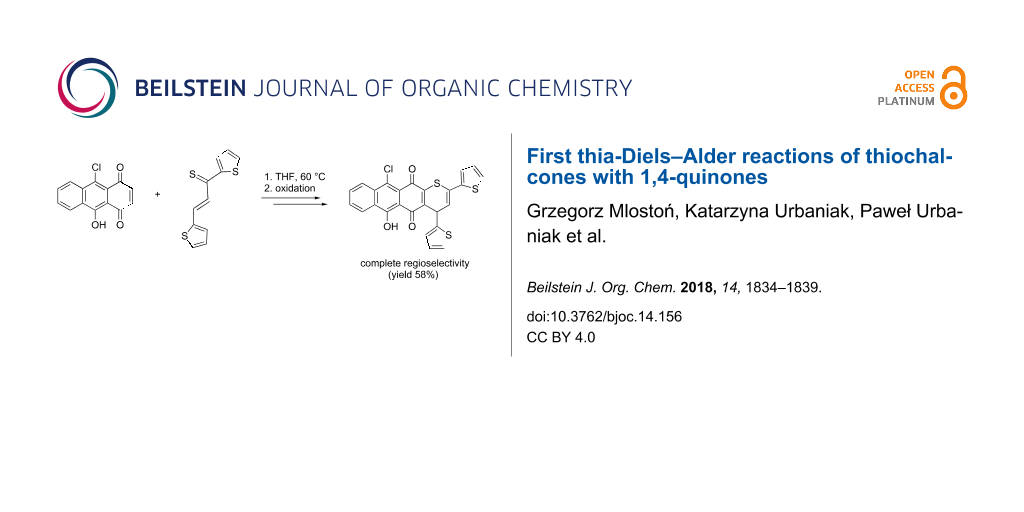
Abstract
Aryl and hetaryl thiochalcones react smoothly with 1,4-quinones in THF solution at 60 °C yielding the corresponding fused 4H-thiopyrans after spontaneous dehydrogenation of the initially formed [4 + 2] cycloadducts. In general, the yields of the isolated products were high. With 5-chloro-10-hydroxy-1,4-anthraquinone, the thia-Diels–Alder reaction occurred with complete regioselectivity. In the case of the reaction of vitamin K3 (menadione) with diphenylthiochalcone, the initial cycloadduct was isolated in 37% yield.

Graphical Abstract
Introduction
Hetero-Diels–Alder reactions are considered to be a powerful methodology widely explored for the synthesis of six-membered heterocycles [1,2] with numerous applications for the construction of complex molecules including naturally occurring products [3,4], drugs [5,6], agrochemicals [7], etc. In addition, asymmetric hetero-Diels–Alder reactions are of current interest [8-10]. Whereas aza- and oxa-Diels–Alder reactions are frequently applied, thia-Diels–Alder reactions are rarely reported. However, aryl and hetaryl thioketones are known to react as ‘superdienophiles’, thereby yielding the corresponding 3,6-dihydro-2H-thiopyrans [11-14].
Despite the fact that thiochalcones exist in solution as mixtures of dimers [15,16], they enter into cycloaddition reactions not only as heterodienes [17-19], but also as heterodipolarophiles [15]. In two recent publications we reported new thia-Diels–Alder reactions of aryl, hetaryl and ferrocenyl-substituted thiochalcones with acetylenic dienophiles, which lead to the corresponding 4H-thiopyrans in a regioselective manner [16,20].
In cycloaddition chemistry, 1,4-quinones are applied widely both as dipolarophiles and dienophiles. In the case of [3 + 2] cycloadditions, reactions can occur chemoselectively with either the C=O or the C=C unit [21-23]. On the other hand, reactions with diverse 1,3-dienes and heterodienes generally occur at the C=C group [24-26]. To the best of our knowledge, thia-Diels–Alder reactions of 1,4-quinones with thiochalcones have not yet been reported.
In the present study, thia-Diels–Alder reactions of aryl and hetaryl thiochalcones with selected 1,4-quinones, such as 1,4-benzoquinone, 1,4-naphthoquinone, and 1,4-anthraquinone were investigated as a route to novel 4H-thiochromene-5,8-dione derivatives.
Results and Discussion
Aryl and hetary lthiochalcones 1a–d are easily obtained by treatment of the corresponding chalcones with Lawesson′s reagent in THF solution [15]. Along with the commercially available 1,4-benzoquinone (2a) and 1,4-naphthoquinone (2b), 1,4-anthraquinone (2c) and 5-chloro-10-hydroxy-1,4-anthraquinone (2d) were prepared from quinizarine according to known procedures [27,28].
First experiments were performed with 2b and thiochalcones 1a–d in THF solutions at 60 °C by starting with equimolar amounts of substrates. After 2 h, completion of the reaction was confirmed by TLC, and, after typical workup, products 4 were obtained as colored solids in high yields (Scheme 1).
![[1860-5397-14-156-i1]](/bjoc/content/inline/1860-5397-14-156-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of aryl/hetarylthiochalcones 1a–d with 1,4-naphthoquinone (2b).
Scheme 1: Reactions of aryl/hetarylthiochalcones 1a–d with 1,4-naphthoquinone (2b).
The 1H NMR analysis revealed that the initially formed [4 + 2] cycloadducts 3 underwent spontaneous oxidation under the reaction conditions. The structures of type 4 were confirmed by spectroscopic methods and elemental analysis. For example, the 1H NMR spectrum of 4a showed two doublets at 5.46 and 6.35 ppm with J = 6.4 Hz attributed to H–C(4) and H–C(3), respectively. In the 13C NMR spectrum, the sp3-C(4) atom absorbs at 40.9 ppm and the signals for the two C=O groups were found at 180.8 and 181.4 ppm, respectively.
In the second series of experiments, thiochalcones 1a–d were subjected to reaction with the symmetrical 1,4-anthraquinone (2c) and its non-symmetrically substituted derivative 2d. In analogy to the reactions with 2b, the expected products 4e–h were isolated in all cases as stable colored solids in high yields (Scheme 2).
![[1860-5397-14-156-i2]](/bjoc/content/inline/1860-5397-14-156-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reactions of thiochalcones 1a–d with 1,4-anthraquinones 2c and 2d.
Scheme 2: Reactions of thiochalcones 1a–d with 1,4-anthraquinones 2c and 2d.
The reactions of thiochalcones 1a–d with 2d require a brief discussion. In these cases, the formation of two regioisomeric cycloadducts could be expected, but the 1H NMR analysis of the crude products showed that only one product was present in each case and, therefore, the studied [4 + 2] cycloaddition reactions occurred with complete regioselectivity. Based on the assumption that the nucleophilic S-atom of the thiochalcone attacks the more electrophilic C-atom, we postulate that compounds 4i–l and not their isomers 5a–d are formed in these reactions. This assumption is supported by the intramolecular H-bonding, which enhances the electrophilicity of C(3) in the dienophile 2d. Finally, the structure of 4k was established by X-ray crystallography (Figure 1). Remarkably, the presence of OH and Cl substituents in products 4i–l results in a bathochromic shift of UV–vis absorptions.
![[1860-5397-14-156-1]](/bjoc/content/figures/1860-5397-14-156-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP plot [29] of the molecular structure of 4k showing the major conformation of the disordered thiophene ring (50% probability ellipsoids; arbitrary numbering of the atoms).
Figure 1: ORTEP plot [29] of the molecular structure of 4k showing the major conformation of the disordered thiop...
The crystal structure of 4k is that of the regioisomer proposed on the basis of reactivity considerations. Since the space group is centrosymmetric, the compound in the crystal is racemic. The S-atom of the thiophene ring is disordered over two unequally occupied positions as a result of slight but opposite directions of envelope puckering of the ring. The hydroxy group forms an intramolecular hydrogen bond with the adjacent quinoid O-atom.
In addition to anthraquinones 2b and 2c, the simple 1,4-benzoquinone (2a) was also tested in the reaction with thiochalcones 1. The reactions performed with 1a and 1b delivered the expected 4H-thiochromene-5,8-diones 4m,n, which were isolated in good yields using flash chromatography, but underwent decomposition under ambient conditions, and none of them could be obtained in analytically pure form (Figure 2).
![[1860-5397-14-156-2]](/bjoc/content/figures/1860-5397-14-156-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Products of the reactions of thiochalcones 1a and 1b with 1,4-benzoquinone (2a) and of 1a with menadione (2e).
Figure 2: Products of the reactions of thiochalcones 1a and 1b with 1,4-benzoquinone (2a) and of 1a with mena...
Vitamin K3 (2e, menadione), which is an important representative of 1,4-naphtoquinones, was also involved in the present study and tested in the reaction with diphenylthiochalcone (1a). In that case, however, a longer reaction time was required, and substantial amounts of decomposition products were formed. The chromatographic workup led to a brownish fraction, containing a single product, which was identified as the [4 + 2] cycloadduct 6 (Figure 2). Its structure was elucidated by 1H NMR spectroscopy, which evidenced the presence of two doublets at 3.76 (J = 4.8 Hz) and 6.50 ppm (J = 4.3 Hz) and a triplet-like signal at 4.18 ppm with J ≈ 4.5 Hz. These signals were attributed to HC(4a), HC(3) and HC(4), respectively. All attempts to prepare an analytically pure sample were unsuccessful. In contrast to all other initially formed cycloadducts, further oxidative conversion to the quinone structure was not possible in this case.
Conclusion
The present study demonstrates that 1,4-quinones are prone dienophiles in reactions with aryl/hetarylthiochalcones. These thia-Diels–Alder reactions led to a new class of 4H-thiopyran derivatives. The presence of a quinone system is an important structural aspect of this class as it is common in many naturally occurring compounds, e.g., dyes such as alizarin, carminic acid, and isoprenoid dyes, as well as drugs such as doxorubicin. In addition, the presence of thiophenyl substituents may be of importance for their applications in materials chemistry.
Experimental
General information: Solvents and chemicals were purchased and used as received without further purification. Products were purified by standard column chromatography on silica gel (230–400 mesh, Merck). Unless stated otherwise, yields refer to analytically pure samples. NMR spectra were recorded with a Bruker Avance III 600 MHz instrument (1H NMR: 600 MHz; 13C NMR: 151 MHz). Chemical shifts are reported relative to solvent residual peaks (1H NMR: δ = 7.26 ppm [CHCl3]; 13C NMR: δ = 77.0 ppm [CDCl3]). IR spectra were registered with a FTIR NEXUS spectrometer (as film or KBr pellets). UV–vis spectra were recorded using a UV–vis JASCO V-630 spectrophotometer. Melting points were determined in capillaries with a Stuart SMP30 apparatus with automatic temperature monitoring.
Starting materials: 1,4-Benzoquinone (2a) and 1,4-naphthoquinone (2b) were commercial reagents and used without further purification. 1,4-Anthraquinone (2c) was prepared from commercial quinizarine by treatment with sodium borohydride according to a literature procedure [27]. 5-Chloro-10-hydroxy-1,4-anthraquinone (2d) was obtained by treatment of quinizarine with thionyl chloride according to the protocol described in [27]. Thiochalcones 1a–d were prepared according to our protocol reported in an earlier publication [16].
General procedure: A solution of 1 mmol of the corresponding thiochalcone 1 and 1 mmol of the 1,4-quinone 2 in 1 mL of dry THF was placed in a thick-walled glass tube, which was closed with a screw cap. The mixtures were heated at 60 °C for 2 h (for 4a–n) or 48 h (for 6). In the last case, progress of the reaction was monitored by TLC, and an additional amount of thiochalcone 1a was added in small portions until menadione (2e) was completely consumed. The solvent was evaporated in vacuo and the crude mixtures were analyzed by 1H NMR first and subsequently purified by flash chromatography using dichloromethane (for 4a–n) or a mixture of petroleum ether and dichloromethane 1:1 (for 6) as the eluents. For products 4a–l, analytically pure samples were obtained by crystallization from petroleum ether with a small amount of dichloromethane. All attempts to obtain analytically pure samples for 4m, 4n and 6 were unsuccessful and the purification procedure led to formation of some decomposition products.
2,4-Diphenyl-4H-benzo[g]thiochromene-5,10-dione (4a): Yield: 340 mg (89%). Red-orange crystals; mp 165 °C (dec.); 1H NMR δ 5.46 (d, JH,H = 6.4 Hz, Ph-CH), 6.35 (d, JH,H = 6.4 Hz, C=CH), 7.24–7.25 (m, 1CHarom), 7.32–7.34 (m, 2CHarom), 7.41–7.43 (m, 3CHarom), 7.52–7.54 (m, 2CHarom), 7.61–7.62 (m, 2CHarom), 7.70–7.74 (m, 2CHarom), 8.10 (dd, JH,H = 7.4 Hz, JH,H = 1.2 Hz, 1CHarom), 8.13 (dd, JH,H = 7.4 Hz, JH,H = 1.2 Hz, 1CHarom) ppm; 13C NMR δ 40.9 (Ph-CH), 121.0, 126.6, 126.7, 127.0, 127.5, 128.4, 128.8, 128.9, 129.0, 133.4, 134.3 (14CHarom, C=CH), 131.7, 132.0, 132.2, 136.4, 137.4, 142.3, 144.54 (6Carom, C=CH), 180.8, 181.4 (2C=O) ppm; IR ν: 3060 (w), 3028 (w), 1653 (vs, 2C=O), 1591 (s), 1562 (m), 1489 (m), 1451 (m), 1334 (m), 1288 (vs), 1152 (m), 1106 (w), 1030 (m), 913 (m), 834 (m), 710 (s), 694 (s) cm−1; UV–vis (CH2Cl2) λmax/nm (lg ε): 243 (4.49), 332 (3.57), 478 (328); anal. calcd for C25H16O2S (380.46): C, 78.92; H, 4.24; S, 8.43; found: C, 78.87; H, 4.24; S, 8.37.
2-Phenyl-4-(thiophen-2-yl)-4H-benzo[g]thiochromene-5,10-dione (4b): Yield: 365 mg (94%). Red-orange crystals; mp 168 °C (dec.); 1H NMR δ 5.80 (d, JH,H = 6.7 Hz, thiophen-2-yl-CH), 6.40 (d, JH,H = 6.7 Hz, C=CH), 6.95 (dd, JH,H = 5.0 Hz, JH,H = 3.7 Hz, 1CHarom), 7.06 (d, JH,H = 3.5 Hz, 1CHarom), 7.19 (dd, JH,H = 5.0 Hz, JH,H = 1.0 Hz, 1CHarom), 7.42–7.46 (m, 3CHarom), 7.63–7.65 (m, 2CHarom), 7.71–7.78 (m, 2CHarom), 8.13 (dd, JH,H = 7.6 Hz, JH,H = 1.1 Hz, 1CHarom), 8.18 (dd, JH,H = 7.6 Hz, JH,H = 1.1 Hz, 1CHarom) ppm; 13C NMR δ 35.2 (thiophen-2-yl-CH), 119.9, 125.3, 125.6, 126.7, 127.0, 127.1, 127.2, 128.8, 129.2, 133.5, 134.4 (12CHarom, C=CH), 128.5, 131.8, 132.1, 135.3, 137.2, 144.2, 144.3 (6Carom, C=CH), 180.6, 181.4 (2C=O) ppm; IR ν: 3075 (w), 3031 (w), 1652 (vs, 2C=O), 1589 (s), 1571 (s), 1489 (m), 1442 (m), 1420 (w), 1331 (m), 1289 (vs), 1220 (m), 1151 (m), 1103 (w), 1033 (w), 919 (w), 831 (m), 767 (m), 704 (s), 695 (s) cm−1; UV–vis (CH2Cl2) λmax/nm (lg ε): 244 (4.50), 338 (3.59), 463 (3.34); anal. calcd for C23H14O2S2 (386.49): C, 71.48; H, 3.65; S, 16.59; found: C, 71.48; H, 3.65; S 16.59.
4-Phenyl-2-(thiophen-2-yl)-4H-benzo[g]thiochromene-5,10-dione (4c): Yield: 355 mg (92%). Red-orange crystals; mp 158 °C (dec.); 1H NMR δ 5.42 (d, JH,H = 6.5 Hz, Ph-CH), 6.41 (d, JH,H = 6.5 Hz, C=CH), 7.01 (dd, JH,H = 5.2 Hz, JH,H = 3.4 Hz, 1CHarom), 7.23–7.25 (m, 1CHarom), 7.28–7.33 (m, 3CHarom), 7.36 (dd, JH,H = 3.4 Hz, JH,H = 1.0 Hz, 1CHarom), 7.50–7.51 (m, 2CHarom), 7.69–7.75 (m, 2CHarom), 8.09 (dd, JH,H = 7.4 Hz, JH,H = 1.3 Hz, 1CHarom), 8.14 (dd, JH,H = 7.4 Hz, JH,H = 1.2 Hz, 1CHarom) ppm; 13C NMR δ 40.6 (Ph-CH), 119.9, 125.3, 125.7, 126.6, 127.0, 127.6, 127.7, 128.4, 129.0, 133.4, 134.2 (12CHarom, C=CH), 125.9, 131.7, 132.1, 136.8, 140.3, 141.8, 143.9 (6Carom, C=CH), 180.6, 181.3 (2C=O) ppm; IR ν: 3085 (w), 3063 (w), 1652 (vs, 2C=O), 1590 (s), 1573 (s), 1490 (m), 1453 (m), 1347 (m), 1335 (m), 1289 (vs), 1230 (m), 1150 (m), 1111 (w), 1074 (m), 839 (m), 880 (m), 815 (m), 710 (s), 696 (s) cm−1; UV–vis (CH2Cl2) λmax/nm (lg ε): 269 (4.42), 473 (3.12); anal. calcd for C23H14O2S2 (386.49): C, 71.48; H, 3.65; S, 16.59; found: C, 71.48; H, 3.70; S, 16.58.
11-Chloro-6-hydroxy-4-phenyl-2-(thiophen-2-yl)-4H-naphtho[2,3-g]thiochromene-5,12-dione (4k): Yield: 285 mg (59%). Dark red crystals; mp 162 °C (dec.); 1H NMR δ 5.78 (d, JH,H = 6.7 Hz, thiophen-2-yl-CH), 6.48 (d, JH,H = 6.8 Hz, C=CH), 6.96 (dd, JH,H = 4.9 Hz, JH,H = 3.8 Hz, 1CHarom), 7.01–7.11 (m, 2CHarom), 7.21 (dd, JH,H = 4.9 Hz, JH,H = 1.0 Hz, 1CHarom), 7.35 (dd, JH,H = 4.9 Hz, JH,H = 1.0 Hz, 1CHarom), 7.40 (dd, JH,H = 4.9 Hz, JH,H = 1.0 Hz, 1CHarom), 7.76–7.80 (m, 1CHarom), 7.87–7.89 (m, 1CHarom), 8.59 (d, JH,H = 8.2 Hz, 1CHarom), 8.63 (d, JH,H = 8.2 Hz, 1CHarom), 14.98 (s, OH) ppm; 13C NMR δ 34.5 (thiophen-2-yl-CH), 118.7, 125.0, 125.5, 125.7, 125.8, 126.2, 127.2, 127.5, 127.8, 130.1, 132.2 (10CHarom, C=CH), 108.8, 122.5, 127.6, 128.5, 134.8, 135.2, 139.9, 143.5, 142.1, 147.4, 162.6 (10Carom, C=CH), 179.0, 184.0 (2C=O) ppm; IR ν: 3022 (w), 2953 (s), 2923 (w), 1655 (s, 2C=O), 1601 (s), 1584 (s), 1565 (s), 1491 (m), 1427 (s), 1397 (s), 1360 (s), 1360 (m), 1333 (m), 1242 (vs), 1170 (m), 902 (s), 852 (m), 832 (m), 758 (m), 699 (vs) cm−1; UV–vis (CH2Cl2) λmax/nm (lg ε): 245 (4.69), 294 (4.32), 487 (3.91); anal. calcd for C27H15ClO3S2 (486.99): C, 66.59; H, 3.10; S, 13.17; found: C, 66.39; H, 3.07; S, 13.06.
(4aSR,10aRS)-10a-Methyl-2,4-diphenyl-4H-benzo[g]thiochromene-5,10-(4aH,10aH)-dione (6): Yield: 145 mg (37%). Yellow-brown viscous oil; 1H NMR δ 1.94 (s, CH3), 3.76 (d, JH,H = 4.8 Hz, 1CH), 4.18 (t, JH,H = 4.5 Hz, 1CH), 6.50 (d, JH,H = 4.3 Hz, 1CH), 7.08–7.12 (m, 1CH), 7.16–7.19 (m, 2CH), 7.30–7.38 (m, 5CH), 7.54–7.57 (m, 2CH), 7.58–7.61 (m, 2CH), 7.79–7.82 (m, 1CH), 7.91–7.93 (m, 1CH) ppm; 13C NMR δ 26.4 (CH3), 42.7 (CH-Ph), 55.9 (Cq), 58.0 (CH-S), 120.0, 126.3, 126.8, 126.9, 127.3, 128.0, 128.5, 128.6, 129.5, 133.8, 134.2 (14CHarom, C=CH), 132.4, 134.4, 134.6, 138.9, 139.6 (4Carom, C=CH), 193.9, 194.2 (2C=O) ppm; IR ν: 3062 (w), 3034 (w), 2957 (w), 1696 (vs) and 1691 (s, 2C=O), 1614 (s), 1489 (m), 1451 (m), 1266 (s), 1013 (m), 979 (m), 761 (s), 692 (s) cm–1; ESIMS (for C26H20O2S): 395 (100, [M − 1]−).
Supporting Information
CCDC-1838975 contains the supplementary crystallographic data for this paper. These data can be obtainded free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/structures.
| Supporting Information File 1: Experimental data for selected compounds 4, details of the crystal structure determination, and copies of 1H and 13C NMR spectra for all products. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Boger, D. L.; Weinreb, S. N., Eds. Hetero Diels–Alder Methodology in Organic Synthesis; Organic Chemistry, A Series of Monographs; Academic Press Inc.: London, 1987; Vol. 47.
Return to citation in text: [1] -
Blond, G.; Gulea, M.; Mamane, V. Curr. Org. Chem. 2016, 20, 2161–2210. doi:10.2174/1385272820666160216000401
Return to citation in text: [1] -
Schmidt, R. R. Acc. Chem. Res. 1986, 19, 250–259. doi:10.1021/ar00128a004
Return to citation in text: [1] -
Heravi, M. M.; Ahmadi, T.; Ghavidel, M.; Heidari, D.; Hamidi, H. RSC Adv. 2015, 5, 101999–102075. doi:10.1039/C5RA17488K
Return to citation in text: [1] -
Gregoritza, M.; Brandl, F. P. Eur. J. Pharm. Biopharm. 2015, 97, 438–453. doi:10.1016/j.ejpb.2015.06.007
Return to citation in text: [1] -
Constantino, A. F.; Francisco, C. S.; Cubides-Roman, D. C.; Lacerda, V., Jr. Curr. Org. Synth. 2018, 15, 84–104. doi:10.2174/1570179414666170517170230
Return to citation in text: [1] -
Funel, J.-A.; Abele, S. Angew. Chem., Int. Ed. 2013, 52, 3822–3863. doi:10.1002/anie.201201636
Return to citation in text: [1] -
Cui, H.-L.; Chouthaiwale, P. V.; Yin, F.; Tanaka, F. Org. Biomol. Chem. 2016, 14, 1777–1783. doi:10.1039/c5ob02393a
Return to citation in text: [1] -
Liu, L.; Kim, H.; Xie, Y.; Farès, C.; Kaib, P. S. J.; Goddard, R.; List, B. J. Am. Chem. Soc. 2017, 139, 13656–13659. doi:10.1021/jacs.7b08357
Return to citation in text: [1] -
Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Eur. J. Org. Chem. 2017, 950–954. doi:10.1002/ejoc.201601307
Return to citation in text: [1] -
Breu, J.; Höcht, P.; Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2861–2873. doi:10.1002/(SICI)1099-0690(199812)1998:12<2861::AID-EJOC2861>3.0.CO;2-C
Return to citation in text: [1] -
Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2875–2883. doi:10.1002/(SICI)1099-0690(199812)1998:12<2875::AID-EJOC2875>3.0.CO;2-N
Return to citation in text: [1] -
Wilker, S.; Erker, G. J. Am. Chem. Soc. 1995, 117, 10922–10930. doi:10.1021/ja00149a015
Return to citation in text: [1] -
Mlostoń, G.; Grzelak, P.; Linden, A.; Heimgartner, H. Chem. Heterocycl. Compd. 2017, 53, 518–525. doi:10.1007/s10593-017-2086-9
Return to citation in text: [1] -
Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. Synthesis 2017, 49, 2129–2137. doi:10.1055/s-0036-1588774
Return to citation in text: [1] [2] [3] -
Mlostoń, G.; Grzelak, P.; Heimgartner, H. J. Sulfur Chem. 2017, 38, 1–10. doi:10.1080/17415993.2016.1230857
Return to citation in text: [1] [2] [3] -
Karakasa, T.; Yamaguchi, H.; Motoki, S. J. Org. Chem. 1980, 45, 927–930. doi:10.1021/jo01294a001
Return to citation in text: [1] -
Saito, T.; Fujii, H.; Hayashibe, S.; Matsushita, T.; Kato, H.; Kobayashi, K. J. Chem. Soc., Perkin Trans. 1 1996, 1897–1903. doi:10.1039/P19960001897
Return to citation in text: [1] -
Hejmanowska, J.; Jasiński, M.; Wojciechowski, J.; Mlostoń, G.; Albrecht, Ł. Chem. Commun. 2017, 53, 11472–11475. doi:10.1039/C7CC06518C
Return to citation in text: [1] -
Mlostoń, G.; Hamera-Fałdyga, R.; Heimgartner, H. J. Sulfur Chem. 2018, 39, 322–331. doi:10.1080/17415993.2018.1427749
Return to citation in text: [1] -
Stegmann, W.; Uebelhart, P.; Heimgartner, H. Helv. Chim. Acta 1983, 66, 2252–2268. doi:10.1002/hlca.19830660736
Return to citation in text: [1] -
Collis, G. E.; Wege, D. Aust. J. Chem. 2003, 56, 903–908. doi:10.1071/CH03060
Return to citation in text: [1] -
Hamadi, N. B.; Msaddek, M. Heterocycl. Commun. 2006, 12, 457–462. doi:10.1515/HC.2006.12.6.457
Return to citation in text: [1] -
Laatsch, H. Z. Naturforsch. 1984, 39b, 244–247.
Return to citation in text: [1] -
Kotera, M.; Lehn, J.-M.; Vigneron, J.-P. Tetrahedron 1995, 51, 1953–1972. doi:10.1016/0040-4020(94)01076-C
Return to citation in text: [1] -
Kotha, S.; Manivannan, E. Indian J. Chem., Sect. B 2002, 41, 808–811.
Return to citation in text: [1] -
Flock, M.; Nieger, M.; Breitmaier, E. Liebigs Ann. Chem. 1993, 451–455. doi:10.1002/jlac.199319930175
Return to citation in text: [1] [2] [3] -
Nor, S. M. M.; Sukari, M. A. H. M.; Azziz, S. S. S. A.; Fah, W. C.; Alimon, H.; Juhan, S. F. Molecules 2013, 18, 8046–8062. doi:10.3390/molecules18078046
Return to citation in text: [1] -
Johnson, C. K. ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, Tennessee, 1976.
Return to citation in text: [1]
| 27. | Flock, M.; Nieger, M.; Breitmaier, E. Liebigs Ann. Chem. 1993, 451–455. doi:10.1002/jlac.199319930175 |
| 16. | Mlostoń, G.; Grzelak, P.; Heimgartner, H. J. Sulfur Chem. 2017, 38, 1–10. doi:10.1080/17415993.2016.1230857 |
| 1. | Boger, D. L.; Weinreb, S. N., Eds. Hetero Diels–Alder Methodology in Organic Synthesis; Organic Chemistry, A Series of Monographs; Academic Press Inc.: London, 1987; Vol. 47. |
| 2. | Blond, G.; Gulea, M.; Mamane, V. Curr. Org. Chem. 2016, 20, 2161–2210. doi:10.2174/1385272820666160216000401 |
| 8. | Cui, H.-L.; Chouthaiwale, P. V.; Yin, F.; Tanaka, F. Org. Biomol. Chem. 2016, 14, 1777–1783. doi:10.1039/c5ob02393a |
| 9. | Liu, L.; Kim, H.; Xie, Y.; Farès, C.; Kaib, P. S. J.; Goddard, R.; List, B. J. Am. Chem. Soc. 2017, 139, 13656–13659. doi:10.1021/jacs.7b08357 |
| 10. | Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Eur. J. Org. Chem. 2017, 950–954. doi:10.1002/ejoc.201601307 |
| 29. | Johnson, C. K. ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, Tennessee, 1976. |
| 7. | Funel, J.-A.; Abele, S. Angew. Chem., Int. Ed. 2013, 52, 3822–3863. doi:10.1002/anie.201201636 |
| 27. | Flock, M.; Nieger, M.; Breitmaier, E. Liebigs Ann. Chem. 1993, 451–455. doi:10.1002/jlac.199319930175 |
| 5. | Gregoritza, M.; Brandl, F. P. Eur. J. Pharm. Biopharm. 2015, 97, 438–453. doi:10.1016/j.ejpb.2015.06.007 |
| 6. | Constantino, A. F.; Francisco, C. S.; Cubides-Roman, D. C.; Lacerda, V., Jr. Curr. Org. Synth. 2018, 15, 84–104. doi:10.2174/1570179414666170517170230 |
| 15. | Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. Synthesis 2017, 49, 2129–2137. doi:10.1055/s-0036-1588774 |
| 3. | Schmidt, R. R. Acc. Chem. Res. 1986, 19, 250–259. doi:10.1021/ar00128a004 |
| 4. | Heravi, M. M.; Ahmadi, T.; Ghavidel, M.; Heidari, D.; Hamidi, H. RSC Adv. 2015, 5, 101999–102075. doi:10.1039/C5RA17488K |
| 27. | Flock, M.; Nieger, M.; Breitmaier, E. Liebigs Ann. Chem. 1993, 451–455. doi:10.1002/jlac.199319930175 |
| 28. | Nor, S. M. M.; Sukari, M. A. H. M.; Azziz, S. S. S. A.; Fah, W. C.; Alimon, H.; Juhan, S. F. Molecules 2013, 18, 8046–8062. doi:10.3390/molecules18078046 |
| 15. | Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. Synthesis 2017, 49, 2129–2137. doi:10.1055/s-0036-1588774 |
| 21. | Stegmann, W.; Uebelhart, P.; Heimgartner, H. Helv. Chim. Acta 1983, 66, 2252–2268. doi:10.1002/hlca.19830660736 |
| 22. | Collis, G. E.; Wege, D. Aust. J. Chem. 2003, 56, 903–908. doi:10.1071/CH03060 |
| 23. | Hamadi, N. B.; Msaddek, M. Heterocycl. Commun. 2006, 12, 457–462. doi:10.1515/HC.2006.12.6.457 |
| 17. | Karakasa, T.; Yamaguchi, H.; Motoki, S. J. Org. Chem. 1980, 45, 927–930. doi:10.1021/jo01294a001 |
| 18. | Saito, T.; Fujii, H.; Hayashibe, S.; Matsushita, T.; Kato, H.; Kobayashi, K. J. Chem. Soc., Perkin Trans. 1 1996, 1897–1903. doi:10.1039/P19960001897 |
| 19. | Hejmanowska, J.; Jasiński, M.; Wojciechowski, J.; Mlostoń, G.; Albrecht, Ł. Chem. Commun. 2017, 53, 11472–11475. doi:10.1039/C7CC06518C |
| 24. | Laatsch, H. Z. Naturforsch. 1984, 39b, 244–247. |
| 25. | Kotera, M.; Lehn, J.-M.; Vigneron, J.-P. Tetrahedron 1995, 51, 1953–1972. doi:10.1016/0040-4020(94)01076-C |
| 26. | Kotha, S.; Manivannan, E. Indian J. Chem., Sect. B 2002, 41, 808–811. |
| 15. | Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. Synthesis 2017, 49, 2129–2137. doi:10.1055/s-0036-1588774 |
| 16. | Mlostoń, G.; Grzelak, P.; Heimgartner, H. J. Sulfur Chem. 2017, 38, 1–10. doi:10.1080/17415993.2016.1230857 |
| 11. | Breu, J.; Höcht, P.; Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2861–2873. doi:10.1002/(SICI)1099-0690(199812)1998:12<2861::AID-EJOC2861>3.0.CO;2-C |
| 12. | Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2875–2883. doi:10.1002/(SICI)1099-0690(199812)1998:12<2875::AID-EJOC2875>3.0.CO;2-N |
| 13. | Wilker, S.; Erker, G. J. Am. Chem. Soc. 1995, 117, 10922–10930. doi:10.1021/ja00149a015 |
| 14. | Mlostoń, G.; Grzelak, P.; Linden, A.; Heimgartner, H. Chem. Heterocycl. Compd. 2017, 53, 518–525. doi:10.1007/s10593-017-2086-9 |
| 16. | Mlostoń, G.; Grzelak, P.; Heimgartner, H. J. Sulfur Chem. 2017, 38, 1–10. doi:10.1080/17415993.2016.1230857 |
| 20. | Mlostoń, G.; Hamera-Fałdyga, R.; Heimgartner, H. J. Sulfur Chem. 2018, 39, 322–331. doi:10.1080/17415993.2018.1427749 |
© 2018 Mlostoń et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)