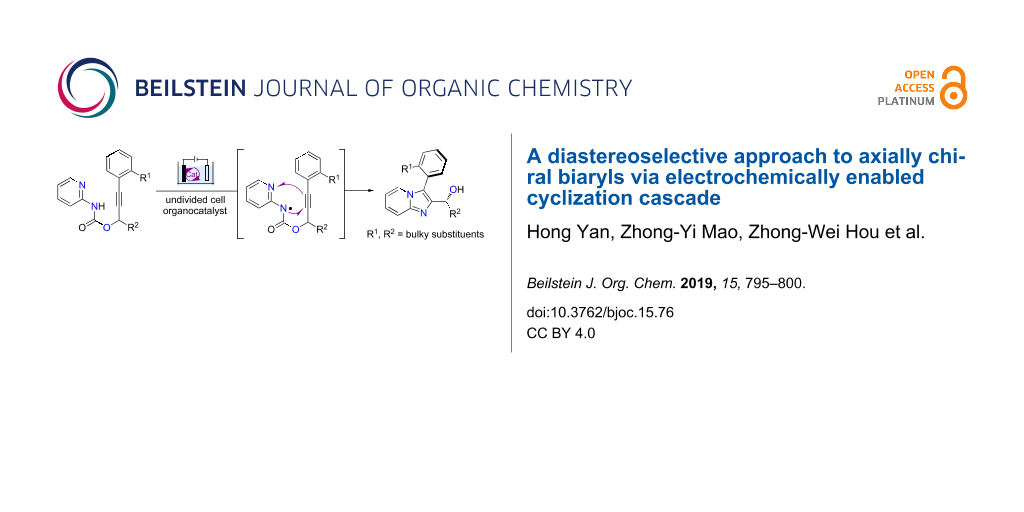
A diastereoselective approach to axially chiral imidazopyridine-containing biaryls has been developed. The reactions proceed through a radical cyclization cascade to construct the biaryls with good to excellent central-to-axial chirality transfer.

Graphical Abstract
Introduction
Axially chiral biaryls are prevalent in natural products, bioactive molecules and organocatalysts [1,2]. Among the many methods that have been developed for the synthesis of chiral biaryls [3-10], reactions that take avantage of the central-to-axial chirality transfer have been less explored [11-14]. In addition, an antroposelective synthesis of imidazopyridine-based biaryls has not been reported.
Nitrogen-centered radicals (NCRs) are attractive reactive intermediates for organic synthesis as they provide opportunities for the efficient construction of C–N bonds [15-19]. Recently, the generation of NCRs through electron transfer-based methods has been attracting attention. Organic electrochemistry is a powerful tool for adding or taking electrons from organic molecules to promote redox reactions because of its reagent-free feature and the tunability of electric current and potential [20-30]. We [31-34] and others [35-41] have studied the reactions of electrochemically generated NCRs. Particularly, we have recently reported an electrochemical synthesis of imidazo-fused N-heteroaromatic compounds via a radical cyclization cascade [31]. Building on this work, we report herein an atroposelective synthesis of imidazopyridine-containing biaryls via central-to-axial chirality transfer (Scheme 1).
![[1860-5397-15-76-i1]](/bjoc/content/inline/1860-5397-15-76-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
The substituents on the phenyl ring (R1) and at the propargylic position (R2) of carbamate 2 were varied to study their effects on the diastereoselectivity (Table 1). The electrolysis was conducted under previously established conditions employing a three-necked round-bottomed flask as the cell, a reticulated vitreous carbon (RVC) anode and a platinum plate cathode [31]. The reaction was carried in refluxing MeCN/H2O (9:1) with tetraarylhydrazine 1 as the redox catalyst, NaHCO3 (2 equiv) as an additive, and Et4NBF4 (1 equiv) as the supporting electrolyte. These investigations indicated that bulky tertiary groups at both R1 and R2 positions were needed to ensure efficient chirality transfer. Hence, carbamate 2a (Table 1, entry 1) bearing a t-Bu group at R1 and R2 positions, respectively, reacted to give imidazopyridine-based biaryl 3a in 68% yield with good diastereoselectivity (14:1 dr). Replacing the t-Bu group at the propargylic position with iPr (Table 1, entry 2) or on the phenyl ring with Ph (Table 1, entry 3) or OiPr (Table 1, entry 4) all resulted in low diastereoselectivity (2:1 to 3:1).
Table 1: Investigation on the effects of substituents on the diastereoselectivity.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-76-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Substrate | Product, yield,b drc |
| 1 | 2a (R1 = t-Bu, R2 = t-Bu) | (±)-3a, 68%, 14:1 dr |
| 2 | 2b (R1 = t-Bu, R2 = iPr) | (±)-3b, 64%, 3:1 dr |
| 3 | 2c (R1 = Ph, R2 = t-Bu) | (±)-3c, 73%, 2:1 dr |
| 4 | 2d (R1 = OiPr, R2 = t-Bu) | (±)-3d, 78%, 3:1 dr |
aReaction conditions: undivided cell, 1 (0.03 mmol), 2 (0.3 mmol), H2O (1 mL), MeCN (9 mL), 3.5 F mol−1. bIsolated yield. cDetermined by 1H NMR analysis of the crude reaction mixture.
The scope of the electrosynthesis was investigated by varying the peripheral substituents of the carbamate substrate 2 (Scheme 2). The pyridyl ring could be substituted at positions 4, 5 and 6 with a range of substituents with diverse electronic properties such as OMe (3e), Br (3f), CF3 (3g), CN (3h), Cl (3i), and Me (3j). Pyridyl rings bearing multiple substituents were also tolerated (3k and 3l). The stereochemistry of the biaryl product was determined by obtaining an X-ray crystal structure of 3k. The t-Bu-substituted phenyl ring on the alkyne moiety containing an extra OMe (3m) or Me (3n and 3o) group was tolerated albeit with reduced diastereoselectivity. The t-Bu group on the phenyl ring and at the propargylic position could be replaced with other bulky tertiary substituents to afford a range of functionalized biaryl products (3p–w). The electrochemical conditions were compatible with several functional groups including aryltrimethylsilane (3p), silyl ether (3q–s), ester (3t) and cylic ketal (3u). Note that all the diastereomers were separable by flash column chromatography.
![[1860-5397-15-76-i2]](/bjoc/content/inline/1860-5397-15-76-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of electrochemical synthesis of axially chiral biaryls. Reaction conditions: undivided cell, 2 (0.3 mmol), H2O (1 mL), MeCN (9 mL), 3.5 F mol−1. aIsolated yield of the major diastereomer. bDetermined by 1H NMR analysis of crude reaction mixture. cCombined yield of the two diastereomers.
Scheme 2: Scope of electrochemical synthesis of axially chiral biaryls. Reaction conditions: undivided cell, 2...
Heating a solution of the major isomer of 3n in MeCN at 80 °C for 4 h did not lead to isomerization, suggesting that the stereoselectivity of the reaction was not controlled by relative thermodynamic stability of the diastereomers. The major isomer of 3c, which contained a sterically less demanding Ph group at the R1 position (cf. Table 1), did not isomerize at room temperature for 1 year. However, heating a solution of this compound in MeCN at 80 °C for 4 h resulted in a mixture of diastereomers in a ratio of 3:1. These results suggest that the sterically demanding substituents at R1 and R2 positions (cf. Table 1) are critical to ensure good stereoselectivity during the product formation and to prevent isomerization after the reaction.
A mechanism for the electrochemical synthesis was proposed based on the results from our previous work [31] and of this work (Scheme 3). The redox catalyst 1 is oxidized at the anode to give radical cation I. In the meanwhile, H2O is reduced at the cathode to afford HO− and H2. The base generated at the cathode deprotonates 2a to give its conjugate base II. The anionic II is oxidized by radical cation I through single electron transfer (SET) to give radical intermediate III, which undergoes a biscyclization to give V. Further oxidation of V followed by hydrolysis of the cyclic carbamate moiety leads to the formation of 3a.
![[1860-5397-15-76-i3]](/bjoc/content/inline/1860-5397-15-76-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanism for the electrochemical synthesis of 3a.
Scheme 3: Proposed reaction mechanism for the electrochemical synthesis of 3a.
Based on the proposed reaction mechanism and the results mentioned above, the cyclization of vinyl radical IV to give V is the atroposelective step. Density functional theory (DFT)-based calculations suggested that the cyclization of IV could be explained by a Curtin–Hammett scenario (Scheme 4) [42]. Specifically, the equilibrium of the conformations IV and IV′ is much faster than their respectively cyclizations to give V and V′. Since TS2 is relatively lower in energy than TS3, V is formed as the major product.
![[1860-5397-15-76-i4]](/bjoc/content/inline/1860-5397-15-76-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Computation investigation on the vinyl radical cyclization. DFT (M06-2X/6-31G*) calculated energetics (kcal mol−1) are Gibbs free energies in MeCN.
Scheme 4: Computation investigation on the vinyl radical cyclization. DFT (M06-2X/6-31G*) calculated energeti...
Conclusion
In summary, we have developed a diastereoselective approach for the synthesis of axially chiral biaryls through an electricity-powered cyclization cascade. The reactions employ easily assembled starting materials and afford functionalized imidazopyridine-based biaryls in good to high yields and diastereoselectivity.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 8.0 MB | Download |
References
-
Bringmann, G.; Gulder, T.; Gulder, T. A. M.; Breuning, M. Chem. Rev. 2011, 111, 563–639. doi:10.1021/cr100155e
Return to citation in text: [1] -
Pu, L. Chem. Rev. 1998, 98, 2405–2494. doi:10.1021/cr970463w
Return to citation in text: [1] -
Tanaka, K. Chem. – Asian J. 2009, 4, 508–518. doi:10.1002/asia.200800378
Return to citation in text: [1] -
Wallace, T. W. Org. Biomol. Chem. 2006, 4, 3197–3210. doi:10.1039/b608470m
Return to citation in text: [1] -
Bringmann, G.; Price Mortimer, A. J.; Keller, P. A.; Gresser, M. J.; Garner, J.; Breuning, M. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. doi:10.1002/anie.200462661
Return to citation in text: [1] -
Ma, G.; Sibi, M. P. Chem. – Eur. J. 2015, 21, 11644–11657. doi:10.1002/chem.201500869
Return to citation in text: [1] -
Link, A.; Sparr, C. Chem. Soc. Rev. 2018, 47, 3804–3815. doi:10.1039/c7cs00875a
Return to citation in text: [1] -
Wang, Y.-B.; Tan, B. Acc. Chem. Res. 2018, 51, 534–547. doi:10.1021/acs.accounts.7b00602
Return to citation in text: [1] -
Yang, H.; Yang, X.; Tang, W. Tetrahedron 2016, 72, 6143–6174. doi:10.1016/j.tet.2016.08.042
Return to citation in text: [1] -
Wencel-Delord, J.; Panossian, A.; Leroux, F. R.; Colobert, F. Chem. Soc. Rev. 2015, 44, 3418–3430. doi:10.1039/c5cs00012b
Return to citation in text: [1] -
Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140–2143. doi:10.1021/jacs.6b11079
Return to citation in text: [1] -
Vorogushin, A. V.; Wulff, W. D.; Hansen, H.-J. J. Am. Chem. Soc. 2002, 124, 6512–6513. doi:10.1021/ja0201505
Return to citation in text: [1] -
Guo, F.; Konkol, L. C.; Thomson, R. J. J. Am. Chem. Soc. 2011, 133, 18–20. doi:10.1021/ja108717r
Return to citation in text: [1] -
Qiu, L.; Kwong, F. Y.; Wu, J.; Lam, W. H.; Chan, S.; Yu, W.-Y.; Li, Y.-M.; Guo, R.; Zhou, Z.; Chan, A. S. C. J. Am. Chem. Soc. 2006, 128, 5955–5965. doi:10.1021/ja0602694
Return to citation in text: [1] -
Kärkäs, M. D. Chem. Soc. Rev. 2018, 47, 5786–5865. doi:10.1039/c7cs00619e
Return to citation in text: [1] -
Zhao, Y.; Xia, W. Chem. Soc. Rev. 2018, 47, 2591–2608. doi:10.1039/c7cs00572e
Return to citation in text: [1] -
Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d
Return to citation in text: [1] -
Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603–1618. doi:10.1039/b613443m
Return to citation in text: [1] -
Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2016, 45, 3069–3087. doi:10.1039/c5cs00852b
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060
Return to citation in text: [1] -
Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. doi:10.1002/anie.201712732
Return to citation in text: [1] -
Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346–2352. doi:10.1021/acs.accounts.7b00287
Return to citation in text: [1] -
Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Angew. Chem., Int. Ed. 2018, 57, 4149–4155. doi:10.1002/anie.201707584
Return to citation in text: [1] -
Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338–352. doi:10.1002/cjoc.201700740
Return to citation in text: [1] -
Tang, S.; Liu, Y.; Lei, A. Chem 2018, 4, 27–45. doi:10.1016/j.chempr.2017.10.001
Return to citation in text: [1] -
Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271
Return to citation in text: [1] -
Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k
Return to citation in text: [1] -
Hou, Z.-W.; Mao, Z.-Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. doi:10.1002/anie.201711876
Return to citation in text: [1] [2] [3] [4] -
Hou, Z.-W.; Yan, H.; Song, J.-S.; Xu, H.-C. Chin. J. Chem. 2018, 36, 909–915. doi:10.1002/cjoc.201800301
Return to citation in text: [1] -
Hou, Z.-W.; Mao, Z.-Y.; Song, J.; Xu, H.-C. ACS Catal. 2017, 7, 5810–5813. doi:10.1021/acscatal.7b02105
Return to citation in text: [1] -
Hou, Z.-W.; Mao, Z.-Y.; Zhao, H.-B.; Melcamu, Y. Y.; Lu, X.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2016, 55, 9168–9172. doi:10.1002/anie.201602616
Return to citation in text: [1] -
Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K. D.; Waldvogel, S. R. J. Am. Chem. Soc. 2017, 139, 12317–12324. doi:10.1021/jacs.7b07488
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2008, 130, 13542–13543. doi:10.1021/ja806259z
Return to citation in text: [1] -
Gieshoff, T.; Schollmeyer, D.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2016, 55, 9437–9440. doi:10.1002/anie.201603899
Return to citation in text: [1] -
Kehl, A.; Breising, V. M.; Schollmeyer, D.; Waldvogel, S. R. Chem. – Eur. J. 2018, 24, 17230–17233. doi:10.1002/chem.201804638
Return to citation in text: [1] -
Lin, M.-Y.; Xu, K.; Jiang, Y.-Y.; Liu, Y.-G.; Sun, B.-G.; Zeng, C.-C. Adv. Synth. Catal. 2018, 360, 1665–1672. doi:10.1002/adsc.201701536
Return to citation in text: [1] -
Zhang, S.; Li, L.; Xue, M.; Zhang, R.; Xu, K.; Zeng, C. Org. Lett. 2018, 20, 3443–3446. doi:10.1021/acs.orglett.8b00981
Return to citation in text: [1] -
Hu, X.; Zhang, G.; Bu, F.; Nie, L.; Lei, A. ACS Catal. 2018, 8, 9370–9375. doi:10.1021/acscatal.8b02847
Return to citation in text: [1] -
Seeman, J. I. J. Chem. Educ. 1986, 63, 42–48. doi:10.1021/ed063p42
Return to citation in text: [1]
| 1. | Bringmann, G.; Gulder, T.; Gulder, T. A. M.; Breuning, M. Chem. Rev. 2011, 111, 563–639. doi:10.1021/cr100155e |
| 2. | Pu, L. Chem. Rev. 1998, 98, 2405–2494. doi:10.1021/cr970463w |
| 20. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 21. | Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060 |
| 22. | Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. doi:10.1002/anie.201712732 |
| 23. | Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346–2352. doi:10.1021/acs.accounts.7b00287 |
| 24. | Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656 |
| 25. | Yan, M.; Kawamata, Y.; Baran, P. S. Angew. Chem., Int. Ed. 2018, 57, 4149–4155. doi:10.1002/anie.201707584 |
| 26. | Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338–352. doi:10.1002/cjoc.201700740 |
| 27. | Tang, S.; Liu, Y.; Lei, A. Chem 2018, 4, 27–45. doi:10.1016/j.chempr.2017.10.001 |
| 28. | Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 29. | Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271 |
| 30. | Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492–2521. doi:10.1039/c3cs60464k |
| 15. | Kärkäs, M. D. Chem. Soc. Rev. 2018, 47, 5786–5865. doi:10.1039/c7cs00619e |
| 16. | Zhao, Y.; Xia, W. Chem. Soc. Rev. 2018, 47, 2591–2608. doi:10.1039/c7cs00572e |
| 17. | Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d |
| 18. | Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603–1618. doi:10.1039/b613443m |
| 19. | Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2016, 45, 3069–3087. doi:10.1039/c5cs00852b |
| 11. | Raut, V. S.; Jean, M.; Vanthuyne, N.; Roussel, C.; Constantieux, T.; Bressy, C.; Bugaut, X.; Bonne, D.; Rodriguez, J. J. Am. Chem. Soc. 2017, 139, 2140–2143. doi:10.1021/jacs.6b11079 |
| 12. | Vorogushin, A. V.; Wulff, W. D.; Hansen, H.-J. J. Am. Chem. Soc. 2002, 124, 6512–6513. doi:10.1021/ja0201505 |
| 13. | Guo, F.; Konkol, L. C.; Thomson, R. J. J. Am. Chem. Soc. 2011, 133, 18–20. doi:10.1021/ja108717r |
| 14. | Qiu, L.; Kwong, F. Y.; Wu, J.; Lam, W. H.; Chan, S.; Yu, W.-Y.; Li, Y.-M.; Guo, R.; Zhou, Z.; Chan, A. S. C. J. Am. Chem. Soc. 2006, 128, 5955–5965. doi:10.1021/ja0602694 |
| 3. | Tanaka, K. Chem. – Asian J. 2009, 4, 508–518. doi:10.1002/asia.200800378 |
| 4. | Wallace, T. W. Org. Biomol. Chem. 2006, 4, 3197–3210. doi:10.1039/b608470m |
| 5. | Bringmann, G.; Price Mortimer, A. J.; Keller, P. A.; Gresser, M. J.; Garner, J.; Breuning, M. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. doi:10.1002/anie.200462661 |
| 6. | Ma, G.; Sibi, M. P. Chem. – Eur. J. 2015, 21, 11644–11657. doi:10.1002/chem.201500869 |
| 7. | Link, A.; Sparr, C. Chem. Soc. Rev. 2018, 47, 3804–3815. doi:10.1039/c7cs00875a |
| 8. | Wang, Y.-B.; Tan, B. Acc. Chem. Res. 2018, 51, 534–547. doi:10.1021/acs.accounts.7b00602 |
| 9. | Yang, H.; Yang, X.; Tang, W. Tetrahedron 2016, 72, 6143–6174. doi:10.1016/j.tet.2016.08.042 |
| 10. | Wencel-Delord, J.; Panossian, A.; Leroux, F. R.; Colobert, F. Chem. Soc. Rev. 2015, 44, 3418–3430. doi:10.1039/c5cs00012b |
| 31. | Hou, Z.-W.; Mao, Z.-Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. doi:10.1002/anie.201711876 |
| 31. | Hou, Z.-W.; Mao, Z.-Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. doi:10.1002/anie.201711876 |
| 35. | Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K. D.; Waldvogel, S. R. J. Am. Chem. Soc. 2017, 139, 12317–12324. doi:10.1021/jacs.7b07488 |
| 36. | Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2008, 130, 13542–13543. doi:10.1021/ja806259z |
| 37. | Gieshoff, T.; Schollmeyer, D.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2016, 55, 9437–9440. doi:10.1002/anie.201603899 |
| 38. | Kehl, A.; Breising, V. M.; Schollmeyer, D.; Waldvogel, S. R. Chem. – Eur. J. 2018, 24, 17230–17233. doi:10.1002/chem.201804638 |
| 39. | Lin, M.-Y.; Xu, K.; Jiang, Y.-Y.; Liu, Y.-G.; Sun, B.-G.; Zeng, C.-C. Adv. Synth. Catal. 2018, 360, 1665–1672. doi:10.1002/adsc.201701536 |
| 40. | Zhang, S.; Li, L.; Xue, M.; Zhang, R.; Xu, K.; Zeng, C. Org. Lett. 2018, 20, 3443–3446. doi:10.1021/acs.orglett.8b00981 |
| 41. | Hu, X.; Zhang, G.; Bu, F.; Nie, L.; Lei, A. ACS Catal. 2018, 8, 9370–9375. doi:10.1021/acscatal.8b02847 |
| 31. | Hou, Z.-W.; Mao, Z.-Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. doi:10.1002/anie.201711876 |
| 32. | Hou, Z.-W.; Yan, H.; Song, J.-S.; Xu, H.-C. Chin. J. Chem. 2018, 36, 909–915. doi:10.1002/cjoc.201800301 |
| 33. | Hou, Z.-W.; Mao, Z.-Y.; Song, J.; Xu, H.-C. ACS Catal. 2017, 7, 5810–5813. doi:10.1021/acscatal.7b02105 |
| 34. | Hou, Z.-W.; Mao, Z.-Y.; Zhao, H.-B.; Melcamu, Y. Y.; Lu, X.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2016, 55, 9168–9172. doi:10.1002/anie.201602616 |
| 31. | Hou, Z.-W.; Mao, Z.-Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. doi:10.1002/anie.201711876 |
© 2019 Yan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)