Abstract
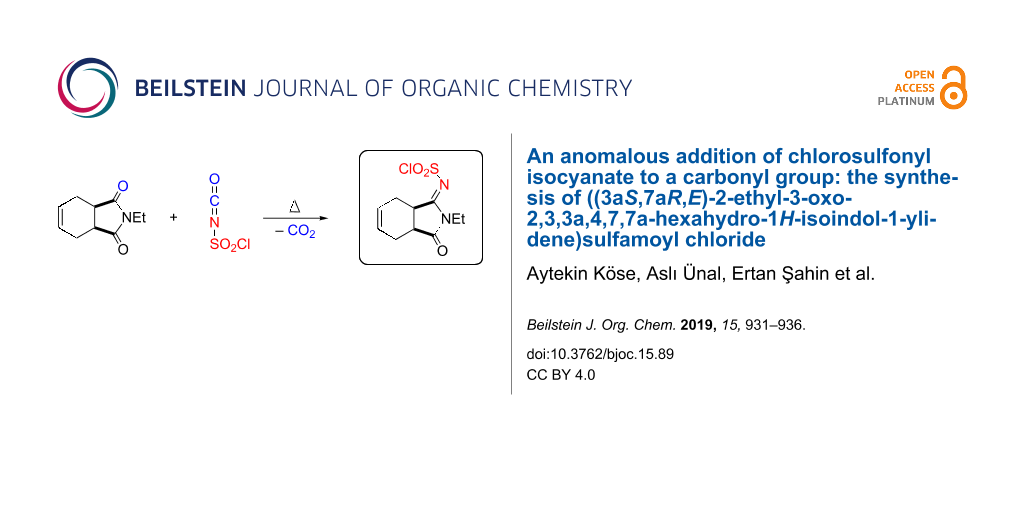
In this study, we developed a new addition reaction of chlorosulfonyl isocyanate (CSI), starting from 2-ethyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione. The addition reaction of CSI with 2-ethyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione resulted in the formation of ylidenesulfamoyl chloride, whose exact configuration was determined by X-ray crystal analysis. We explain the mechanism of product formation supported by theoretical calculations.

Graphical Abstract
Introduction
Since its identification in 1959 [1], chlorosulfonyl isocyanate (CSI, 1) continues to be the most reactive isocyanate to date. CSI is relatively more reactive than alkylsulfonyl isocyanate in olefin additions [2]. Its highly reactive nature is due to the polarization of the allene double bond by the highly electronegative chlorosulfonyl group. CSI reacts with unsaturated systems to yield either N-chlorosulfonyl-β-lactams 3 or unsaturated N-chlorosulfonyl amides 4 (Scheme 1).
![[1860-5397-15-89-i1]](/bjoc/content/inline/1860-5397-15-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The reaction of CSI with olefins.
Scheme 1: The reaction of CSI with olefins.
β-Lactams 3 generally predominate and in many cases are the exclusive products and they serve as key substances in a variety of chemical transformations. When chlorosulfonyl amides 4 are produced, they may be converted to other compounds. Graf [1] proposed that the reaction of CSI with unsaturated systems proceeds via the direct formation of dipolar intermediate 5 which may undergo a ring closure to form 3 or a hydrogen transfer to afford 4 (Figure 1).
![[1860-5397-15-89-1]](/bjoc/content/figures/1860-5397-15-89-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The dipolar intermediate formed in the reaction of CSI with olefins.
Figure 1: The dipolar intermediate formed in the reaction of CSI with olefins.
A wide variety of reactions has been reported for CSI, among which its addition to unsaturated systems has proven to be the most interesting and synthetically useful [3]. The addition of CSI to unsaturated systems and its further reactions have been examined by various groups [4-11]. On the other hand, examples for the reaction of carbonyl group containing compounds with CSI are limited in the literature. In particular, compounds containing an amide or pyrone skeletal structure were used in these studies. These reactions were reported by the research groups of Reynolds [12], Sattur [13], Jiménez [14] and Schwarz [15]. In our continuing studies on the synthesis of isoindole derivatives, we have included the addition of CSI to unsaturated systems and obtained unexpected results. When the reactions were performed by heating without any solvent, the condensation product imine was the sole product. In this paper, we present for the first time a unique example describing the addition of CSI to a system comprising both independent double bonds and imide functional groups. The mechanism for the addition of CSI to a carbonyl group is explained by theoretical computations.
Results and Discussion
Our starting material was 2-ethyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (9), which we have synthesized in previous studies [16,17]. Imide 9 was synthesized via the cycloaddition of 3-sulfone to maleic anhydride. The reaction of ethylamine with anhydride 8 in the presence of a toluene/triethylamine mixture (3:1) produced imide 9 in 80% yield (Scheme 2).
![[1860-5397-15-89-i2]](/bjoc/content/inline/1860-5397-15-89-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
To synthesize a new lactam derivative of isoindole-1,3-dione, we investigated the reaction of imide 9 with CSI in toluene at room temperature. Nevertheless, the starting imide 9 did not react with CSI, and we could not obtain the expected products lactam 11 and amide 12. Next, we studied the reaction of 9 with CSI by heating without solvent, which produced a very interesting product 10 that contained a N-chlorosulfonylimine group (Scheme 3).
![[1860-5397-15-89-i3]](/bjoc/content/inline/1860-5397-15-89-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The synthesis of ylidene sulfamoyl chloride 10.
Scheme 3: The synthesis of ylidene sulfamoyl chloride 10.
Upon examination, the most conspicuous features in the 1H and 13C NMR spectra of 10 were the change in the molecular symmetry and the chemical shifts of the carbonyl groups in the molecule. If the amide compound 12 had formed, there would be a change in the symmetry of the molecule, but the 1H and 13C NMR spectra do not support the structure of amide 12. The results of double resonance experiments clearly indicated that the HC=CH double bond was located between two CH2 groups (C-4 and C-7), similar to those present in the starting compound. Additionally, there was no signal of the three different carbonyl carbons in the 13C NMR spectrum of 10. On the other hand, the presence of double bond protons, three (CH2) groups, two CH protons, and one methyl (CH3) group, such as those in the starting compound, reveal that the reaction takes place on the carbonyl groups in the imide ring. While the 1H and 13C NMR spectra support a structure, we were not sure what kind of structure was indicated based on the spectral data. We determined the exact structure of 10 by X-ray crystal analysis (Figure 2) [18].
![[1860-5397-15-89-2]](/bjoc/content/figures/1860-5397-15-89-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Molecular structure of racemic molecule 10 (asymmetric unit). Thermal ellipsoids are drawn at the 30% probability level. (b) Geometric parameter with H-bonded geometry. Hydrogen bonds are drawn as dashed lines. (c) Stacking motif with the unit cell viewed downward along the c-axis. Dashed lines indicate C–H∙∙∙O interactions.
Figure 2: (a) Molecular structure of racemic molecule 10 (asymmetric unit). Thermal ellipsoids are drawn at t...
In addition to determining its structure, we performed X-ray crystal analysis of molecule 10 to identify the possible interactions. The structure has a racemic form, with the atoms of racemate 10 labeled and the polymeric H-bonding geometries shown in Figure 2a. We provide crystallographic data and structural refinement details in the experimental section. Compound 10 crystallizes in the monoclinic space group C2/c. The C–C (cyclohexene) distances are in the typical single bond range [1.491(3)–1.356(3) Å]. The C=C double bonds are in the cyclohexene units range between 1.307–1.316(3) Å and the S=O bonds are between 1.417–1.414(3) Å. The conformation is defined by steric effects, which force a fold of the cyclohexene rings relative to the mean plane through the pyrrolidine group. For both enantiomers, the cyclohexene ring has a twist-boat conformation and the pyrrolidine rings are in half-chair conformation. The structures contain four asymmetric carbon atoms and the stereogenic centers are as follows: C1(R), C6(S), C11(R), and C16(S), where the N-chlorosulfonyl group attached to the carbonyl atom changes the stoichiometry. In the solid state, compound 10 is stabilized via effective intramolecular H-bonds. Interactions between C9–H∙∙∙O5 [D∙∙∙A = 3.331(3) Å], C5–H∙∙∙O6 [D∙∙∙A = 3.529(3) Å], C19–H∙∙∙O3 [D∙∙∙A = 3.316(3) Å], C16–H∙∙∙O5 [D∙∙∙A = 3.315(3) Å], and C9–H∙∙∙O4 [D∙∙∙A = 3.313(3) Å] contribute to the formation of a stable structure (Figure 2a and Figure 2b).
Based on the structure of the product, we propose the reaction mechanism shown in Scheme 4. First, CSI reacts with the carbonyl carbon in the imide ring to form a four-membered urethane ring. Afterwards the imine is formed by the release of carbon dioxide from the molecule.
![[1860-5397-15-89-i4]](/bjoc/content/inline/1860-5397-15-89-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism for the formation of ylidenesulfamoyl chloride 10.
Scheme 4: Mechanism for the formation of ylidenesulfamoyl chloride 10.
We performed theoretical computations to better understand the reaction mechanism shown in Scheme 4 (Figure 3). For this purpose, we employed density functional theory (DFT) calculations and performed geometric optimizations using the B3LYP functional [19-22]. We computed the vibrational frequencies to characterize each stationary structure. In all the computations, we utilized Pople’s polarized triple-ζ split valence basis set with diffuse functions, 6-311++G(d,p) [23-25]. All the computations were performed using the Gaussian 09 program package [26]. The energies of all the structures are on the B3LYP/6-311G++(d,p) level, and the zero-point vibrational energy (ZPVE) corrections are all at the DFT level. Throughout this study, all the relative energies refer to the ZPVE-corrected energies. For the transition state (TS) between species A and B, we use the notation A/B throughout the article.
![[1860-5397-15-89-3]](/bjoc/content/figures/1860-5397-15-89-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Relative energy profile of the reaction mechanism shown in Scheme 4.
Figure 3: Relative energy profile of the reaction mechanism shown in Scheme 4.
Figure 3 shows the relative energy profile for the reaction mechanism shown in Scheme 4. The rate-determining steps for the formation of 10, 11, and 12 are the transition states 9/14, 9/11, and 9/12, respectively. The difference between the reaction barriers for the formations of 10 and 11 is 3.6 kcal/mol, whereas that for the formations of 10 and 12 is 14.1 kcal/mol. Hence, the formation of 10 is kinetically more favorable. This computational result is consistent with the experimental observations.
Conclusion
For the first time, we have demonstrated the addition of chlorosulfonyl isocyanate to the system comprising both independent double bonds and imide functional groups. The mechanism for the addition of CSI to a carbonyl group is explained by theoretical computations. Supported by theoretical calculations, we determined the reaction mechanism of the addition product. Such an addition reaction is one of the unique examples that define the addition of CSI. Furthermore, the chemical transformation of chlorosulfonyl isocyanate to the related compounds is currently under investigation.
Experimental
General
All reagents and substrates were purchased from commercial sources and used without further purification. Solvents were purified and dried by standard procedures before use. 1H and 13C NMR spectra were recorded on Varian 400 and Bruker 400 spectrometers. Elemental analyses were performed on a Leco CHNS-932 instrument. The melting points were measured with Gallenkamp melting point devices. X-ray crystallography was performed using a Rigaku R-AXIS RAPID IP diffractometer. HRMS: electron-spray technique (M+/M−) from the solution in MeOH (Waters LCT PremierTM XE UPLC/MS TOF (Manchester, UK)). All the computations were performed using the Gaussian 09 program package. The energies of all the structures are on the B3LYP/6-311G++(d,p) level.
((3aS,7aR,E)-2-Ethyl-3-oxo-2,3,3a,4,7,7a-hexahydro-1H-isoindol-1-ylidene)sulfamoyl chloride (10): The synthesis of 2-ethyl-3-oxo-2,3,3a,4,7,7a-hexahydro-1H-isoindol-1-ylidene)sulfamoyl chloride started from (3aR,7aS)-2-ethyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (0.50 g, 2.79 mmol). The starting material was put into a round bottomed flask and N2 was passed throughout the flask. Chlorosulfonyl isocyanate (CSI, 0.73 mL, 8.37 mmol) was added and reaction mixture was stirred at 80 °C for 4 h. At the end of this time, it was cooled to rt and diluted with EtOAc. Unreacted CSI and solvent were removed in vacuo and the residue was dissolved in CH2Cl2. It was filtered via a column and concentrated in vacuo. The residue was crystallized from CH2Cl2/hexane to obtain ((3aS,7aR,E)-2-ethyl-3-oxo-2,3,3a,4,7,7a-hexahydro-1H-isoindol-1-ylidene)sulfamoyl chloride (10, 0.233 g, colorless crystalline solid, 30% yield). 1H NMR (400 MHz, CDCl3) δ 5.93–5.89 (m, 1H, A of AB system, H6), 5.86–5.81 (m, 1H B of AB system, H5), 4.09 (m, 1H, H7a) 3.68 (q, J = 7.2 Hz 2H, N-CH2-), 3.10 (m, 1H, H3a), 2.70–2.61 (m, 2H, 2 × H4), 2.47–2.40 (m, 1H, H7(axial)), 2.34–2.26 (m, 1H, H7(equatorial)), 1.17 (t, 3H, -CH3, J = 7.2 Hz.); 13C NMR (100 MHz, CDCl3) δ 179.7 (C1), 178.1 (C3), 127.6 (C6), 126.0 (C5), 39.5 (C7a), 37.9 (C3a), 36.5 (N-CH2-), 25.5 (C7), 22.4 (C4), 12.6 (-CH3); mp 68–70 °C; HRMS (APCI): [M + H]+ calcd for C10H13ClN2O3S, 276.7350; found, 277.0434.
Supporting Information
| Supporting Information File 1: Theoretical computations, experimental procedures, copies of 1H and 13C NMR spectra, X-ray diffraction and HRMS analysis. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Graf, R. Chem. Ber. 1959, 92, 509–513. doi:10.1002/cber.19590920237
Return to citation in text: [1] [2] -
Clauβ, K. Justus Liebigs Ann. Chem. 1969, 722, 110–121. doi:10.1002/jlac.19697220111
Return to citation in text: [1] -
Graf, R. Angew. Chem., Int. Ed. Engl. 1968, 7, 172–182. doi:10.1002/anie.196801721
Return to citation in text: [1] -
Moriconi, E. J.; Jalandoni, C. C. J. Org. Chem. 1970, 35, 3796–3800. doi:10.1021/jo00836a047
Return to citation in text: [1] -
Vorbrüggen, H. Tetrahedron Lett. 1968, 9, 1631–1634. doi:10.1016/s0040-4039(01)99018-5
Return to citation in text: [1] -
Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 2046–2048. doi:10.1021/ja01087a034
Return to citation in text: [1] -
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Return to citation in text: [1] -
Moriconi, E. J.; Kelly, J. F. Tetrahedron Lett. 1968, 9, 1435–1439. doi:10.1016/s0040-4039(01)98973-7
Return to citation in text: [1] -
Bestian, H.; Biener, H.; Clauss, K.; Heyn, H. Justus Liebigs Ann. Chem. 1968, 718, 94–100. doi:10.1002/jlac.19687180109
Return to citation in text: [1] -
Paquette, L. A.; Wyvratt, M. J.; Allen, G. R., Jr. J. Am. Chem. Soc. 1970, 92, 1763–1765. doi:10.1021/ja00709a060
Return to citation in text: [1] -
Moriconi, E. J.; Crawford, W. C. J. Org. Chem. 1968, 33, 370–378. doi:10.1021/jo01265a075
Return to citation in text: [1] -
Van Allan, J. A.; Chang, S. C.; Reynolds, G. A. J. Heterocycl. Chem. 1974, 11, 195–198. doi:10.1002/jhet.5570110216
Return to citation in text: [1] -
Rao, K. R.; Nageswar, Y. V. D.; Srinivasan, T. N.; Sattur, P. B. Synth. Commun. 1988, 18, 877–880. doi:10.1080/00397918808057857
Return to citation in text: [1] -
Castellanos, L.; Duque, C.; Zea, S.; Espada, A.; Rodríguez, J.; Jiménez, C. Org. Lett. 2006, 8, 4967–4970. doi:10.1021/ol062087k
Return to citation in text: [1] -
Bartsch, H.; Schwarz, O. Arch. Pharm. (Weinheim, Ger.) 1982, 315, 545–551. doi:10.1002/ardp.19823150612
Return to citation in text: [1] -
Tan, A.; Koc, B.; Şahin, E.; Kishali, N. H.; Kara, Y. Synthesis 2011, 1079–1084. doi:10.1055/s-0030-1258466
Return to citation in text: [1] -
Tan, A.; Kazancıoğlu, M. Z.; Aktaş, D.; Gündoğdu, Ö.; Şahin, E.; Kishali, N. H.; Kara, Y. Turk. J. Chem. 2014, 38, 629–637. doi:10.3906/kim-1310-30
Return to citation in text: [1] -
SHELXS-97, SHELXL-97 Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785
Return to citation in text: [1] -
Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200–1211. doi:10.1139/p80-159
Return to citation in text: [1] -
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001
Return to citation in text: [1] -
Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213–222. doi:10.1007/bf00533485
Return to citation in text: [1] -
McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980
Return to citation in text: [1] -
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2013.
Return to citation in text: [1]
| 4. | Moriconi, E. J.; Jalandoni, C. C. J. Org. Chem. 1970, 35, 3796–3800. doi:10.1021/jo00836a047 |
| 5. | Vorbrüggen, H. Tetrahedron Lett. 1968, 9, 1631–1634. doi:10.1016/s0040-4039(01)99018-5 |
| 6. | Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 2046–2048. doi:10.1021/ja01087a034 |
| 7. | Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811 |
| 8. | Moriconi, E. J.; Kelly, J. F. Tetrahedron Lett. 1968, 9, 1435–1439. doi:10.1016/s0040-4039(01)98973-7 |
| 9. | Bestian, H.; Biener, H.; Clauss, K.; Heyn, H. Justus Liebigs Ann. Chem. 1968, 718, 94–100. doi:10.1002/jlac.19687180109 |
| 10. | Paquette, L. A.; Wyvratt, M. J.; Allen, G. R., Jr. J. Am. Chem. Soc. 1970, 92, 1763–1765. doi:10.1021/ja00709a060 |
| 11. | Moriconi, E. J.; Crawford, W. C. J. Org. Chem. 1968, 33, 370–378. doi:10.1021/jo01265a075 |
| 3. | Graf, R. Angew. Chem., Int. Ed. Engl. 1968, 7, 172–182. doi:10.1002/anie.196801721 |
| 23. | Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213–222. doi:10.1007/bf00533485 |
| 24. | McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980 |
| 25. | Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955 |
| 2. | Clauβ, K. Justus Liebigs Ann. Chem. 1969, 722, 110–121. doi:10.1002/jlac.19697220111 |
| 15. | Bartsch, H.; Schwarz, O. Arch. Pharm. (Weinheim, Ger.) 1982, 315, 545–551. doi:10.1002/ardp.19823150612 |
| 18. | SHELXS-97, SHELXL-97 Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. |
| 14. | Castellanos, L.; Duque, C.; Zea, S.; Espada, A.; Rodríguez, J.; Jiménez, C. Org. Lett. 2006, 8, 4967–4970. doi:10.1021/ol062087k |
| 19. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 20. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785 |
| 21. | Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200–1211. doi:10.1139/p80-159 |
| 22. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 13. | Rao, K. R.; Nageswar, Y. V. D.; Srinivasan, T. N.; Sattur, P. B. Synth. Commun. 1988, 18, 877–880. doi:10.1080/00397918808057857 |
| 12. | Van Allan, J. A.; Chang, S. C.; Reynolds, G. A. J. Heterocycl. Chem. 1974, 11, 195–198. doi:10.1002/jhet.5570110216 |
| 16. | Tan, A.; Koc, B.; Şahin, E.; Kishali, N. H.; Kara, Y. Synthesis 2011, 1079–1084. doi:10.1055/s-0030-1258466 |
| 17. | Tan, A.; Kazancıoğlu, M. Z.; Aktaş, D.; Gündoğdu, Ö.; Şahin, E.; Kishali, N. H.; Kara, Y. Turk. J. Chem. 2014, 38, 629–637. doi:10.3906/kim-1310-30 |
© 2019 Köse et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)