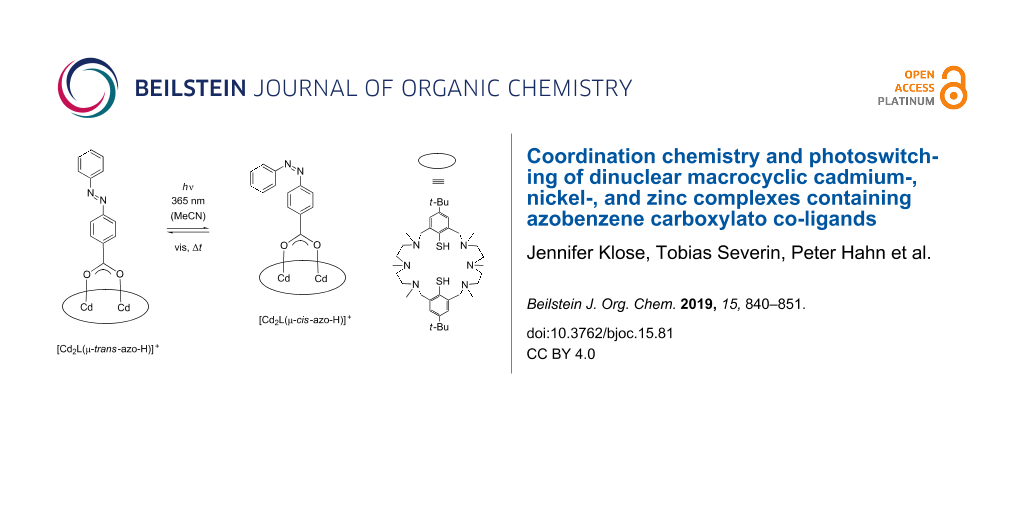
Abstract
The synthesis of mixed-ligand complexes of the type [M2L(μ-L')]+, where L represents a 24-membered macrocyclic hexaaza-dithiophenolate ligand, L' is an azobenzene carboxylate co-ligand, and M = Cd(II), Ni(II) or Zn(II), is reported. A series of new complexes were synthesized, namely [M2L(μ-L')]+ (L' = azo-H, M = Cd (1), Ni (2); L' = azo-OH, M = Zn (3), Ni (4); L' = azo-NMe2, M = Zn (5), Cd (6), Ni (7); L' = azo-CO2Me, M = Cd (8), Ni (9)), and characterized by elemental analysis, electrospray ionization mass spectrometry (ESIMS), IR, UV–vis and NMR spectroscopy (for diamagnetic Zn and Cd complexes) and X-ray single crystal structure analysis. The crystal structures of 3' and 5–8 display an isostructural series of compounds with bridging azobenzene carboxylates in the trans form. The paramagnetic Ni complexes 2, 4 and 7 reveal a weak ferromagnetic exchange interaction with magnetic exchange coupling constant values between 21 and 23 cm−1 (H = −2JS1S2). Irradiation of 1 with λ = 365 nm reveals a photoisomerization of the co-ligand from the trans to the cis form.

Graphical Abstract
Introduction
The macrocyclic N6S2 donor ligand H2L is an effective dinucleating ligand that supports a large number of mixed ligand complexes of the type [M2L(μ-O2CR)]+ [1], where M is a divalent or trivalent metal ion (M = Mn2+, Fe2+, Co2+, Co3+, Ni2+, Zn2+ and Cd2+) and μ-O2CR is a μ1,3-bridging carboxylato ligand (Figure 1). The encapsulation of a functional carboxylate ion by the bowl-shaped [M2L] fragment can drastically alter its structural properties and reactivities. Prominent examples are the cis-bromination of α,β-unsaturated carboxylato ligands [2], regioselective Diels–Alder reactions of encapsulated dienoate ligands [3], and the stabilization of unusual co-ligand conformations, respectively [4]. The physicochemical properties of the bound guest molecules are also greatly affected. Thus, complexation of naphthalene diimide carboxylato ligands leads to a substantial (>95%) quenching of the diimide fluorescence [5], and incorporation of a Fe(CpCO2H)2 unit leads to a significant anodic shift of the metallocene’s redox potential [6].
![[1860-5397-15-81-1]](/bjoc/content/figures/1860-5397-15-81-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Left: Mixed ligand complexes of the type [M2L(μ-L')]n+ supported by the macrocyclic ligand H2L (M = divalent or trivalent transition metal). Right: Perspective view of the bowl-shaped structure of the corresponding carboxylato complexes.
Figure 1: Left: Mixed ligand complexes of the type [M2L(μ-L')]n+ supported by the macrocyclic ligand H2L (M =...
The facile formation of such compounds and their unusual properties led us to synthesize derivatives bearing azobenzene carboxylates as bridging co-ligands. A series of mixed-ligand complexes of the type [M2L(μ-L')]+, where M = Zn(II), Ni(II), or Cd(II), L' = p-azobenzene carboxylate (azo-H), p'-hydroxy-p-azobenzene carboxylate (azo-OH), p'-dimethylamine-p-azobenzene carboxylate (azo-NMe2), and p'-(methoxycarbonyl)azobenzene-p-oxymethylcarboxylic acid (azo-CO2Me) were synthesized (Figure 2). Their preparation and structural characterization by NMR, IR, UV–vis spectroscopy, SQUID magnetometry and X-ray crystallography is reported herein. Preliminary results concerning the photoisomerization of compound 1 are reported as well.
![[1860-5397-15-81-2]](/bjoc/content/figures/1860-5397-15-81-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesized compounds and their labels.
Figure 2: Synthesized compounds and their labels.
Results and Discussion
Synthesis of ligands and metal complexes
Scheme 1 shows the synthetic procedures. The yellow-orange colored zinc complexes (3, 5) were obtained directly from stoichiometric complexation reactions between H2L·6HCl, Zn(OAc)2·2H2O, and the corresponding azobenzene carboxylate ion (prepared in situ from the free acid by deprotonation with NEt3 as a base) in methanol. The green-brown nickel (2, 4, 7 and 9) and red-orange colored cadmium complexes 1, 6 and 8, on the other hand, were prepared by substitution reactions involving the known chlorido-bridged [Ni2L(μ-Cl)]+ or [Cd2L(μ-Cl)]+ precursors and the respective azobenzene carboxylates. All complexes were isolated as ClO4− salts with yields between 76% and 91%. The perchlorate salts are stable in air both in solution and in the solid state, and are very soluble in a range of common polar organic solvents (CH3CN, EtOH, MeOH). The new compounds gave satisfactory elemental analyses and were characterized by mass spectrometry, spectroscopic methods (IR, UV–vis, 1H and 13C NMR spectroscopy) and in case of 3' and 5–8 also by X-ray crystal structure analysis.
![[1860-5397-15-81-i1]](/bjoc/content/inline/1860-5397-15-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Characterization of complexes
IR and NMR spectroscopy
Table 1 lists selected analytical data for the synthesized compounds. The infrared spectra of 1–9 were recorded in the 4000–400 cm−1 spectral range. The IR spectra display in each case two strong bands, one between 1620–1590 cm−1 and the other in the 1419–1395 cm−1 range. These bands can be assigned to the asymmetric (νas(RCO2−)) and symmetric stretching vibrations (νs(RCO2−)) of the carboxylato co-ligands [1]. Coordination is implied by the fact that the νas(RCO2−) frequencies are significantly red-shifted by 78–103 cm−1 to lower wavenumbers. A bathochromic shift of 14–32 cm−1 is also clearly observed for the νs(RCO2–) frequencies. Notice that Δ = νas(RCO2−) − νs(RCO2−) is invariably larger than 180 cm−1, a clear indication that the co-ligand is in the bridging mode [7-9]. Another prominent feature is the intense band around 1100 cm–1, which can be readily assigned to a vibration of the ClO4− ion (ν3F4). An unambiguous assignment of the N=N vibration of the azo group was not possible due to the large number of overlapping bands in the fingerprint region.
Table 1: Selected infrared data for compounds 1–9.a
| complex | νas RCO2− [cm−1] | νs RCO2− [cm−1] | Δν = (νas – νs) [cm−1] |
| 1 [Cd2L(µ-azo-H)]+ | 1599 | 1395 | 204 |
| 2 [Ni2L(µ-azo-H)]+ | 1602 | 1401 | 201 |
| 3 [Zn2L(µ-azo-OH)]+ | 1600 | 1404 | 196 |
| 4 [Ni2L(µ-azo-OH)]+ | 1590 | 1407 | 183 |
| 5 [Zn2L(µ-azo-NMe2)]+ | 1599 | 1398 | 201 |
| 6 [Cd2L(µ-azo-NMe2)]+ | 1598 | 1401 | 197 |
| 7 [Ni2L(µ-azo-NMe2)]+ | 1599 | 1400 | 199 |
| 8 [Cd2L(µ-azo-CO2Me)]+ | 1614 | 1412 | 202 |
| 9 [Ni2L(µ-azo-CO2Me)]+ | 1620 | 1419 | 201 |
aThe IR data refer to the solid ClO4− salts.
To determine their structures in solution the diamagnetic zinc and cadmium complexes were subjected to 1H and 13C NMR spectroscopic studies. Table 2, and Tables S4 and S5 (Supporting Information File 1) list the 1H and 13C NMR spectroscopic data for complexes 1, 3, 5, 6 and 8. The data for [Zn2L(μ-OAc)]+ and [Cd2L(μ-OAc)]+ have been reported previously and are included for comparative purposes [10,11]. Figure 3 displays the 1H NMR spectrum of [Cd2L(μ-azo-NMe2)]+ (6) in CD3CN which is representative for all complexes. Only one set of signals is observed, showing that all compounds exist as single isomers in solution. Thus, the six N-methyl groups give rise to two singlets (one for the methyl protons on the benzylic nitrogen atoms (NBzCH3) and one for the methyl protons on the central amine nitrogen of the linking diethylenetriamine units (NCH3)). Note that the four aromatic protons (ArH) and the tert-butyl protons [C(CH3)3] appear as singlets. The remaining six signals can be assigned to the methylene protons of the linking diethylenetriamine chains (two doublets for the benzylic CH2 and four multiplets for linking CH2 groups). The 1H NMR data are indicative of a C2v symmetric structure of the [Cd2L]2+ fragment in solution, as in [Cd2L(μ-Cl)]+ and other carboxylato-bridged Cd2 complexes supported by this macrocycle [10,12]. The remaining four signals (two doublets and one singlet for aromatic and one singlet for aliphatic CH3 protons) can be attributed to the azobenzene co-ligand. These were readily assigned with the aid of APT, HSQC, HMBC and COSY spectra. The 13C NMR spectrum of complex 6 is also in agreement with C2v symmetry revealing only 11 signals (seven for the aliphatic and four for the aromatic carbon atoms) for the 38 carbon atoms of the [Cd2L]2+ fragment.
![[1860-5397-15-81-3]](/bjoc/content/figures/1860-5397-15-81-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectrum of 6 in CD3CN at 295 K (1.0–8.0 ppm). The resonances and assignments are listed in Table 2 and Table S4 in Supporting Information File 1.
Figure 3: 1H NMR spectrum of 6 in CD3CN at 295 K (1.0–8.0 ppm). The resonances and assignments are listed in Table 2...
Table 2: Comparison of 1H NMR resonances of the azobenzene co-ligands of the zinc (3, 5) and cadmium complexes (1, 6, 8).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-81-i4.svg?max-width=637&scale=1.0)
|
||||
|
[ZnL(OAc)]b [4,10]
[CdL(OAc)]b [4] |
1c | 3d | 5c, 6b | 8c |
|
0.85 (C2H3)
0.98 (C2H3) |
7.48–7.52 (C9H) | 6.86 C8H |
3.08 (C10H3)
3.06 (C10H3) |
3.60 (OC12H2) |
| 7.48–7.52 (C8H) | 7.31 C3H |
6.74 (C8H)
6.81 (C8H) |
3.93 (OC11H3) | |
| 7.48–7.52 (C3H) | 7.46 C4H |
7.34 (C3H)
7.51 (C3H) |
6.44 (C3H) | |
| 7.63 (C4H) | 7.67 C7H |
7.49 (C4H)
7.51 (C4H) |
7.72 (C4H) | |
| 7.86 (C7H) |
7.79 (C7H)
7.77 (C7H) |
7.91 (C8H) | ||
| 8.17 (C7H) | ||||
aNMR data correspond to the ClO4− salts. bSolvent: CD3CN. cSolvent: CD2Cl2. dSolvent: (CD3)2SO.
X-ray crystallography
Further confirmation regarding the composition and structures of the complexes were obtained by single crystal X-ray diffraction studies. Single crystals could be obtained for the tetraphenylborates 3' and 8 and perchlorate salts 5–7. The molecular structures of the complexes are displayed in Figures 4–8. Selected bond lengths are given in Table S6, Supporting Information File 1.
Description of the crystal structures
[Zn2L(μ-azo-OH)][Zn2L(μ-azo-O)]·BPh4·4MeCN·3H2O (3'·4MeCN·3H2O): Suitable crystals of [Zn2L(μ-azo-OH)]ClO4 (3) could not be obtained. However, the addition of NaBPh4 to a solution of 3 in MeCN led to crystallization of [Zn2L(μ-azo-OH)][Zn2L(μ-azo-O)]·BPh4·4MeCN·3H2O (3'·4MeCN·3H2O). The asymmetric unit contains one [Zn2L(μ-azo-OH)]+ cation (Figure 4), one [Zn2L(μ-azo-O)] neutral complex (comprising a doubly deprotonated co-ligand), one BPh4− anion, and MeCN and H2O solvate molecules. Both complexes are structurally very similar and the corresponding bond lengths lie within narrow ranges (Table S6 in Supporting Information File 1). The difference between the two molecules concerns the twisting angle between the carboxylate group and the azobenzene ring. Thus, in the [Zn2L(μ-azo-OH)]+ cation the carboxylato group is twisted out of the azobenzene plane by an angle of 38.0°. In the neutral [Zn2L(μ-azo-O)] complex this angle is only 5.8°. Note that the azobenzene co-ligands are not planar. They are twisted by 8–10° about the C–N=N–C linkages, as in other structures. The Zn–carboxylato bond lengths reveal no anomalies and are similar to those in [Zn2L(μ-OAc)]+. There are no π–π stacking interactions between the azobenzene moieties. However, the [Zn2L(μ-azo-OH)]+ and [Zn2L(μ-azo-O)] complexes are connected by a OH···O hydrogen bond of length 2.46 Å (O3a···O3b, not shown in Figure 4).
![[1860-5397-15-81-4]](/bjoc/content/figures/1860-5397-15-81-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Structure of the [Zn2L(μ-azo-OH)]+ cation in crystals of [Zn2L(μ-azo-OH)][Zn2L(μ-azo-O)]·4MeCN·3H2O (3'·4MeCN·3H2O). Hydrogen atoms omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level (color codes: C dark grey, H pale gray, N green/or blue, O red, S yellow, metals flesh (Zn, Cd) or green (Ni)).
Figure 4: Structure of the [Zn2L(μ-azo-OH)]+ cation in crystals of [Zn2L(μ-azo-OH)][Zn2L(μ-azo-O)]·4MeCN·3H2O...
[Zn2L(μ-azo-NMe2)]ClO4·1.5MeCN (5·1.5MeCN): Crystals of [Zn2L(μ-azo-NMe2)]ClO4·1.5MeCN (5·1.5MeCN) grown by recrystallization from MeCN are triclinic, space group P![[Graphic 2]](/bjoc/content/inline/1860-5397-15-81-i5.svg?max-width=637&scale=1.18182) . The asymmetric unit comprises a [Zn2L(μ-azo-NMe2)]+ cation, a ClO4– anion, and MeCN solvate molecules. The [Zn2L]2+ unit is isostructural with 3'. The co-ligand binds again via its carboxylato function in a μ1,3-bridging mode to give a Zn···Zn distance of 3.440 Å, which is typical for carboxylato-bridged Zn complexes supported by the macrocyclic N6S2 donor ligand. The average Zn–S, Zn–N and Zn–O distances are at 2.536 Å, 2.305 Å, and 2.049 Å, respectively. These values compare well with those in 3' and other [Zn2L(μ-carboxylato)]+ complexes. The [Zn2L(μ-azo-NMe2)]+ complexes in 5 assemble in pairs (Figure 5) most likely via π···π stacking interactions, as manifested by the distance of 3.34 Å between the planes through the azobenzene moieties.
. The asymmetric unit comprises a [Zn2L(μ-azo-NMe2)]+ cation, a ClO4– anion, and MeCN solvate molecules. The [Zn2L]2+ unit is isostructural with 3'. The co-ligand binds again via its carboxylato function in a μ1,3-bridging mode to give a Zn···Zn distance of 3.440 Å, which is typical for carboxylato-bridged Zn complexes supported by the macrocyclic N6S2 donor ligand. The average Zn–S, Zn–N and Zn–O distances are at 2.536 Å, 2.305 Å, and 2.049 Å, respectively. These values compare well with those in 3' and other [Zn2L(μ-carboxylato)]+ complexes. The [Zn2L(μ-azo-NMe2)]+ complexes in 5 assemble in pairs (Figure 5) most likely via π···π stacking interactions, as manifested by the distance of 3.34 Å between the planes through the azobenzene moieties.
![[1860-5397-15-81-5]](/bjoc/content/figures/1860-5397-15-81-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Space filling representation of the packing of two symmetry-related [Zn2L(μ-azo-NMe2)]+ cations in crystals of 5·1.5MeCN. The ClO4− anions and solvate molecules are omitted for clarity.
Figure 5: Space filling representation of the packing of two symmetry-related [Zn2L(μ-azo-NMe2)]+ cations in ...
[Cd2L(μ-Azo-NMe2)]ClO4·0.5MeOH (6·0.5MeOH): Crystals of the title compound were grown from MeOH. The asymmetric unit comprises a [Cd2L(μ-azo-NMe2)]+ cation (with the carboxylate co-ligand disordered over two positions), a ClO4− anion, and MeOH solvate molecules. The [Cd2L]2+ unit is isostructural with that in [Cd2L(μ-OAc)]+ and the compounds above, with the typical bowl-shaped structure of the [Cd2L]2+ fragment and a bridging carboxylate function (Figure 6). The average Cd–S, Cd–N and Cd–O distances at 2.663 Å, 2.428 Å, and 2.258 Å are longer than in 3', as one might expect from the larger ionic radius of Cd2+. The Cd···Cd distance is at 3.399 Å. Virtually the same values are observed in [Cd2L(μ-OAc)]+ [10]. As in 5 π–π stacking of the azo-carboxylato co-ligands occurs (Figure 7). The shortest distance between two carbon atoms of adjacent benzene rings is at 3.41 Å.
![[1860-5397-15-81-6]](/bjoc/content/figures/1860-5397-15-81-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Structure of the [Cd2L(μ-azo-NMe2)]+ cation in crystals of [Cd2L(μ-azo-NMe2)]ClO4·0.5MeOH (6·0.5MeOH). Only one orientation of the disordered azobenzene carboxylato co-ligand is displayed. Hydrogen atoms omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level.
Figure 6: Structure of the [Cd2L(μ-azo-NMe2)]+ cation in crystals of [Cd2L(μ-azo-NMe2)]ClO4·0.5MeOH (6·0.5MeO...
![[1860-5397-15-81-7]](/bjoc/content/figures/1860-5397-15-81-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Space filling representation of the packing of four [Cd2L(μ-azo-NMe2)]+ cations in crystals of 6·0.5MeOH. H atoms, ClO4− anions and solvate molecules are omitted for clarity. The C atoms of the azobenzene moieties are in pale blue.
Figure 7: Space filling representation of the packing of four [Cd2L(μ-azo-NMe2)]+ cations in crystals of 6·0....
[Ni2L(μ-Azo-NMe2)]ClO4·xEtOH (7·ClO4·xEtOH; x ≈ 4): Crystals of [Ni2L(μ-azo-NMe2)]ClO4.xEtOH obtained from a mixed ethanol/acetonitrile solvent system are triclinic, space group P![[Graphic 3]](/bjoc/content/inline/1860-5397-15-81-i6.svg?max-width=637&scale=1.18182) . The asymmetric unit comprises two crystallographically independent [Ni2L(μ-azo-NMe2)]+ cations, two ClO4– ions and 8 EtOH solvate molecules. The latter were found to be highly disordered and were therefore removed by the SQUEEZE procedure implemented in PLATON. Removing the EtOH molecules led to a total solvent accessible void of 1500 Å3, in good agreement with the space needed by ca. eight ethanol solvate molecules. The two [Ni2L(μ-azo-NMe2)]+ cations are structurally very similar as illustrated in Figure 8. In cation A, the co-ligand is nearly flat and coplanar with the Ni2 carboxylato plane. In cation B, the benzene rings are tilted about the azo group and the Ni2carboxlyato plane by angles of ≈32.3° and 37.5°, respectively. Interestingly, no π–π stacking occurs in this structure.
. The asymmetric unit comprises two crystallographically independent [Ni2L(μ-azo-NMe2)]+ cations, two ClO4– ions and 8 EtOH solvate molecules. The latter were found to be highly disordered and were therefore removed by the SQUEEZE procedure implemented in PLATON. Removing the EtOH molecules led to a total solvent accessible void of 1500 Å3, in good agreement with the space needed by ca. eight ethanol solvate molecules. The two [Ni2L(μ-azo-NMe2)]+ cations are structurally very similar as illustrated in Figure 8. In cation A, the co-ligand is nearly flat and coplanar with the Ni2 carboxylato plane. In cation B, the benzene rings are tilted about the azo group and the Ni2carboxlyato plane by angles of ≈32.3° and 37.5°, respectively. Interestingly, no π–π stacking occurs in this structure.
![[1860-5397-15-81-8]](/bjoc/content/figures/1860-5397-15-81-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Structures of the two crystallographically independent [Ni2L(μ-azo-NMe2)]+ cations A (left) and B (right) in crystals of [Ni2L(μ-azo-NMe2)]ClO4·8EtOH (7·8EtOH). Only one orientation of the disordered azo-carboxylato co-ligand is displayed. Hydrogen atoms omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level.
Figure 8: Structures of the two crystallographically independent [Ni2L(μ-azo-NMe2)]+ cations A (left) and B (...
[Cd2L(μ-azo-CO2Me)]BPh4.MeCN (8·MeCN): The crystal structure determination of 8·MeCN confirms that azo-CO2Me is also attached in the bridging mode. Figure 9 shows an ORTEP representation of the structure of the [Cd2L(μ-azo-CO2Me)]+ cation in 8. The [Cd2L]2+ fragment is isostructural with that in 6, but the co-ligand protrudes laterally out of the binding pocket of the [Cd2L]2+ fragment, most likely due to steric constraints exerted by the surrounding NMe2 groups. We have observed similar effects in a stearato bridged complex, where the surrounding alkyl groups dictate the coordination mode of the carboxylato ligand [13]. The average Cd–S, Cd–N and Cd–O distances at 2.670, 2.429, and 2.258 Å, respectively, are all very similar to those in 6. The Cd···Cd distance is at 3.405 Å. Again, π–π stacking of the azo-carboxylato co-ligand occurs, the shortest distance between the two planes through the co-ligands amounts to 3.45 Å.
![[1860-5397-15-81-9]](/bjoc/content/figures/1860-5397-15-81-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Left: ORTEP representation of the molecular structure of the [Cd2L(μ-azo-CO2Me)]+ cation in crystals of 8·MeCN. Right: Space filling representation of the packing of two [Cd2L(μ-azo-CO2Me)]+ cations in crystals of 8·MeCN. H atoms, BPh4− anions and solvate molecules are omitted for clarity.
Figure 9: Left: ORTEP representation of the molecular structure of the [Cd2L(μ-azo-CO2Me)]+ cation in crystal...
Magnetic properties of nickel complexes
The paramagnetic nickel complexes 2, 4, and 7 were further investigated by temperature dependent magnetic susceptibility measurements between 2 and 300 K in an applied external field of 0.5 T (or 1 T) by using a MPMS 7XL SQUID magnetometer (Quantum Design). Figure 10 shows the susceptibility data in the form of μeff versus T plots for the three complexes.
![[1860-5397-15-81-10]](/bjoc/content/figures/1860-5397-15-81-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Plots of the effective magnetic moment μeff for 2 (open circles), 4 (open squares), and 7 (open triangles) at H = 0.5 T (2, 7) and 1.0 T (4). The solid lines represent the best theoretical fit to Equation 1 (see text). Experimental and calculated values are provided as Supporting Information File 1.
Figure 10: Plots of the effective magnetic moment μeff for 2 (open circles), 4 (open squares), and 7 (open tri...
For complex 2, the effective magnetic moment increases from 4.86 μB (per dinuclear complex) at 300 K to a maximum value of 5.42 μB at 20 K, and then drops to 4.93 μB at 2 K. The complexes 4 and 7 behave in a similar fashion. At 300 K, μeff is at ≈4.87 per dinuclear complex which increases to maximum values of 5.34 μB (4) and 5.44 μB (7) at 20 K. On further lowering of the temperature, these values decrease again to 4.54 μB and 4.90 μB at 2 K. This behavior implies that the electron spins on the two NiII (S = 1) ions are coupled by an intramolecular ferromagnetic exchange interaction. This leads to an Stotal = 2 ground state, in agreement with other carboxylato-bridged compounds supported by L2− [14-16]. The ferromagnetic exchange interaction can be rationalized in terms of the Goodenough–Kanamori rules for superexchange [17], and recent DFT calculations, which revealed that a dominant ferromagnetic exchange interaction is propagated via the thiolato bridges [13].
The magnitude of the exchange interactions was determined by least-squares fitting of the experimental magnetic susceptibility data to the appropriate spin Hamiltonian (Equation 1) [18-20] including the isotropic HDvV (Heisenberg Dirac van Vleck) exchange interaction, the single-ion zero-field splitting of Ni(II) and the single-ion Zeeman interactions using a full-matrix diagonalization approach [21]. The experimental susceptibility data were fitted to Equation 1 over the temperature range 2–300 K, assuming identical D and g values for the two Ni(II) ions in each one of the complexes, but it should be mentioned that temperature-dependent magnetic susceptibility measurements do not allow a concise determination of the magnitude and sign of D [22].
![[1860-5397-15-81-i3]](/bjoc/content/inline/1860-5397-15-81-i3.svg?max-width=590&scale=1.18182)
By taking into account zero-field splitting and temperature-independent paramagnetism (TIP), reasonable fits of the experimental data were possible, yielding J = +22.6 cm−1, g = 2.21 and D = |4.18| cm−1 for 2. Fitting the experimental data of 4 and 7 led to very similar values, namely J = +21.79 cm–1, g = 2.17, D = |3.64| cm–1 and J = +21.70 cm–1, g = 2.22 and D = |4.67| cm–1, respectively. In each case the low-temperature fit was significantly improved by the inclusion of the D parameter, but as stated above, the D values should be taken rather indicative than definite [23]. The magnetic parameters for the azo-carboxylate complexes are in good agreement with those of nickel(II)-carboxylate complexes supported by L2−. For [Ni2L(μ-O2C(CH2)10SH)](ClO4), for example, values of J = +23.0 cm−1, g = 2.20 and D = |2.59| cm−1 were determined [13].
UV–vis spectroscopy
All new compounds were further investigated by electronic absorption spectroscopy. The electronic absorption spectra were recorded in acetonitrile (1, 2 and 5–9) or dimethyl sulfoxide (3, 4) solution in the 190 to 1200 nm spectral range. Figure 11 shows a representative set of spectra recorded for Hazo-H and the corresponding Cd(II) complex [Cd2(L)(μ-azo-H)]+ (1). The spectrum of the chlorido-bridged complex [Cd2L(μ-Cl)]+ is also shown for comparison. Table 3 lists the corresponding data. A comparison reveals that the electronic absorption spectrum of [Cd2L(μ-azo-H)]+ can (at least in the 260–450 nm range) in first order be traced to a superposition of the absorptions of the Cd-bound amino-thiophenolato ligand (absorbing at 260 and 300 nm) and the azobenzene co-ligand (absorbing at 325 and 441 nm). Note, that the band at 322 nm for free Hazo-H appears as a shoulder around 325 nm in 1. The slight red-shift may be a consequence of deprotonation and coordination to the Cd2+ ions. The spectrum of the deprotonated azoH– ion, for comparison, absorbs at 329 nm. The absorption bands above 310 nm can thus be attributed to the π–π* and n–π* transitions of the trans-configured azobenzene chromophore, consistent with literature reports [24,25]. The absorptions below 310 nm (λ = 260 and 300 nm) are attributable to the π–π* transitions within the thiophenolate units of the [Cd2L]2+ fragment, which may be further differentiated as p- and α-bands of the parent benzene chromophore utilizing Clar’s notation [26], assuming that the electron-donating alkyl and thiol substituents exert a bathochromic effect.
![[1860-5397-15-81-11]](/bjoc/content/figures/1860-5397-15-81-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: UV–vis spectra of Hazo-H (red line), [Cd2L(μ-Cl)](ClO4) (black line) and [Cd2L(μ-azo-H)]ClO4 (1, blue line) in acetonitrile. Concentration of solutions: 10−4 M.
Figure 11: UV–vis spectra of Hazo-H (red line), [Cd2L(μ-Cl)](ClO4) (black line) and [Cd2L(μ-azo-H)]ClO4 (1, bl...
Table 3: Selected UV–vis data for compounds 1–9.a
| Complex/co-ligand | λmax [nm] (ε [M–1 cm–1])a |
| [Cd2L(µ-Cl)]+,b | 194 (63477), 262 (8926), 304 (24784) |
| [Ni2L(µ-Cl)]+,b | 194 (51524), 265 (13994), 304 (12283), 645 (48), 920 (65), 999 (85) |
| [Zn2L(µ-OAc)]+,b | 192 (55860), 208 sh (41434), 261 (13740), 288 (24464) |
| Hazo-Hb | 190 (45246), 228 (10477), 322 (18707), 441 (757) |
| Hazo-OHc | 194 (432), 243 (209), 352 (371) |
| Hazo-NMe2b | 193 (44362), 253 sh (13356), 280 (12545), 311 (11315), 436 (15104) |
| Hazo-CO2Meb | 190 (40583), 232 (9820), 255 (9915), 352 (16597), 439 (1380) |
| 1 [Cd2L(µ-azo-H)]+,b | 192 (104317), 260 (19844), 300 (42752), 325 sh (23011), 441 (882) |
| 2 [Ni2L(µ-azo-H)]+,b | 195 (88412), 276 sh (24155), 312 (33230), 328 (34994), 439 (1180), 647 (39), 1117 (66) |
| 3 [Zn2L(µ-azo-OH)]+,c | 259 (24979), 292 (24378), 366 (28535), 450 (1970) |
| 4 [Ni2L(µ-azo-OH)]+,c | 258 (27612), 311 (20738), 341 (29144), 364 (31733), 454 (2099), 645 (115), 1113 (76) |
| 5 [Zn2L(µ-azo-NMe2)]+,b | 195 (83650), 261 (27080), 285 (31050), 382 (11790), 434 (16000) |
| 6 [Cd2L(µ-azo-NMe2)]+,b | 193 (129340), 261 (30525), 298 (38705), 381 (12432), 434 (16292) |
| 7 [Ni2L(µ-azo-NMe2)]+,b | 194 (114417), 267 (37868), 301 sh (24544), 328 sh (20921), 380 (16567), 431 (19801), 650 (221), 1112 (186) |
| 8 [Cd2L(µ-azo-CO2Me)]+,b | 193 (113128), 261 (29102), 300 (33143), 370 (6018), 447 sh (2949) |
| 9 [Ni2L(µ-azo-CO2Me)]+,b | 194 (113002), 261 (35911), 308 (28168), 334 (32443), 360 sh (29137), 448 (3186), 645 sh (470), 1113 (153) |
aThe complexes were isolated as ClO4− salts. bSolvent: CH3CN or cDMSO. Concentration of solutions: 10−4 M.
The spectral properties of the nickel complexes differ from those of the zinc and cadmium counterparts in that they exhibit two additional Laporte-forbidden but spin-allowed d–d transitions around 645 and 1120 nm, typical for carboxylato-bridged Ni2 complexes supported by H2L (Figure 12) [5]. They are assigned as 3A2g(F)→ T1g(F) (ν2) and 3A2g(F) → 3T1g(P) (ν1) of an octahedral nickel(II) (S = 1) ion (in pure Oh symmetry) [5]. The third spin-allowed 3A2g(F) → 3T2g(F) (ν3) transition is expected around 440 nm, but is obscured by the stronger RS → Ni2+ charge transfer and π→π* transitions in this region.
![[1860-5397-15-81-12]](/bjoc/content/figures/1860-5397-15-81-12.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: UV–vis spectra of Hazo-NMe2 (red line), [Ni2L(μ-Cl)](ClO4) (black line) and [Ni2L(μ-azo-NMe2)]ClO4 (7, blue line) in acetonitrile at 230–700 nm and 550–1300 nm (inset). Concentration of solutions: 10−4 M.
Figure 12: UV–vis spectra of Hazo-NMe2 (red line), [Ni2L(μ-Cl)](ClO4) (black line) and [Ni2L(μ-azo-NMe2)]ClO4 (...
We carried out orienting irradiation experiments with regard to a possible trans to cis photoisomerization of the bound azobenzene-carboxylate co-ligands. The diamagnetic Cd2 complex [Cd2L(μ-azoH)]ClO4 (1) was selected for these studies. Note that this complex (with the azobenzene moiety in the trans form) is distinguished by a strong absorption band (shoulder) around 325 nm (ε = 23000 M–1cm–1) that originates from the π→π* transition in trans azo-H. The much weaker n→π* band appears in the visible at 450 nm. Dilute anhydrous acetonitrile solutions of complex 1 were placed in a standard quartz cuvette and irradiated at 365 nm for varying amounts of time (1 to 30 s) with a 14 W/cm2 UV LED lamp and thereafter immediately placed in the chamber of a UV–vis spectrometer. Figure 13 shows the corresponding UV–vis spectra, which were recorded directly after sample preparation.
![[1860-5397-15-81-13]](/bjoc/content/figures/1860-5397-15-81-13.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: UV–vis spectra of solutions of [Cd2L(μ-azo-H)]ClO4 (1) in acetonitrile irradiated with a UV LED lamp (365 nm) for 1 to 30 s after irradiation. Concentration of solutions: 10−5 M.
Figure 13: UV–vis spectra of solutions of [Cd2L(μ-azo-H)]ClO4 (1) in acetonitrile irradiated with a UV LED lam...
As can be seen, the intensity of the π→π* band at 325 nm for the trans-azobenzene carboxylate decreases with increasing irradiation time, while the intensity of the n→π* band increases with increasing irradiation time. A blue shift of the n–π* band by ≈10 nm is evident. The π→π* transitions for the thiophenolate group at 260 and 300 nm are also affected, being blue-shifted to 256 and 296 nm, respectively, and significantly reduced (≈260 nm) or increased (≈300 nm) in intensity. Isosbestic points at 220, 240, 280 and 380 nm are also clearly discernible indicative of the presence of a single equilibrium. On the basis of these data, we conclude that the trans-form of the bound co-ligand converts to its cis-form as indicated in Scheme 2. Further studies show, that the meta-stable cis-form relaxes back thermally to the more stable trans from (Figures S50 and S51 in Supporting Information File 1). Thus, the cis–trans isomerization appears to be not constrained by the [Cd2L]2+ fragment, as revealed by the similar half-lives (τ1/2 cis–trans free deprotonated azobenzene carboxylate: ≈80 h; τ1/2 cis–trans Cd complex ≈73 h. In view of the fact that the azobenzene carboxylate is only partially buried in the binding pocket of the [Cd2L]2+ complex this is not surprising.
![[1860-5397-15-81-i2]](/bjoc/content/inline/1860-5397-15-81-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cis/trans isomerization process of the bound azo-carboxylato co-ligand in [Cd2L(μ-azo-H)]ClO4 (1) in acetonitrile mediated by irradiation with 365 nm UV light. The amino-thiophenolato ligand is shown as an ellipse for clarity.
Scheme 2: Cis/trans isomerization process of the bound azo-carboxylato co-ligand in [Cd2L(μ-azo-H)]ClO4 (1) i...
Conclusion
A series of dinuclear macrocyclic [M2L(μ-L')]+ complexes co-ligated by various azobenzene-carboxylates has been synthesized and characterized in solution and solid state. The diamagnetic Cd(II) and Zn(II) and the paramagnetic Ni(II) complexes were found to be isostructural, with the co-ligands bound via bridging carboxylate functions, a bowl-shaped structure of the supporting [M2L]2+ entity, and trans-configured azo groups. NMR and UV–vis studies clearly show that the complexes retain their mixed-ligand nature in the solution state. Irradiation of the Cd complex 1 with a LED UV lamp leads to a trans-to-cis isomerization which appears to proceed within the coordination sphere of the [Cd2L]2+ fragment. The switch from the trans to the cis form induces significant change of the π–π transitions of the supporting N6S2 macrocycle, which may be indicative of some increased π–π- (or charge transfer) interactions between the aromatic rings of the electron rich amino thiophenolato macrocycle and the electron poor azobenzene-carboxylato ligands. There are little, if any, differences in the time-scales for thermal relaxation of the free and Cd-bound azobenzene-carboxylate systems, suggesting that the cavity of [Cd2L]2+ does not sterically constrain the photoisomerization process.
Experimental
Unless otherwise noted all preparations were carried out under a protective atmosphere of nitrogen by using standard Schlenk techniques [27]. Compounds H2L [28], [Ni2L(μ-Cl)]ClO4 [29], [Cd2L(μ-Cl)]ClO4, and p-hydroxyazobenzene-4-carboxylic acid [30] and p'-carbomethoxy-azobenzene-p-oxymethylcarboxylic acid [31-33] were prepared as described in the literature. All other reagents were purchased from commercial vendors and used without further purification. Melting points were determined in open glass capillaries and are uncorrected. The IR spectra were recorded as KBr disks using a Bruker Tensor 27 FTIR spectrometer. UV–vis spectra were recorded on a Jasco V-670 UV–vis/near-IR spectrophotometer. Elemental analyses were carried out with a Vario EL – elemental analyzer. NMR spectra were recorded on a Bruker Fourier 300 spectrometer or Avance DRX 400 at 298 K. 1H and 13C chemical shifts refer to solvent signals. ESIMS spectra were recorded on a Bruker Daltronics Esquire 3000 Plus spectrometer. Temperature-dependent magnetic susceptibility measurements on powdered solid samples were carried out using a MPMS 7XL Squid magnetometer (Quantum Design) over a temperature range of 2–300 K at an applied magnetic field of 0.5 and 1.0 Tesla. The observed susceptibility data were corrected for underlying diamagnetism. The synthesis of the complexes are shown in Supporting Information File 1.
X-ray crystallography. Single crystals of 3', 5–8 suitable for X-ray crystallography were selected and mounted on the tip of a glass fiber. The data sets were collected at 213(2) K or 200 K (7) using a STOE IPDS-2T diffractometer equipped with graphite monochromated Mo Kα radiation (0.71073 Å). Table S7 in Supporting Information File 1 lists selected crystallographic data. The intensity data were processed with the program STOE X-AREA [34] Structures were solved by direct methods [35] and refined by full-matrix least-squares on the basis of all data against F2 using SHELXL-97 [36]. PLATON was used to search for higher symmetry [37]. H atoms were placed in calculated positions and treated isotropically using the 1.2-fold Uiso value of the parent atom except methyl protons, which were assigned the 1.5-fold Uiso value of the parent C atoms. Unless otherwise noted, all non-hydrogen atoms were refined anisotropically. ORTEP-3 was used for the artwork of the structures [38]. All non-hydrogen atoms were refined anisotropically.
In the crystal structure of 3'·4MeCN·3H2O one MeCN molecule was found to be disordered over two positions (at half occupancy). It was not possible to locate H atoms for the H2O solvate molecules, and so no H2O hydrogen atoms were included in this structure. In the crystal structures of 5 and 6 the co-ligands were found to be disordered over two positions. This disorder could be successfully modelled by using appropriate DFIX, DANG and AFIX instructions implemented in SHELXL. The site occupancy factors of the two orientations were initially refined, but were fixed at 0.65 and 0.35 (or 0.50 and 0.50 for 3') in the final refinement cycle. The occupancy factor of a MeCN (for 5) and a MeOH solvate molecule (for 6) was set at 0.5 to keep the Ueq values reasonable. For 7, additional solvent molecules occupy interstitial spaces that are generated by packing, and were found to be highly disordered. The routine SQUEEZE was applied to remove diffuse electron density [39].
CCDC-1892644 (3'), -1892646 (5) -1892647 (6), -1892648 (8) and -1892649 (7) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Irradiation experiments. Irradiation experiments were performed on dry acetonitrile solutions of complex 1 and azo-H (concentration = 10−5 M) in a quartz cuvette at room temperature. An OmniCure LX500 system and a 14 W/cm2 UV LED lamp (365 nm) from Excelitas Technologies was used. After irradiation, the solutions were immediately placed in the chamber of the JASCO V-670 UV–vis/near-IR spectrophotometer for subsequent spectroscopic investigations.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 3.7 MB | Download |
Acknowledgements
We are thankful to Prof. Dr. H. Krautscheid for providing facilities for X-ray crystallographic measurements. Financial support from the University of Leipzig and the German Science Foundation (DFG) through the SPP 2102 programm (“Light controlled reactivity of metal complexes”) is gratefully acknowledged.
References
-
Lehmann, U.; Klingele, J.; Lozan, V.; Steinfeld, G.; Klingele, M. H.; Käss, S.; Rodenstein, A.; Kersting, B. Inorg. Chem. 2010, 49, 11018–11029. doi:10.1021/ic101574a
Return to citation in text: [1] [2] -
Steinfeld, G.; Lozan, V.; Kersting, B. Angew. Chem. 2003, 115, 2363–2365. doi:10.1002/ange.200351131
Angew. Chem., Int. Ed. 2003, 42, 2261–2263. doi:10.1002/anie.200351131
Return to citation in text: [1] -
Käss, S.; Gregor, T.; Kersting, B. Angew. Chem. 2006, 118, 107–110. doi:10.1002/ange.200500683
Angew. Chem., Int. Ed. 2006, 45 , 101–104. doi:10.1002/anie.200500683
Return to citation in text: [1] -
Käss, S.; Kersting, B. Eur. J. Inorg. Chem. 2012, 2389–2401. doi:10.1002/ejic.201200036
Return to citation in text: [1] [2] [3] -
Lee, K. A.; Lozan, V.; Langford, S. J.; Kersting, B. Dalton Trans. 2009, 7481–7485. doi:10.1039/b907785e
Return to citation in text: [1] [2] [3] -
Lozan, V.; Buchholz, A.; Plass, W.; Kersting, B. Chem. – Eur. J. 2007, 13, 7305–7316. doi:10.1002/chem.200700107
Return to citation in text: [1] -
Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 1997.
Return to citation in text: [1] -
Deacon, G. B.; Philipp, R. J. Coord. Chem. Rev. 1980, 33, 227–250. doi:10.1016/s0010-8545(00)80455-5
Return to citation in text: [1] -
Martini, D.; Pellei, M.; Pettinari, C.; Skelton, B. W.; White, A. H. Inorg. Chim. Acta 2002, 333, 72–82. doi:10.1016/s0020-1693(02)00772-7
Return to citation in text: [1] -
Lozan, V.; Kersting, B. Eur. J. Inorg. Chem. 2005, 504–512. doi:10.1002/ejic.200400603
Return to citation in text: [1] [2] [3] [4] -
Lach, J.; Hahn, T.; Kersting, B.; Kortus, J. Eur. J. Inorg. Chem. 2012, 2381–2388. doi:10.1002/ejic.201101402
Return to citation in text: [1] -
Gressenbuch, M.; Kersting, B. Eur. J. Inorg. Chem. 2007, 90–102. doi:10.1002/ejic.200600318
Return to citation in text: [1] -
Börner, M.; Blömer, L.; Kischel, M.; Richter, P.; Salvan, G.; Zahn, D. R. T.; Siles, P. F.; Fuentes, M. E. N.; Bufon, C. C. B.; Grimm, D.; Schmidt, O. G.; Breite, D.; Abel, B.; Kersting, B. Beilstein J. Nanotechnol. 2017, 8, 1375–1387. doi:10.3762/bjnano.8.139
Return to citation in text: [1] [2] [3] -
Lozan, V.; Loose, C.; Kortus, J.; Kersting, B. Coord. Chem. Rev. 2009, 253, 2244–2260. doi:10.1016/j.ccr.2008.08.016
Return to citation in text: [1] -
Journaux, Y.; Glaser, T.; Steinfeld, G.; Lozan, V.; Kersting, B. Dalton Trans. 2006, 1738–1748. doi:10.1039/b513717a
Return to citation in text: [1] -
Lach, J.; Jeremies, A.; Breite, D.; Abel, B.; Mahns, B.; Knupfer, M.; Matulis, V.; Ivashkevich, O. A.; Kersting, B. Inorg. Chem. 2014, 53, 10825–10834. doi:10.1021/ic500683f
Return to citation in text: [1] -
Ginsberg, A. P.; Martin, R. L.; Brookes, R. W.; Sherwood, R. C. Inorg. Chem. 1972, 11, 2884–2889. doi:10.1021/ic50118a006
Return to citation in text: [1] -
Kahn, O. Molecular Magnetism; VCH Verlagsgesellschaft: Weinheim, New York, 1993.
Return to citation in text: [1] -
O’Connor, C. J. Magnetochemistry-Advances in Theory and Experimentation; Progress in Inorganic Chemistry; John Wiley & Sons: New Jersey, 2007.
Return to citation in text: [1] -
Janiak, C.; Klapötke, T.; Meyer, H.-J.; Alsfasser, R. In Moderne Anorganische Chemie; Riedel, E., Ed.; de Gruyter: Berlin, 2007.
Return to citation in text: [1] -
Azuah, R. T.; Kneller, L. R.; Qiu, Y.; Tregenna-Piggott, P. L. W.; Brown, C. M.; Copley, J. R. D.; Dimeo, R. M. J. Res. Natl. Inst. Stand. Technol. 2009, 114, 341–358. doi:10.6028/jres.114.025
Return to citation in text: [1] -
Herchel, R.; Boča, R.; Krzystek, J.; Ozarowski, A.; Durán, M.; van Slageren, J. J. Am. Chem. Soc. 2007, 129, 10306–10307. doi:10.1021/ja0725807
Return to citation in text: [1] -
Nanda, K. K.; Addison, A. W.; Paterson, N.; Sinn, E.; Thompson, L. K.; Sakaguchi, U. Inorg. Chem. 1998, 37, 1028–1036. doi:10.1021/ic9712434
Return to citation in text: [1] -
Casimiro, L.; Groppi, J.; Baroncini, M.; La Rosa, M.; Credi, A.; Silvi, S. Photochem. Photobiol. Sci. 2018, 17, 734–740. doi:10.1039/c8pp00062j
Return to citation in text: [1] -
Klajn, R. Pure Appl. Chem. 2010, 82, 2247–2279. doi:10.1351/pac-con-10-09-04
Return to citation in text: [1] -
Hesse, M.; Meier, H.; Zeeh, B., Eds. Spektroskopische Methoden in der organischen Chemie; Georg Thieme Verlag: Stuttgart, Germany, 2005. doi:10.1055/b-002-46985
Return to citation in text: [1] -
Herzog, S.; Dehnert, J. Z. Chem. 1964, 4, 1–11. doi:10.1002/zfch.19640040102
Return to citation in text: [1] -
Kersting, B.; Steinfeld, G. Chem. Commun. 2001, 1376–1377. doi:10.1039/b103050g
Return to citation in text: [1] -
Gressenbuch, M.; Lozan, V.; Steinfeld, G.; Kersting, B. Eur. J. Inorg. Chem. 2005, 2223–2234. doi:10.1002/ejic.200500022
Return to citation in text: [1] -
Fujita, K.; Fujiwara, S.; Yamada, T.; Tsuchido, Y.; Hashimoto, T.; Hayashita, T. J. Org. Chem. 2017, 82, 976–981. doi:10.1021/acs.joc.6b02513
Return to citation in text: [1] -
Pospíšil, J.; Potáček, M. Tetrahedron 2007, 63, 337–346. doi:10.1016/j.tet.2006.10.074
Return to citation in text: [1] -
Chen, S.; Ling, A.; Zhang, H.-L. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2759–2768. doi:10.1002/pola.26683
Return to citation in text: [1] -
Sahu, S.; Sinha, N.; Bhutia, S. K.; Majhi, M.; Mohapatra, S. J. Mater. Chem. B 2014, 2, 3799–3808. doi:10.1039/c3tb21669a
Return to citation in text: [1] -
Stoe, Cie, IPDS and X-RED32 2008.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 1990, 46, 467–473. doi:10.1107/s0108767390000277
Return to citation in text: [1] -
SHELXL-97, Computer program for crystal structure refinement; University of Göttingen: Göttingen, Germany, 1997.
Return to citation in text: [1] -
PLATON − A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2000.
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/s0021889897003117
Return to citation in text: [1] -
Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x
Return to citation in text: [1]
| 28. | Kersting, B.; Steinfeld, G. Chem. Commun. 2001, 1376–1377. doi:10.1039/b103050g |
| 29. | Gressenbuch, M.; Lozan, V.; Steinfeld, G.; Kersting, B. Eur. J. Inorg. Chem. 2005, 2223–2234. doi:10.1002/ejic.200500022 |
| 30. | Fujita, K.; Fujiwara, S.; Yamada, T.; Tsuchido, Y.; Hashimoto, T.; Hayashita, T. J. Org. Chem. 2017, 82, 976–981. doi:10.1021/acs.joc.6b02513 |
| 1. | Lehmann, U.; Klingele, J.; Lozan, V.; Steinfeld, G.; Klingele, M. H.; Käss, S.; Rodenstein, A.; Kersting, B. Inorg. Chem. 2010, 49, 11018–11029. doi:10.1021/ic101574a |
| 5. | Lee, K. A.; Lozan, V.; Langford, S. J.; Kersting, B. Dalton Trans. 2009, 7481–7485. doi:10.1039/b907785e |
| 14. | Lozan, V.; Loose, C.; Kortus, J.; Kersting, B. Coord. Chem. Rev. 2009, 253, 2244–2260. doi:10.1016/j.ccr.2008.08.016 |
| 15. | Journaux, Y.; Glaser, T.; Steinfeld, G.; Lozan, V.; Kersting, B. Dalton Trans. 2006, 1738–1748. doi:10.1039/b513717a |
| 16. | Lach, J.; Jeremies, A.; Breite, D.; Abel, B.; Mahns, B.; Knupfer, M.; Matulis, V.; Ivashkevich, O. A.; Kersting, B. Inorg. Chem. 2014, 53, 10825–10834. doi:10.1021/ic500683f |
| 39. | Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x |
| 4. | Käss, S.; Kersting, B. Eur. J. Inorg. Chem. 2012, 2389–2401. doi:10.1002/ejic.201200036 |
| 17. | Ginsberg, A. P.; Martin, R. L.; Brookes, R. W.; Sherwood, R. C. Inorg. Chem. 1972, 11, 2884–2889. doi:10.1021/ic50118a006 |
| 3. |
Käss, S.; Gregor, T.; Kersting, B. Angew. Chem. 2006, 118, 107–110. doi:10.1002/ange.200500683
Angew. Chem., Int. Ed. 2006, 45 , 101–104. doi:10.1002/anie.200500683 |
| 10. | Lozan, V.; Kersting, B. Eur. J. Inorg. Chem. 2005, 504–512. doi:10.1002/ejic.200400603 |
| 37. | PLATON − A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2000. |
| 2. |
Steinfeld, G.; Lozan, V.; Kersting, B. Angew. Chem. 2003, 115, 2363–2365. doi:10.1002/ange.200351131
Angew. Chem., Int. Ed. 2003, 42, 2261–2263. doi:10.1002/anie.200351131 |
| 13. | Börner, M.; Blömer, L.; Kischel, M.; Richter, P.; Salvan, G.; Zahn, D. R. T.; Siles, P. F.; Fuentes, M. E. N.; Bufon, C. C. B.; Grimm, D.; Schmidt, O. G.; Breite, D.; Abel, B.; Kersting, B. Beilstein J. Nanotechnol. 2017, 8, 1375–1387. doi:10.3762/bjnano.8.139 |
| 38. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/s0021889897003117 |
| 10. | Lozan, V.; Kersting, B. Eur. J. Inorg. Chem. 2005, 504–512. doi:10.1002/ejic.200400603 |
| 11. | Lach, J.; Hahn, T.; Kersting, B.; Kortus, J. Eur. J. Inorg. Chem. 2012, 2381–2388. doi:10.1002/ejic.201101402 |
| 4. | Käss, S.; Kersting, B. Eur. J. Inorg. Chem. 2012, 2389–2401. doi:10.1002/ejic.201200036 |
| 10. | Lozan, V.; Kersting, B. Eur. J. Inorg. Chem. 2005, 504–512. doi:10.1002/ejic.200400603 |
| 35. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 1990, 46, 467–473. doi:10.1107/s0108767390000277 |
| 7. | Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 1997. |
| 8. | Deacon, G. B.; Philipp, R. J. Coord. Chem. Rev. 1980, 33, 227–250. doi:10.1016/s0010-8545(00)80455-5 |
| 9. | Martini, D.; Pellei, M.; Pettinari, C.; Skelton, B. W.; White, A. H. Inorg. Chim. Acta 2002, 333, 72–82. doi:10.1016/s0020-1693(02)00772-7 |
| 4. | Käss, S.; Kersting, B. Eur. J. Inorg. Chem. 2012, 2389–2401. doi:10.1002/ejic.201200036 |
| 36. | SHELXL-97, Computer program for crystal structure refinement; University of Göttingen: Göttingen, Germany, 1997. |
| 1. | Lehmann, U.; Klingele, J.; Lozan, V.; Steinfeld, G.; Klingele, M. H.; Käss, S.; Rodenstein, A.; Kersting, B. Inorg. Chem. 2010, 49, 11018–11029. doi:10.1021/ic101574a |
| 31. | Pospíšil, J.; Potáček, M. Tetrahedron 2007, 63, 337–346. doi:10.1016/j.tet.2006.10.074 |
| 32. | Chen, S.; Ling, A.; Zhang, H.-L. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2759–2768. doi:10.1002/pola.26683 |
| 33. | Sahu, S.; Sinha, N.; Bhutia, S. K.; Majhi, M.; Mohapatra, S. J. Mater. Chem. B 2014, 2, 3799–3808. doi:10.1039/c3tb21669a |
| 6. | Lozan, V.; Buchholz, A.; Plass, W.; Kersting, B. Chem. – Eur. J. 2007, 13, 7305–7316. doi:10.1002/chem.200700107 |
| 10. | Lozan, V.; Kersting, B. Eur. J. Inorg. Chem. 2005, 504–512. doi:10.1002/ejic.200400603 |
| 12. | Gressenbuch, M.; Kersting, B. Eur. J. Inorg. Chem. 2007, 90–102. doi:10.1002/ejic.200600318 |
| 21. | Azuah, R. T.; Kneller, L. R.; Qiu, Y.; Tregenna-Piggott, P. L. W.; Brown, C. M.; Copley, J. R. D.; Dimeo, R. M. J. Res. Natl. Inst. Stand. Technol. 2009, 114, 341–358. doi:10.6028/jres.114.025 |
| 13. | Börner, M.; Blömer, L.; Kischel, M.; Richter, P.; Salvan, G.; Zahn, D. R. T.; Siles, P. F.; Fuentes, M. E. N.; Bufon, C. C. B.; Grimm, D.; Schmidt, O. G.; Breite, D.; Abel, B.; Kersting, B. Beilstein J. Nanotechnol. 2017, 8, 1375–1387. doi:10.3762/bjnano.8.139 |
| 18. | Kahn, O. Molecular Magnetism; VCH Verlagsgesellschaft: Weinheim, New York, 1993. |
| 19. | O’Connor, C. J. Magnetochemistry-Advances in Theory and Experimentation; Progress in Inorganic Chemistry; John Wiley & Sons: New Jersey, 2007. |
| 20. | Janiak, C.; Klapötke, T.; Meyer, H.-J.; Alsfasser, R. In Moderne Anorganische Chemie; Riedel, E., Ed.; de Gruyter: Berlin, 2007. |
| 5. | Lee, K. A.; Lozan, V.; Langford, S. J.; Kersting, B. Dalton Trans. 2009, 7481–7485. doi:10.1039/b907785e |
| 26. | Hesse, M.; Meier, H.; Zeeh, B., Eds. Spektroskopische Methoden in der organischen Chemie; Georg Thieme Verlag: Stuttgart, Germany, 2005. doi:10.1055/b-002-46985 |
| 5. | Lee, K. A.; Lozan, V.; Langford, S. J.; Kersting, B. Dalton Trans. 2009, 7481–7485. doi:10.1039/b907785e |
| 13. | Börner, M.; Blömer, L.; Kischel, M.; Richter, P.; Salvan, G.; Zahn, D. R. T.; Siles, P. F.; Fuentes, M. E. N.; Bufon, C. C. B.; Grimm, D.; Schmidt, O. G.; Breite, D.; Abel, B.; Kersting, B. Beilstein J. Nanotechnol. 2017, 8, 1375–1387. doi:10.3762/bjnano.8.139 |
| 24. | Casimiro, L.; Groppi, J.; Baroncini, M.; La Rosa, M.; Credi, A.; Silvi, S. Photochem. Photobiol. Sci. 2018, 17, 734–740. doi:10.1039/c8pp00062j |
| 25. | Klajn, R. Pure Appl. Chem. 2010, 82, 2247–2279. doi:10.1351/pac-con-10-09-04 |
| 22. | Herchel, R.; Boča, R.; Krzystek, J.; Ozarowski, A.; Durán, M.; van Slageren, J. J. Am. Chem. Soc. 2007, 129, 10306–10307. doi:10.1021/ja0725807 |
| 23. | Nanda, K. K.; Addison, A. W.; Paterson, N.; Sinn, E.; Thompson, L. K.; Sakaguchi, U. Inorg. Chem. 1998, 37, 1028–1036. doi:10.1021/ic9712434 |
© 2019 Klose et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)