Abstract
Ferrocenyl thioketones reacted with donor–acceptor cyclopropanes in dichloromethane at room temperature in the presence of catalytic amounts of Sc(OTf)3 yielding tetrahydrothiophene derivatives, products of formal [3 + 2]-cycloaddition reactions, in moderate to high yields. In all studied cases, dimethyl 2-arylcyclopropane dicarboxylates reacted with the corresponding aryl ferrocenyl thioketones in a completely diastereoselective manner to form single products in which (C-2)-Ar and (C-5)-ferrocenyl groups were oriented in a cis-fashion. In contrast, the same cyclopropanes underwent reaction with alkyl ferrocenyl thioketones to form nearly equal amounts of both diastereoisomeric tetrahydrothiophenes. A low selectivity was also observed in the reaction of a 2-phthalimide-derived cyclopropane with ferrocenyl phenyl thioketone.

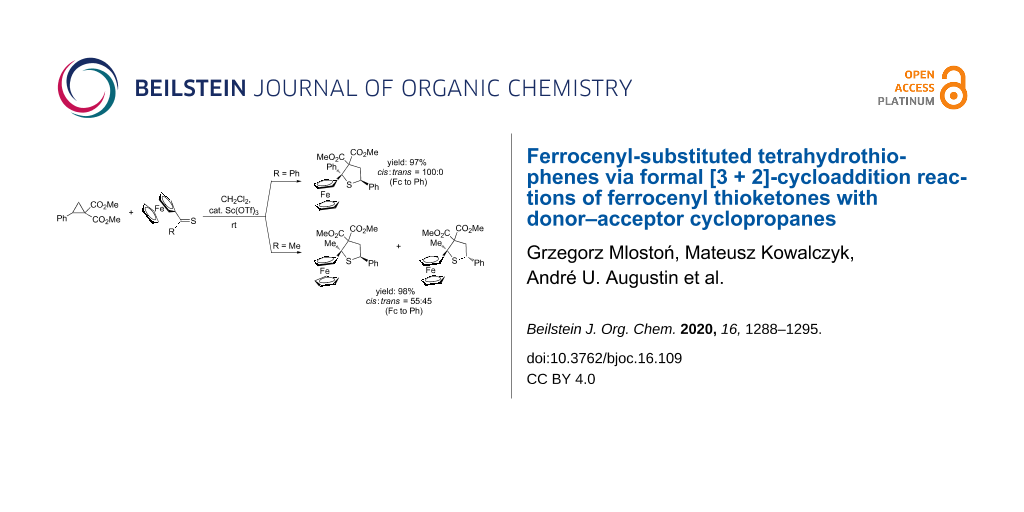
Graphical Abstract
Introduction
Functionalized tetrahydrothiophenes constitute an important group of five-membered sulfur heterocycles; many of them, both chiral and achiral, with biotin as the best-known representative, form the key motif in numerous compounds of great practical importance [1,2]. The development of chemo- and diastereoselective syntheses for these compounds is thus a challenging problem. An elegant and highly efficient method for the construction of the tetrahydrothiophene ring is based on 1,3-dipolar cycloadditions of in-situ-generated thiocarbonyl S-methanides (thiocarbonyl ylides) with electron-deficient ethylenic dipolarophiles. This method was extensively developed by Huisgen and co-workers in the 1980s [3-5]. In the course of these studies, a non-orthodox stepwise mechanism of the 1,3-dipolar cycloaddition was established by experiments performed with the sterically crowded thiocarbonyl S-methanide 1, derived from 2,2,4,4-tetramethyl-3-thioxocyclobutanone and extremely electron-deficient ethylenes 2 such as (E)- and (Z)-dialkyl dicyanobutenoates (R = CO2Me) [6], tetracyanoethylene (R = CN) [7] or (E)- and (Z)-1,2-bis(trifluoromethyl)ethylene-1,2-dicarbonitrile (R = CF3) [8]. Both five-membered spirotetrahydrothiophenes 3 and seven-membered S,N-heterocycles (ketene imines) 4 were observed in the course of these reactions (Scheme 1). The latter products were trapped with suitable nucleophiles (R = CO2Me) or even isolated and identified by means of spectroscopic methods (R = CF3).
![[1860-5397-16-109-i1]](/bjoc/content/inline/1860-5397-16-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of spirotetrahydrothiophenes 3 via non-concerted [3 + 2]-cycloadditions of thiocarbonyl ylide 1 with electron-deficient ethylenes 2. Cyclic ketene imines 4 are also formed as products of formal [4 + 3]-cycloadditions.
Scheme 1: Synthesis of spirotetrahydrothiophenes 3 via non-concerted [3 + 2]-cycloadditions of thiocarbonyl y...
In a recent work, an alternative, efficient and useful method for the synthesis of highly functionalized tetrahydrothiophenes of type 6 was reported [9] (Scheme 2). Under Lewis acid catalysis, formal [3 + 2]-cycloadditions of aromatic and cycloaliphatic thioketones (also thionoesters) with donor–acceptor cyclopropanes 5 (D–A cyclopropanes) were realized.
![[1860-5397-16-109-i2]](/bjoc/content/inline/1860-5397-16-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formal [3 + 2]-cycloadditions of thioketones and [4 + 3]-cycloadditions of thiochalcones with donor–acceptor cyclopropanes 5 leading to tetrahydrothiophenes 6 and tetrahydrothiepines 7, respectively.
Scheme 2: Formal [3 + 2]-cycloadditions of thioketones and [4 + 3]-cycloadditions of thiochalcones with donor...
In contrast, thiochalcones (α,β-unsaturated aromatic thioketones) were shown to react under similar conditions with cyclopropanes 5 yielding exclusively seven-membered tetrahydrothiepines 7 as products of the formal [4 + 3]-cycloaddition [10] (Scheme 2).
In a series of our recent publications, ferrocenyl/aryl and ferrocenyl/alkyl thioketones were demonstrated to be attractive substrates for the preparation of six- and five-membered sulfur heterocycles via [4 + 2]- and [3 + 2]-cycloadditions, respectively [11-15]. Notably, in contrast to aryl/alkyl thioketones (e.g., thioacetophenone), their ferrocenyl analogs of type 8 (e.g., ferrocenyl phenyl thioketone (8a), diferrocenyl thioketone (8b), and ferrocenyl methyl thioketone (8c)) were stable compounds at ambient conditions and could be used with no special precautions. In general, ferrocene has been considered as an ‘exceptional compound’ [16,17] and in our hands ferrocenyl-functionalized sulfur heterocycles, e.g., thiiranes and 1,3-dithiolanes, have found applications for the synthesis of compounds relevant for medicinal [18] and materials chemistry, and electrochemical studies [19].
In continuation of our studies on organic sulfur compounds and the mechanisms of their reactions, the main goal of the present work was the examination of the formal [3 + 2]-cycloaddition reactions of ferrocenyl-substituted thioketones 8 with D–A cyclopropanes 5, aimed at the synthesis of hitherto unreported, ferrocenyl-substituted tetrahydrothiophene dicarboxylates (thiolanes) of type 9.
Results and Discussion
In analogy to experiments described in our earlier publication [9], the test reaction was performed with dimethyl 2-phenylcyclopropane dicarboxylate (5a) and ferrocenyl phenyl thioketone (8a) in CH2Cl2 at room temperature using aluminum chloride (AlCl3) as a catalyst. The reaction was monitored by TLC, and was shown to be complete after 1 h. The crude reaction mixture was examined by 1H NMR, revealing the formation of a single product with characteristic signals of both CO2Me groups located at 3.38 and 3.81 ppm. After chromatographic separation the expected tetrahydrothiophene 9a was isolated in only 23% yield. As the next model substrate, the sterically crowded diferrocenyl thioketone (8b) was tested as a structural analog of thiobenzophenone, which was widely applied in studies involving aromatic thioketones [3-5]. However, in contrast to 8a, the reaction of 8b with 5a was unsuccessful. This observation prompted us to replace AlCl3 by scandium triflate (Sc(OTf)3), which is also known to be an efficient catalyst in various reactions of D–A cyclopropanes [9,10,20]. This time, the reaction was complete after 1 h and the expected 2,2-diferrocenyl-substituted tetrahydrothiophene 9b was isolated chromatographically in about 28% yield (Scheme 3, Table 1). This experiment was successfully repeated, again using Sc(OTf)3 instead of AlCl3, in further experiments of ferrocenyl thioketones 8 with differently substituted cyclopropanes 5. Again using Sc(OTf)3, we repeated the experiment with 8a, which this time led to the isolation of 9a in an excellent yield of 98% (Table 1).
![[1860-5397-16-109-i3]](/bjoc/content/inline/1860-5397-16-109-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formal [3 + 2]-cycloadditions of dimethyl 2-substituted cyclopropane-1,1-dicarboxylates 5a–g with ferrocenyl thioketones 8a–g, leading to dimethyl tetrahydrothiophene 3,3-dicarboxylates 9a–n (Table 1).
Scheme 3: Formal [3 + 2]-cycloadditions of dimethyl 2-substituted cyclopropane-1,1-dicarboxylates 5a–g with f...
Table 1: Ferrocenyl-substituted tetrahydrothiophenes 9a–n obtained in reactions of D–A cyclopropanes 5a–h with ferrocenyl thioketones 8a–g catalyzed with Sc(OTf)3.
|
compound
9 |
substituent
Ar |
substituent
R |
ratio of diastereoisomers | yield of isolated products (%) |
|
a
b c d e f g h i j k l m n |
Ph
Ph Ph Ph Ph β-naphthyl β-naphthyl 4-Me-C6H4 4-MeO-C6H4 4-Br-C6H4 4-CF3-C6H4 Ph Ph phthalimid-1-yl |
Ph
Fca β-naphthyl Me n-Pr Ph β-naphthyl Ph Ph Ph Ph thien-2-yl fur-2-yl Ph |
100: 0
– 100:0 55:45 52:48 100:0 100:0 100:0 100:0 100:0 100:0 100:0 60:40 60:40 |
98
28 65 98 97 30 31 85 79 93 95 58 96 34 |
aFc = ferrocenyl.
In analogy to 8a, the similarly substituted ferrocenyl (β-naphthyl) thioketone (8c) reacted with 5a in a diastereoselective manner yielding the expected product 9c in good yield (65%) as the sole isolated product. Notably, in all reactions performed with aryl-substituted cyclopropanes 5a–f and with thioketones 8a,c,f, the desired tetrahydrothiophenes 9a,c,f–l were formed with complete diastereoselectivity, leading to a single isomer. In order to establish the structure of the isomers, a single crystal obtained for compound 9c was studied by X-ray diffraction analysis which showed, that the Ph(C-2) group and Fc(C-5) substituent were mutually cis-oriented (Figure 1). Tentatively, the same configuration was also attributed to all tetrahydrothiophenes 9a,f–l that were formed as single isomers (Table 1).
![[1860-5397-16-109-1]](/bjoc/content/figures/1860-5397-16-109-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Thermal ellipsoid plots of the molecular structures of cis-9c and trans-9d drawn using 50% probability displacement ellipsoids. The terminology cis and trans referred to the relative orientation of Ph and Fc groups.
Figure 1: Thermal ellipsoid plots of the molecular structures of cis-9c and trans-9d drawn using 50% probabil...
However, the diastereoselectivity changed in reactions that were conducted with alkyl ferrocenyl thioketones 8d–e with 5a. Thus, the reaction with 8d led to a 55:45 mixture of two isomeric products in nearly quantitative yield (98%). Subsequently, they were carefully separated by preparative thin layer chromatography (PTC) on silica using a mixture of petroleum ether and ethyl acetate as an eluent. The less polar fraction formed the major product and the slightly more polar one was isolated and identified as the minor isomer of 9d. In the course of crystallization from hexane the less polar fraction gave single crystals suitable for the X-ray diffraction analysis, which unambiguously confirmed that in this molecule the Ph(C-5) and Fc(C-2) groups were trans-oriented and for that reason, this isomer was described as trans-9d (Figure 1).
Analogously, the reaction of ferrocenyl n-propyl thioketone (8e) with 5a led to a 52:48 mixture of trans- and cis-isomers of 9e, which were isolated in a total yield of 97% and identified without further separation. Moreover, a mixture of nearly equal amounts of isomeric trans-9m and cis-9m was also observed in the reaction of 5a with ferrocenyl fur-2-yl thioketone (8g). The reaction of the phthalimide-derived cyclopropane 5g with thioketone 8a led to a 4:1 mixture of both isomers cis- and trans-9n. Based on these observations it was difficult to explain the complete diastereoselectivity of tetrahydrothiophene formation observed in the reactions of aryl ferrocenyl-substituted thioketones 8a,c,f with cyclopropanes 5a–f bearing aryl groups. Tentatively, a repulsive interaction of aryl groups rather than steric hindrance of the bulky ferrocenyl unit could be postulated. Remarkably, ferrocenyl fur-2-yl thioketone (8g) was an exception and delivered a 60:40 mixture of trans- and cis-9m.
The mechanistic interpretation of the efficient, formal [3 + 2]-cycloadditions of D–A cyclopropanes 5 with ferrocenyl thioketones 8 in the presence of a Lewis acid was based on the assumption that the coordination of the catalyst by two ester groups activated the cyclopropane ring and allowed a nucleophilic attack of the C=S group on the benzylic position of the cyclopropane derivative (Scheme 4).
![[1860-5397-16-109-i4]](/bjoc/content/inline/1860-5397-16-109-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Plausible mechanism for the formal [3 + 2]-cycloadditions of ferrocenyl thioketones 8 with D–A cyclopropanes 5.
Scheme 4: Plausible mechanism for the formal [3 + 2]-cycloadditions of ferrocenyl thioketones 8 with D–A cycl...
The subsequent ring-closure of the zwitterionic intermediate 10 led to the formation of the tetrahydrothiophene derivative 9. This process formally resembled the [3 + 2]-cycloadditions of thiocarbonyl S-methanides with an activated C–C double bond, which also led to tetrahydrothiophenes [6-8]. Nevertheless, the key step involved the formation of the reactive, zwitterionic intermediate 10. It seemed that repulsive interactions of the aryl groups Ar (from cyclopropane 5) and R (from thioketone 8) controlled the diastereoselective ring-closure to the five-membered ring leading in these cases to the formation of cis-9 (Ar to Fc) as a single isomer. A similar reaction pathway with a zwitterionic intermediate analogous to 10, generated in the presence of a Lewis acid, was proposed for the reaction of cycloaliphatic 3-thioxo-2,2,4,4-tetramethylcyclobutanone with D–A cyclopropanes [21].
Conclusion
The present study showed once more that ferrocenyl/aryl and ferrocenyl/alkyl thioketones 8 are versatile and useful building blocks for a simple and efficient preparation of ferrocenyl-functionalized five-membered sulfur heterocycles. They were shown to react easily with donor–acceptor (D–A) cyclopropanes in the presence of scandium triflate, Sc(OTf)3 as a catalyst, yielding highly functionalized tetrahydrothiophene derivatives of type 9. These formal [3 + 2]-cycloaddition reactions occurred via a nucleophilic attack of the sulfur atom on the activated cyclopropane ring at the most reactive benzylic position. The formation of the five-membered ring occurred regioselectively and the expected tetrahydrothiophene-3,3-carboxylates were the products. The studied reactions displayed an interesting stereoselectivity and, in the case of 2,5-diaryl-substituted products 9, both aryl groups were exclusively located at the opposite sides of the ring plane. The described reactions supplement the recently reported synthetic applications of alkyl/ferrocenyl thioketones as attractive substrates for the synthesis of chiral ferrocene derivatives [22] and ferrocenyl-substituted platinathiiranes [23].
It should be also emphasized that the present study also demonstrated the growing potential of donor–acceptor cyclopropanes [24-28] as unique building blocks for current organic synthesis and especially for the efficient and highly stereoselective preparation of the relevant five-membered sulfur heterocycles derived from tetrahydrothiophene.
Experimental
General information: Solvents and chemicals were purchased and used as received without further purification. Products were purified by standard column chromatography on silica gel. Yields refer to analytically pure samples. NMR spectra were recorded with a Bruker Avance III 600 MHz instrument (1H NMR: 600 MHz; 13C NMR: 151 MHz). Chemical shifts are reported relative to solvent residual peaks (1H NMR: δ = 7.26 ppm [CHCl3]; 13C NMR: δ = 77.0 ppm [CDCl3]). IR spectra were recorded with a Cary 630 FTIR (Agilent Technologies) spectrometer (as film). Melting points were determined in capillaries with a Melt Temp II apparatus.
Starting materials: D–A cyclopropanes 5a–g were obtained following the reported procedure [28]. Ferrocenyl thioketones 8a–g were obtained by thionation of corresponding ferrocenyl ketones [29] by treatment with Lawesson’s reagent [30]. Ferrocenyl β-naphthyl thioketone (8b) obtained from ferrocenyl(β-naphthyl) ketone [31] is reported for the first time (see Supporting Information File 1).
General procedure: A solution of 0.3 mmol of the corresponding cyclopropane 5 in 5 mL of dichloromethane was stirred for 5 min. Then, 0.5 mmol of the corresponding ferrocenyl thioketone 8 and a catalytic amount (ca. 5 mg) of Sc(OTf)3 was added to the stirred solution. The mixture was stirred at room temperature for 1 h. The progress of the reaction was monitored by TLC. The solvent was evaporated in vacuo and the crude mixture was purified by flash chromatography using dichloromethane as the eluent. Analytically pure samples of tetrahydrothiophenes 9 were obtained by crystallization from petroleum ether or hexane with a small amount of dichloromethane.
The diastereoselectivity of the studied reactions was determined by integration of the crude 1H NMR. Preliminary purification of crude mixtures by a short-column chromatography was necessary to remove traces of iron particles formed as a side product after partial decomposition of ferrocenyl containing substrates and/or products formed under reaction conditions.
Dimethyl 2-ferrocenyl-2,5-diphenyltetrahydrothiophene-3,3-dicarboxylate (cis-9a): Yield: 159 mg (98%); red crystals; mp 192–193°C; 1H NMR δ 2.65 (dd, JH,H = 13.9 Hz, JH,H = 4.3 Hz, 1H, HC(4)), 3.44 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.56 (dd, JH,H = 13.9 Hz, JH,H = 12.7 Hz, 1H, HC(4)), 3.51–3.60 (m, 1H, HC(Fc)), 4.00–4.02 (m, 1HC(Fc)), 4.07 (s, 5HC(Fc)), 4.27–4.29 (m, 1HC(Fc)), 4.68–4.70 (m, 1 HC(Fc)), 4.81 (dd, JH,H = 12.6 Hz, JH,H = 4.3 Hz, HC(5)), 7.31–7.39 (m, 2 arom. HC), 7.40–7.46 (m, 4 arom. HC), 7.62–7.65 (m, 2 arom. HC), 8.21–8.24 (m, 2 arom. HC); 13C NMR δ (C(4)-not found), 48.1, 48.4 (2OCH3), 52.4 (C(5)), 67.8 (C(2)), 71.1 (C(3)), 68.7, 69.2, 69.9, 71.0, 73.7 (for 9 HC(Fc)), 97.0 (C(Fc)), 126.6, 127.1, 127.9, 128.0, 128.8, 129.0 (for 10 arom. HC), 138.9, 144.2 (2 arom. C), 169.0, 170.3 (2 C=O); IR (cm−1) ν: 1737 brs (2C=O), 1492 m, 1444 m, 1429 m, 1258 m, 1239 s, 1073 m, 814 m, 760 m, 697 vs, 497 vs; Anal. calcd for C30H28FeO4S (540.45): C, 66.67; H, 5.22; S, 5.93; found: C, 66.58; H, 5.24; S, 5.99.
Dimethyl 2,2-diferrocenyl-5-phenyl tetrahydrothiophene-3,3-dicarboxylate (9b): Yield: 54 mg (28%); red crystals; mp 170 °C (dec.); 1H NMR δ 2.91 (dd, JH,H = 14.0 Hz, JH,H = 6.2 Hz, 1H, HC(4)), 3.36 (s, 3H, OCH3), 3.41 (dd, JH,H = 15.8 Hz, JH,H = 12.4 Hz, 1H, HC(4)), 3.65 (s, 3H, OCH3), 4.05–4.07 (m, 1H, HC(Fc)), 4.07–4.09 (m, 1H, HC(Fc)), 4.16–4.18 (m, 1H, HC(Fc)), 4.23–4.27 (m, 7H, 7HC(Fc)), 4.31 (s, 5H, HC(Fc)), 4.48–4.50 (m, 1H, HC(Fc)), 4.56–4.58 (m, 1H, HC(Fc)), 4.68–4.70 (m, 1H, HC(Fc)), 5.51 (dd, JH,H = 11.2 Hz, JH,H = 6.3 Hz, 1H, HC(5)), 7.34–7.37 (m, 1arom. HC), 7.44–7.48 (m, 2arom. HC), 7.78 (m, 2arom. HC); 13C NMR δ 47.9 (C(4)), 49.5 (C(5)), 51.9, 52.6 (2OCH3), 65.8, 66.2, 66.9, 67.6, 67.7, 69.6, 69.7, 70.4, 73.1 (for 18 HC(Fc), 73.4, 94.3 (C(2) and C(3), 100.0 (2C(Fc)), 127.4, 128.2, 128.6 (5 arom. HC), 141.2 (arom. C), 169.0, 169.1 (2 C=O); IR (cm−1) ν: 1727 brs (2C=O), 1431 m, 1259 s, 1164 s, 1107 m, 1000 m, 818 s, 760 m, 696 s, 479 vs; anal. calcd for C34H32Fe2O4S (648.37): C, 62.98; H, 4.97; S, 4.94; found: C, 62.68; H, 4.93; S, 4.88.
Dimethyl 2-ferrocenyl-5-phenyl-2-(naphth-2-yl)tetrahydrothiophene-3,3-di-carboxylate (cis-9c): Yield: 115 mg (65%); yellow crystals; mp 210–211 °C; single crystals were obtained from hexane solution by slow evaporation at rt; 1H NMR δ 2.69 (dd, JH,H = 13.8 Hz, JH,H = 4.3 Hz, 1H, HC(4)), 3.41 (s, 3H, OCH3), 3.47 (s, 3H, OCH3), 3.57 (s, 1H, HC(Fc)), 3.62 (t, JH,H = 13.1 Hz, 1H, CH), 4.01 (s, 1H, HC(Fc)), 4.09 (s, 5 HC(Fc)), 4.31 (s, 1H, HC(Fc)), 4.77 (s, 1H, HC(Fc)), 4.86 (dd, JH,H = 12.6 Hz, JH,H = 4.3 Hz, 1H, HC(5)), 7.36–7.40 (m, 1 arom., HC), 7.44–7.48 (m, 2 arom. HC), 7.51–7.54 (m, 2 arom. HC), 7.66 (m, 2 arom. HC), 7.87 (d, JH,H = 8.6 Hz, 1 arom., HC), 7.88–7.92 (m, 1 arom., HC), 7.95–7.99 (m, 1 arom., HC), 8.36 (d, JH,H = 8.6 Hz, 1arom., HC), 8.74 (s, 1 arom., HC); 13C NMR δ 48.1 (C(5)), 48.5 (C(4)), 52.5, 52.6 (2OCH3), 67.8, 68.7, 69.2, 69.9, 71.2 (for 9 HC(Fc)), 71.0, 73.5 (C(2) and C(3)) 97.4 (2 C(Fc)), 125.8, 125.9, 126.1, 127.2, 127.3, 127.9, 128.0, 128.1, 128.6, 128.8 (10 arom. HC), 132.1, 132.8, 138.8, 141.8 (4 arom. C), 168.9, 170.3 (2 C=O); IR (cm−1) ν: 1738 brs (2C=O), 1429 m, 1239 s, 1215 s, 1170 m, 1053 m, 810 s, 758 m, 704 s, 480 vs; anal. calcd for C34H30FeO4S (590.51): C, 69.15; H, 5.12; S, 5.43; found: C, 67.16; H, 5.01; S, 5.47.
Dimethyl 2-ferrocenyl-2-phenyl-5-methyltetrahydrothiophene-3,3-dicarboxylate (9d). Obtained as a 55:45 mixture of isomers. The trans- (major) and cis- (minor) isomers (Ph to Fc) were separated by PLC (silica, PE/ethyl acetate). Yields: cis-isomer, yellow crystals, 66 mg (more polar fraction, 44%); mp 148–150 °C, trans-isomer, yellow crystals, 74 mg (less polar fraction, 54%); mp 126–128 °C; single crystals of trans-9d were obtained from hexane/CH2Cl2 solution by slow evaporation at rt; 1H NMR (cis-9d) δ 2.31 (s, 3H, CH3); 2.54 (dd, JH,H = 13.8 Hz, JH,H = 5.3 Hz, 1H, HC(4)); 3.22 (dd, JH,H = 13.8 Hz, JH,H = 12.3 Hz, 1H, HC(4)); 3.46 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 4.09–4.10 (m, 1H, HC(Fc)); 4.12–4.13 (m, 1H, HC(Fc)); 4.22 (s, 5H, HCH(Fc)); 4.23–4.25 (m, 1H, HC(Fc)); 4.55–4.56 (m, 1H, HC(Fc)); 4.78 (dd, JH,H = 12.3 Hz, JH,H = 5.3 Hz, 1H, HC(5)); 7.30–7.34 (m, 1 arom. HC); 7.39–7.43 (m, 2 arom. HC); 7.54–7.58 (m, 2 arom. HC); 13C NMR (cis-9d) δ 25.9 (CH3); 43.9 (C(4)); 47.9 (C(5)); 52.0, 52.6 (2OCH3); 60.0 (C(2)); 68.1, 68.2, 68.8, 69.1, 70.7 (for 9 HC(Fc)); 70.6 (C(3)); 96.5 (C(Fc)); 127.5, 127.6, 128.7 (5 arom. HC); 140.0 (arom. C); 168.5, 169.7 (2C=O); IR (cm−1) ν: 1731 brvs (2C=O); 1494 m, 1453 m, 1436 m, 1248 vs, 1207 m, 1157 vs, 1105 m, 1038 s, 829 m, 766 s, 702 vs; anal. calcd for C25H26FeO4S (478.38): C, 62.77; H, 5.48; S, 6.70; found: C, 62.69; H, 5.52; S, 6.63.
1H NMR (trans-9d) δ 2.28 (s, 3H, CH3); 2.79 (dd, JH,H = 14.1 Hz, JH,H = 10.7 Hz, 1H, HC(4)); 3.12 (dd, JH,H = 14.1 Hz, JH,H = 7.1 Hz, 1H, HC(4)); 3.47 (s, 3H, OCH3); 3.66 (s, 3H, OCH3); 4.19–4.21 (m, 2H, HC(Fc)); 4.23 (s, 5H, 5HC(Fc)); 4.40–4.42 (m, 1H, HC(Fc)); 4.56–4.57 (m, 1H, HC(Fc)); 5.28 (dd, JH,H = 10.7 Hz, JH,H = 7.1 Hz, 1H, HC(5)); 7.30–7.32 (m, 1 arom. HC); 7.39–7.42 (m, 2 arom. HC); 7.57–7.59 (m, 2 arom. HC); 13C NMR (trans-9d) δ 31.3 (CH3); 47.1 (C(4)); 48.6 (C(5)); 52.2, 52.3 (2OCH3); 60.1 (C(2)); 67.6 (C(3)); 68.5, 68.8, 69.1, 69.3, 71.1 (for 9HC(Fc)); 89.9 (C(Fc)); 127.3, 127.9, 128.6 (5 arom. HC); 142.6 (arom. C); 168.9, 169.5 (2C=O); IR (cm−1) ν: 1737 vs, 1720 vs (2C=O); 1492 m, 1453 m, 1427 m, 1258 vs, 1220 s, 1204 m, 1106 m, 1105 m, 1023 m, 993 m, 829 m, 766 s, 703 vs; anal. calcd for C25H26FeO4S (478.38): C, 62.77; H, 5.48; S, 6.70; found: C, 62.70; H, 5.46; S, 6.59.
Dimethyl 2-ferrocenyl-5-phenyl-2-(thien-2-yl)tetrahydrothiophene-3,3-dicarboxy-late (trans-9l): Yield: 95 mg (58%); yellow crystals; mp 210 °C (dec.); 1H NMR δ 2.65 (dd, JH,H = 14.0 Hz, JH,H = 4.5 Hz, 1H, HC(4)), 3.43 (pseudo-t, JH,H = 13.9 Hz, 1H, HC(4)), 3.48 (s, 3H, OCH3), 3.52 (s, 3H, OCH3), 4.10 (s, 5H, 5HC(Fc)), 4.14 (s, 2H, 2HC(Fc)), 4.30 (s, 1H, HC(Fc)), 4.68 (s, 1H, HC(Fc)), 5.02 (dd, JH,H = 13.6 Hz, JH,H = 4.5 Hz, 1H, HC(5)), 7.05–7.07 (m, 1 arom. HC), 7.21–7.23 (m, 1 arom. HC), 7.34–7.38 (m, 1 arom. HC), 7.42–7.45 (m, 2 arom., HC), 7.55–7.57 (m, 1 arom. HC), 7.60–7.63 (m, 2 arom. HC); 13C NMR δ 47.3 (C(4)), 49.0 (C(5)), 52.2, 52,5 (2OCH3), 67.9, 69.0, 69.4, 70.4, 70.7 (for 9 HC(Fc)), 66.3, 74.2 (2 arom. C), 94.6 (1 C(Fc)), 123.4, 125,8, 126.8, 127.9, 128.0, 128.8 (for 8 arom. HC), 138.7, 150.3 (2 arom. C), 168.4, 169.5 (2C=O); IR (cm−1) ν: 1733 brs (2C=O), 1427 m, 1235 s, 1146 s, 1045 m, 1032 m, 818 m, 766 s, 691 vs, 506 m, 488 s; HRMS–EI (m/z): [M]+ calcd. for [C28H26FeO4S2]+, 546.0621; found: 546.0629.
Supporting Information
CCDC-1992864 and CCDC-1992865 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/structures.
| Supporting Information File 1: Experimental data for selected compounds 9, details of the crystal structure determination, and the original 1H and 13C NMR spectra for all products. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Benetti, S.; De Risi, C.; Pollini, G. P.; Zanirato, V. Chem. Rev. 2012, 112, 2129–2163. doi:10.1021/cr200298b
Return to citation in text: [1] -
De Clercq, P. J. Chem. Rev. 1997, 97, 1755–1792. doi:10.1021/cr950073e
Return to citation in text: [1] -
Huisgen, R.; Fulka, C.; Kalwinsch, I.; Xingya, L.; Mloston, G.; Moran, J. R.; Pröbstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701
Return to citation in text: [1] [2] -
Mlostoń, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532.
Return to citation in text: [1] [2] -
Mlostoń, G.; Heimgartner, H. In 1,3-Dipolar cycloaddition chemistry towards heterocycles and natural products; Padwa, A.; Pearson, W. H., Eds.; Wiley: New York,, 2002; pp 315–360. doi:10.1002/0471221902.ch5
Return to citation in text: [1] [2] -
Huisgen, R.; Mlostoń, G.; Giera, H.; Langhals, E. Tetrahedron 2002, 58, 507–519. doi:10.1016/s0040-4020(01)01147-4
Return to citation in text: [1] [2] -
Huisgen, R.; Mloston, G.; Langhals, E. J. Org. Chem. 1986, 51, 4085–4087. doi:10.1021/jo00371a039
Return to citation in text: [1] [2] -
Huisgen, R.; Mlostoń, G.; Langhals, E.; Oshima, T. Helv. Chim. Acta 2002, 85, 2668–2685. doi:10.1002/1522-2675(200209)85:9<2668::aid-hlca2668>3.0.co;2-3
Return to citation in text: [1] [2] -
Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293–14296. doi:10.1002/anie.201708346
Return to citation in text: [1] [2] [3] -
Augustin, A. U.; Merz, J. L.; Jones, P. G.; Mlostoń, G.; Werz, D. B. Org. Lett. 2019, 21, 9405–9409. doi:10.1021/acs.orglett.9b03623
Return to citation in text: [1] [2] -
Kowalski, K.; Karpowicz, R.; Mlostoń, G.; Miesel, D.; Hildebrandt, A.; Lang, H.; Czerwieniec, R.; Therrien, B. Dalton Trans. 2015, 44, 6268–6276. doi:10.1039/c5dt00246j
Return to citation in text: [1] -
Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Eur. J. Org. Chem. 2017, 950–954. doi:10.1002/ejoc.201601307
Return to citation in text: [1] -
Mlostoń, G.; Hamera-Fałdyga, R.; Linden, A.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 1421–1427. doi:10.3762/bjoc.12.136
Return to citation in text: [1] -
Mlostoń, G.; Hamera-Fałdyga, R.; Jeske, M.; Godziszewska, M.; Urbaniak, K.; Heimgartner, H. J. Sulfur Chem. 2018, 39, 47–63. doi:10.1080/17415993.2017.1363206
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Zimmer, R.; Reissig, H.-U.; Heimgartner, H. ChemistrySelect 2018, 3, 11724–11728. doi:10.1002/slct.201803096
Return to citation in text: [1] -
Astruc, D. Eur. J. Inorg. Chem. 2017, 6–29. doi:10.1002/ejic.201600983
Return to citation in text: [1] -
Heinze, K.; Lang, H. Organometallics 2013, 32, 5623–5625. doi:10.1021/om400962w
Return to citation in text: [1] -
Mlostoń, G.; Hamera-Fałdyga, R.; Celeda, M.; Heimgartner, H. Org. Biomol. Chem. 2018, 16, 4350–4356. doi:10.1039/c8ob01022f
Return to citation in text: [1] -
Mloston, G.; Hamera-Fałdyga, R.; Heimgartner, H. J. Sulfur Chem. 2018, 38, 267–278. doi:10.1080/17415993.2017.1415339
Return to citation in text: [1] -
Petzold, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2019, 58, 6225–6229. doi:10.1002/anie.201814409
Return to citation in text: [1] -
Augustin, A. U.; Busse, M.; Jones, P. G.; Werz, D. B. Org. Lett. 2018, 20, 820–823. doi:10.1021/acs.orglett.7b03961
Return to citation in text: [1] -
Cai, Z.-J.; Liu, C.-X.; Wang, Q.; Gu, Q.; You, S.-L. Nat. Commun. 2019, 10, 4168. doi:10.1038/s41467-019-12181-x
Return to citation in text: [1] -
Gröber, S.; Matczak, P.; Domagała, S.; Weisheit, T.; Görls, H.; Düver, A.; Mlostoń, G.; Weigand, W. Materials 2019, 12, 2832. doi:10.3390/ma12172832
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n
Return to citation in text: [1] -
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886
Return to citation in text: [1] -
Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/c3cs60238a
Return to citation in text: [1] -
Werz, D. B.; Biju, A. T. Angew. Chem., Int. Ed. 2020, 59, 3385–3398. doi:10.1002/anie.201909213
Return to citation in text: [1] -
Kreft, A.; Lücht, A.; Grunenberg, J.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2019, 58, 1955–1959. doi:10.1002/anie.201812880
Return to citation in text: [1] [2] -
Mlostoń, G.; Hamera, R.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2125–2133. doi:10.1080/10426507.2015.1071817
Return to citation in text: [1] -
Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71
Return to citation in text: [1] -
Rodriquez-Cendejas, C. D.; Liebeskind, L. S.; Peña-Cabrera, E. ARKIVOC 2005, No. vi, 250–265. doi:10.3998/ark.5550190.0006.621
Return to citation in text: [1]
| 24. | Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n |
| 25. | Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886 |
| 26. | Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/c3cs60238a |
| 27. | Werz, D. B.; Biju, A. T. Angew. Chem., Int. Ed. 2020, 59, 3385–3398. doi:10.1002/anie.201909213 |
| 28. | Kreft, A.; Lücht, A.; Grunenberg, J.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2019, 58, 1955–1959. doi:10.1002/anie.201812880 |
| 22. | Cai, Z.-J.; Liu, C.-X.; Wang, Q.; Gu, Q.; You, S.-L. Nat. Commun. 2019, 10, 4168. doi:10.1038/s41467-019-12181-x |
| 23. | Gröber, S.; Matczak, P.; Domagała, S.; Weisheit, T.; Görls, H.; Düver, A.; Mlostoń, G.; Weigand, W. Materials 2019, 12, 2832. doi:10.3390/ma12172832 |
| 1. | Benetti, S.; De Risi, C.; Pollini, G. P.; Zanirato, V. Chem. Rev. 2012, 112, 2129–2163. doi:10.1021/cr200298b |
| 2. | De Clercq, P. J. Chem. Rev. 1997, 97, 1755–1792. doi:10.1021/cr950073e |
| 8. | Huisgen, R.; Mlostoń, G.; Langhals, E.; Oshima, T. Helv. Chim. Acta 2002, 85, 2668–2685. doi:10.1002/1522-2675(200209)85:9<2668::aid-hlca2668>3.0.co;2-3 |
| 6. | Huisgen, R.; Mlostoń, G.; Giera, H.; Langhals, E. Tetrahedron 2002, 58, 507–519. doi:10.1016/s0040-4020(01)01147-4 |
| 7. | Huisgen, R.; Mloston, G.; Langhals, E. J. Org. Chem. 1986, 51, 4085–4087. doi:10.1021/jo00371a039 |
| 8. | Huisgen, R.; Mlostoń, G.; Langhals, E.; Oshima, T. Helv. Chim. Acta 2002, 85, 2668–2685. doi:10.1002/1522-2675(200209)85:9<2668::aid-hlca2668>3.0.co;2-3 |
| 7. | Huisgen, R.; Mloston, G.; Langhals, E. J. Org. Chem. 1986, 51, 4085–4087. doi:10.1021/jo00371a039 |
| 21. | Augustin, A. U.; Busse, M.; Jones, P. G.; Werz, D. B. Org. Lett. 2018, 20, 820–823. doi:10.1021/acs.orglett.7b03961 |
| 6. | Huisgen, R.; Mlostoń, G.; Giera, H.; Langhals, E. Tetrahedron 2002, 58, 507–519. doi:10.1016/s0040-4020(01)01147-4 |
| 3. | Huisgen, R.; Fulka, C.; Kalwinsch, I.; Xingya, L.; Mloston, G.; Moran, J. R.; Pröbstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701 |
| 4. | Mlostoń, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532. |
| 5. | Mlostoń, G.; Heimgartner, H. In 1,3-Dipolar cycloaddition chemistry towards heterocycles and natural products; Padwa, A.; Pearson, W. H., Eds.; Wiley: New York,, 2002; pp 315–360. doi:10.1002/0471221902.ch5 |
| 3. | Huisgen, R.; Fulka, C.; Kalwinsch, I.; Xingya, L.; Mloston, G.; Moran, J. R.; Pröbstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701 |
| 4. | Mlostoń, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532. |
| 5. | Mlostoń, G.; Heimgartner, H. In 1,3-Dipolar cycloaddition chemistry towards heterocycles and natural products; Padwa, A.; Pearson, W. H., Eds.; Wiley: New York,, 2002; pp 315–360. doi:10.1002/0471221902.ch5 |
| 9. | Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293–14296. doi:10.1002/anie.201708346 |
| 10. | Augustin, A. U.; Merz, J. L.; Jones, P. G.; Mlostoń, G.; Werz, D. B. Org. Lett. 2019, 21, 9405–9409. doi:10.1021/acs.orglett.9b03623 |
| 20. | Petzold, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2019, 58, 6225–6229. doi:10.1002/anie.201814409 |
| 16. | Astruc, D. Eur. J. Inorg. Chem. 2017, 6–29. doi:10.1002/ejic.201600983 |
| 17. | Heinze, K.; Lang, H. Organometallics 2013, 32, 5623–5625. doi:10.1021/om400962w |
| 19. | Mloston, G.; Hamera-Fałdyga, R.; Heimgartner, H. J. Sulfur Chem. 2018, 38, 267–278. doi:10.1080/17415993.2017.1415339 |
| 30. | Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71 |
| 11. | Kowalski, K.; Karpowicz, R.; Mlostoń, G.; Miesel, D.; Hildebrandt, A.; Lang, H.; Czerwieniec, R.; Therrien, B. Dalton Trans. 2015, 44, 6268–6276. doi:10.1039/c5dt00246j |
| 12. | Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Eur. J. Org. Chem. 2017, 950–954. doi:10.1002/ejoc.201601307 |
| 13. | Mlostoń, G.; Hamera-Fałdyga, R.; Linden, A.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 1421–1427. doi:10.3762/bjoc.12.136 |
| 14. | Mlostoń, G.; Hamera-Fałdyga, R.; Jeske, M.; Godziszewska, M.; Urbaniak, K.; Heimgartner, H. J. Sulfur Chem. 2018, 39, 47–63. doi:10.1080/17415993.2017.1363206 |
| 15. | Mlostoń, G.; Urbaniak, K.; Zimmer, R.; Reissig, H.-U.; Heimgartner, H. ChemistrySelect 2018, 3, 11724–11728. doi:10.1002/slct.201803096 |
| 9. | Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293–14296. doi:10.1002/anie.201708346 |
| 31. | Rodriquez-Cendejas, C. D.; Liebeskind, L. S.; Peña-Cabrera, E. ARKIVOC 2005, No. vi, 250–265. doi:10.3998/ark.5550190.0006.621 |
| 10. | Augustin, A. U.; Merz, J. L.; Jones, P. G.; Mlostoń, G.; Werz, D. B. Org. Lett. 2019, 21, 9405–9409. doi:10.1021/acs.orglett.9b03623 |
| 28. | Kreft, A.; Lücht, A.; Grunenberg, J.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2019, 58, 1955–1959. doi:10.1002/anie.201812880 |
| 9. | Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293–14296. doi:10.1002/anie.201708346 |
| 18. | Mlostoń, G.; Hamera-Fałdyga, R.; Celeda, M.; Heimgartner, H. Org. Biomol. Chem. 2018, 16, 4350–4356. doi:10.1039/c8ob01022f |
| 29. | Mlostoń, G.; Hamera, R.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2125–2133. doi:10.1080/10426507.2015.1071817 |
© 2020 Mlostoń et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)